

Abstract
A number of important cellular processes, such as transcriptional regulation, recombination, replication, repair, and DNA modification, are performed by DNA binding proteins. Of particular interest are transcription factors (TFs), which through their sequence-specific interactions with DNA binding sites, modulate gene expression in a manner required for normal cellular growth and differentiation, and also for response to environmental stimuli. Despite their importance, the DNA binding specificities of most DNA binding proteins still remain unknown, since prior technologies aimed at identifying DNA-protein interactions have been laborious, not highly scalable, or have required limiting biological reagents. Recently a new DNA microarray-based technology, termed protein binding microarrays (PBMs), has been developed that allows rapid, high-throughput characterization of the in vitro DNA binding site sequence specificities of TFs, other DNA binding proteins or synthetic compounds. DNA binding site data from PBMs combined with gene annotation data, comparative sequence analysis, and gene expression profiling, can be used to predict what genes are regulated by a given TF, what the functions are of a given TF and its predicted target genes, and how that TF may fit into the cell’s transcriptional regulatory network.
Keywords: Protein binding microarray, DNA binding site motif, Protein-DNA interactions, DNA binding specificity
1 Introduction
The interactions between transcription factors (TFs) and their DNA binding sites are an integral part of the regulatory networks within cells. These interactions control critical steps in development and responses to environmental stresses, and in humans their dysfunction can contribute to the progression of various diseases. Much progress has been made recently in the accumulation and analysis of mRNA transcript profiles and genome-wide location profiles [1, 2]. However, there is still much to be understood about the transcriptional regulatory networks that govern these gene expression profiles.
One step along the way to developing a parallel methodology for characterizing the sequence specificity of DNA binding domains has been the use of 96-well plates for determining the “binding site signatures” of selected domains displayed on phage. Streptavidin-coated 96-well plates were bound by biotin-tagged sequences degenerate in two of three positions of a triplet binding site. Binding was measured in a semi-quantitative manner using ELISA, and the resulting data for the 12 degenerate sequences were compiled to generate a binding site signature [3]. Another version of this methodology, employing luciferase fusion proteins, has been employed recently [4]. The primarily limitation to such a binding site signature analysis is that one needs to start with a consensus or near-consensus sequence. In addition, a problem with simply compiling data from such partially degenerate sequences, is that it assumes that all the base pairs of the DNA recognition site are acting in a completely independent fashion, when in reality there may be synergistic or destructive interference between different positions of a recognition site [5-8]. Therefore, the resulting binding site signatures may not accurately reflect the actual DNA binding specificity.
The development of DNA microarrays [9, 10] has revolutionized mRNA expression analysis, and along with whole-genome sequencing of microbial and eukaryotic genomes has enabled various functional genomic technologies and systems-oriented analyses. Other array-based technologies include protein microarrays [11] for analysis of protein-protein interactions and interactions between proteins and small molecules, and microarrays of small molecules [12] for analysis of protein-ligand interactions.
DNA microarray-based readout of chromatin immunoprecipitation, also known as ‘ChIP-chip’ or ‘genome-wide location analysis’, is currently the most widely used method for identifying in vivo genomic binding sites for transcription factors (TFs) in a high-throughput manner [13-16]. However, ChIP has some inherent caveats that can make the determination of a TF’s DNA binding specificity difficult [17]. Indeed, some ChIP experiments do not result in significant enrichment of bound fragments in the immunoprecipitated (IPed) sample, and thus do not permit identification of the DNA sites bound in vivo [17, 18]. Another recently developed method that takes advantage of DNA microarrays for the identification of in vivo binding sites of TFs utilizes tethered DNA adenine methyltransferase (Dam) [19]. This approach has been used to identify in vivo binding sites in Drosophila [20] and Arabidopsis [21]. However, it does not permit high resolution mapping of binding sites, because methylation by the tethered Dam can extend over a few kb from the TF binding site [19].
Although in vitro selections have permitted the sampling of a large number of potential DNA binding sequences [22], the resulting sites provide only a partial view of the DNA binding specificity of the protein, as typically only the highest affinity binding sites are retained. It is possible that lower affinity DNA sites are functionally significant in transcriptional regulation of gene expression. For example, lower affinity sites may be responsible for the differences in function of two TFs that bind with high affinity to the same site (such as the Drosophila homeodomain proteins even-skipped and fushi-tarazu, or the murine homeodomain proteins Hmx1 and Nkx2.5) [23, 24]. Although highly quantitative, surface plasmon resonance is not currently scalable to a large number of samples [25].
2 Development of Protein Binding Microarrays
Bulyk and colleagues have recently developed a new, highly parallel in vitro microarray technology, termed protein binding microarrays (PBMs), for high-throughput characterization of the sequence specificities of DNA-protein interactions. In PBM experiments, a DNA binding protein of interest is expressed with an epitope tag; this tag serves a dual purpose: (1) it allows for purification of the expressed DNA binding protein, and (2) the epitope-tagged DNA binding protein is then applied to a dsDNA microarray. The protein-bound microarray is washed gently to remove any nonspecifically bound protein, and then stained with a primary antibody specific for the epitope tag (Fig. 1).
Figure 1. Schema of protein binding microarray experiments.
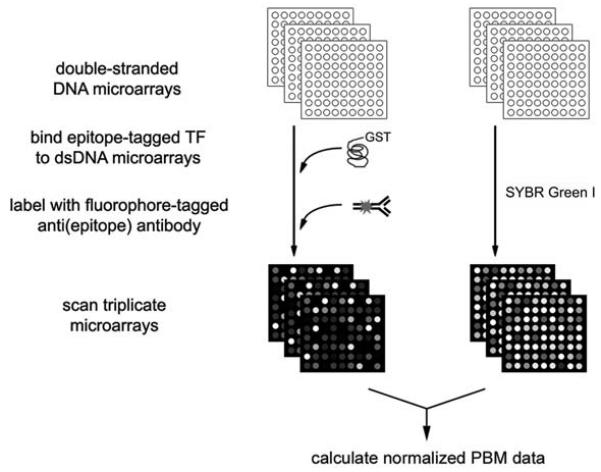
(Reproduced from [26] with permission from Nature Publishing Group.)
Shown in Figure 2 is an example of a PBM in which a GST-tagged yeast TF was bound to a microarray printed with PCR products representing essentially all intergenic regions in the Saccharomyces cerevisiae yeast genome. Through PBM experiments using these whole-genome yeast intergenic microarrays, Bulyk and colleagues identified the DNA binding site sequence specificities of the yeast TFs Abf1, Rap1, and Mig1 (Fig. 3). For Abf1 and Rap1, DNA binding site motifs derived from the PBM data were highly similar to binding site motifs derived from ChIP-chip data [17]. Moreover, analysis of the Mig1 PBM data resulted in a match to the known binding site motif for Mig1 [26], while analysis of the ChIP-chip data [17] did not. In addition to previously identified targets, Abf1, Rap1, and Mig1 bound to numerous putative new target intergenic regions, many of which were upstream of previously uncharacterized open reading frames. Comparative sequence analysis indicated that many of these newly identified sites are highly conserved across five sequenced sensu stricto yeast species and thus are likely to be functional in vivo binding sites that potentially are utilized in a condition-specific manner [26].
Figure 2. Example of a PBM in which a GST-tagged yeast TF was bound to a whole-genome yeast intergenic microarray printed with PCR products.

(a) Whole-genome yeast intergenic microarray bound by Rap1. The fluorescence intensities of the spots are shown in false-color, with white indicating saturated signal intensity, red indicating high signal intensity, green indicating moderate signal intensity, and blue indicating low signal intensity. (b) Zoom-in on a portion of the whole-genome yeast intergenic microarray bound by Rap1. (Reproduced from [26] with permission from Nature Publishing Group.)
Figure 3. DNA binding site motifs determined from PBMs compared to motifs derived from ChIP-chip data and from TRANSFAC.
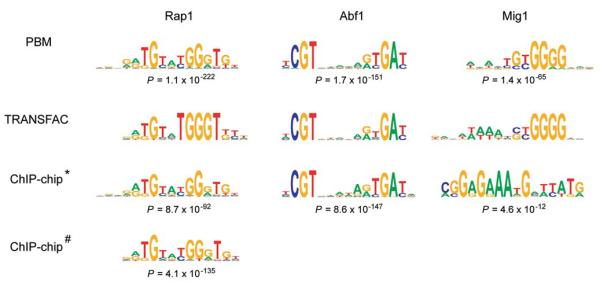
Sequence logos were generated essentially as described previously [53]. Group specificity scores were calculated as described in [49]. “*” indicates Rap1, Abf1, and Mig1 ChIP-chip data from Lee et al. [17], and “#” indicates Rap1 ChIP-chip data from Lieb et al. [13]. Although the Mig1 binding site motif derived from the ChIP-chip data has a statistically significant group specificity score, it is not a match to either the TRANSFAC or PBM Mig1 motif. (Reproduced from [26] with permission from Nature Publishing Group.)
Importantly, the PBM technology allows the determination of the binding site specificities of known or predicted TFs in a single day, starting from the purified TF. Moreover, as with other microarray experiments, the PBM technology is highly scalable, allowing many PBM experiments to be performed in parallel. The PBM experiments themselves are neither time-intensive nor laborious; a single person can perform PBM experiments on a few TFs per day.
The PBM technology has several key advantages over high-throughput in vitro selection (a.k.a. SAGE-SELEX) methodology [27]. First, PBM data are more quantitative, since the signal within each spot on the microarray corresponds to numerous DNA-protein binding events. In addition, non-binding sequences can be identified. Finally, PBMs can provide an extensive, if not complete, reference table of each DNA binding site sequence variant and its relative preference; the number of sequence variants examined is limited only by the number of features on the microarray.
3 Proteins for Examination by PBMs
The Bulyk Lab has successfully used TFs epitope-tagged with GST in PBM experiments using yeast intergenic microarrays (Figs. 2 and 3) [26], and also TFs expressed with the FLAG tag in PBM experiments using microarrays spotted with short synthetic dsDNAs [26]. The size of GST, combined with the use of a polyclonal Alexa488-conjugated anti-GST antibody, likely contributes to the high signal intensities achieved in those PBM experiments. Nevertheless, since GST can self-dimerize [28], other epitope tags may be preferable.
Another group has recently performed PBM experiments using the N-terminal domain of the Drosophila TF Extradenticle directly labeled with the fluorophore Cy3 at a unique cysteine [29]. Yet another group, using directly labeled TFs for binding to dsDNA microarrays, found that the TF Jun C-terminally labeled with Cy5-dC-puromycin was capable of interacting with its protein partner Fos, while Jun labeled at internal lysines did not bind to Fos [30]. Although direct labeling of the protein obviates the need for an antibody staining step, care must be taken to ensure that the incorporated fluorophore does not interfere with DNA binding or any protein-protein interactions necessary for DNA binding. Another possibility for labeling the protein would be to express it as a fusion to a fluorescent molecule, such as green fluorescent protein (GFP).
Overexpression of proteins in E. coli is frequently performed, particularly when fairly large collections of proteins are being examined, because it is an inexpensive expression system that can produce high yields from relatively small cell culture volumes. Even though post-translational modifications may be important for native protein function, many biochemical studies of TFs, and in particular their DNA binding specificities, are performed on proteins expressed in and purified from E. coli. However, certain proteins, particularly those larger than ∼80 kDa, may be difficult to overexpress in E. coli [31]. Alternatively, one could use an expression system that is biologically more similar to the organism whose DNA binding protein is being examined; for example, eukaryotic TFs could be expressed either in vivo in mammalian or insect cells, or in vitro in rabbit reticulocyte lysates. Finally, if a full-length protein is difficult to produce, one can attempt to increase the chances of successful expression and purification by instead expressing just its DNA binding domain plus any necessary protein-protein interaction domain(s).
4 Resources Required for Protein Binding Microarray Experiments
It is important to keep in mind the nature of the protein under examination; if a TF is being examined, then one needs to be sure to use dsDNA microarrays. Bulyk and colleagues have implemented PBMs on two different microarray platforms: robotically printed microarrays, and in situ synthesized oligonucleotide microarrays. Each of these two different platforms has its own unique advantages and disadvantages that include both technical and community accessibility issues, as discussed below.
The robotically printed dsDNA microarrays allow one to ensure that the material spotted onto the glass microarray slides is indeed double-stranded. Synthesis of oligonucleotides for use in PCRs or primer extension reactions can be performed or ordered by almost any lab. Although the cost of synthesis of a large set of oligonucleotides, and in some cases subsequent PCRs, can be great, the sequences present on the microarrays can be determined by the individual investigator and thus microarrays custom-designed for a particular research topic can be made fairly readily. This is accomplished by purifying the dsDNAs, after which there is sufficient material to print thousands of microarrays, which will reduce the long-term per-experiment costs for examining DNA binding proteins in the PBM experiments.
Moreover, most researchers have access to DNA microarraying facilities, if not at their own institution, then through another institution that provides microarraying services for a fee. For production of the whole-genome yeast intergenic DNA microarrays used in PBM experiments [26] as shown in Figure 2, an OmniGrid® 100 microarrayer (Genomic Solutions, Ann Arbor, MI) equipped with Stealth 3 pins (Telechem International, Sunnyvale, CA) was used to spot DNA onto Corning® GAPS II or UltraGAPS 25 × 75 mm amino-silane coated glass slides (Fisher Scientific). Approximately 0.7 nl DNA solution was deposited at each spot. Other slide types can potentially be used. Bulyk and colleagues have found that Corning® GAPS II and UltraGAPS slides result in low slide background in both PBM experiments and staining with SYBR Green I.
Likewise, DNA microarray scanners are readily available in most departments or institutions. A ScanArray 5000 microarray scanner (Perkin Elmer, Boston, MA), which is equipped with a variety of laser and filter sets and permits microarrays to be scanned at a range of different laser power intensities or photomultiplier tube (PMT) gain settings, was used in PBM experiments [26] as shown in Figure 2.
Nevertheless, not all users may have access to robotically printed dsDNA microarrays for use in PBM experiments. In addition, ultimately one might wish to have more features per microarray than typical microarraying robots can print onto standard 1×3 inch glass slides. Thus, Bulyk and colleagues have further developed the PBM technology using a commercially available microarray platform, available from Agilent Technologies, Inc., that already has the capacity to synthesize at least ∼44,000 features per microarray. Note that Agilent microarrays are created by ink-jet synthesis as 60mer single-stranded oligonucleotide microarrays [32], which subsequently need to be double-stranded (see next section) for use in PBMs to examine dsDNA binding proteins. Other platforms offer even higher densities; for example, Nimblegen uses micromirror arrays to synthesize microarrays of long oligonucleotides [33-35] at densities of up to ∼760,000 features per microarray. Alternatively, in-house micromirror array synthesizers could be used to create high-density oligonucleotide microarrays. One group recently synthesized such self-hairpinning high-density oligonucleotide microarrays for use in PBM experiments [29].
5 Design of Double-Stranded DNAs to Use in Protein Binding Microarray Experiments
The key choice to be made in choosing what DNAs to print onto slides for use in PBM experiments is whether one wishes to synthesize a relatively low complexity microarray for directed experimentation on one or a small family of DNA binding proteins [36], or to synthesize a higher complexity microarray [26, 29, 37-39] for examination of a broader set of proteins. In certain situations one might be able to restrict oneself to lower complexity microarrays that would be less expensive to produce if one were manufacturing the microarrays in-house, instead of having them synthesized by a commercial vendor.
Here I describe the design and synthesis of microarrays spotted with PCR products representing essentially all intergenic regions of the S. cerevisiae yeast genome, as the resulting microarrays can be used broadly [26], including for analysis of uncharacterized proteins, and have been described in multiple publications [13, 15, 17, 18]. These microarrays were printed with PCR products ∼60-1500 bp long, covering essentially all noncoding regions of the S. cerevisiae yeast genome [15]. These whole-genome yeast intergenic microarrays were used in PBM experiments in order to identify the DNA binding site specificities of the S. cerevisiae TFs Rap1, Abf1, and Mig1 [26].
Microarrays spotted with coding regions are also expected to aid in identifying the sequence-specific binding properties of DNA binding proteins, despite the fact that it is currently thought that most in vivo regulatory sites will be located in non-protein-coding regions. Since PBM experiments are an in vitro technology, as long as there is sufficient sequence space represented on the DNA microarrays, one can expect to be able to derive a good approximation of the DNA binding site motif from the PBM data. Indeed, it is actually not necessary to utilize microarrays spotted with amplicons representing genomic regions from the same genome as the DNA binding protein of interest, but rather one can use microarrays spotted with a different genome’s sequence. Nevertheless, one could use a genome-specific microarray, such as a promoter microarray [40] or a CpG island microarray [41], as long as such microarrays covered a sufficient amount of binding site sequence space.
The use of microarrays spotted with PCR products has the advantage of covering much sequence space with relatively few spots. However, inherent in those arrays are two key limitations. First, a single intergenic region may be bound once or multiple times at high, medium, or low affinity, depending upon the number and type(s) of candidate binding sites present within a given spotted intergenic region. Currently the measured fluorescence intensity of a spot cannot distinguish between these possibilities. Second, given the variation in probe lengths on the intergenic microarrays, a spot with a single binding site embedded in a long sequence will receive a less significant P-value than a spot with an identical binding site embedded in a shorter sequence [42].
Therefore, one may wish to consider instead using a microarray synthesized with short synthetic dsDNAs [26, 29, 36, 38, 39]. Such dsDNAs can be made from single-stranded oligonucleotides either by primer extension [26, 36-38, 42] or by self-hairpinning [29, 38]. Bulyk and colleagues have performed successful PBMs using microarrays spotted with synthetic dsDNAs ranging from ∼35 to ∼60 base pairs [26, 36, 38]. Another group has performed PBMs using microarrays synthesized in situ with hairpinned 34-mer oligos containing a 14 bp double-stranded hairpin region [29].
Recently, “all k-mer”-style synthetic DNA microarrays have been described [29, 38, 39] for use in PBMs, allowing the analysis of the binding profile for all k-mers up to k=8 to 10 [29] or k=10 to 12 [38, 39] on a single 1×3 inch microarray. Such coverage of binding site sequence space can be accomplished by the synthesis of high-density oligonucleotide arrays [29, 37] or by a compact universal DNA design [38, 39]. Briefly, with the use of high-density arrays, each individual k-mer can be situated on a distinct feature or spot on the array. However, since the number of possible k-mers can become very large for longer motifs, the number of such required spots can become greater than the number of spots that can be manufactured by robotic printing on a single 1×3 inch microarray [10, 32]. Therefore, instead of devoting a unique spot to each k-mer, one can instead employ a compact representation of k-mers [38, 39]. In a compact universal design, for a given double-stranded DNA of length l significantly longer than the motif width k, each spot will contain l-k+1 k-mers, when k-mers are considered in an overlapping fashion [38, 39]. The key difference distinguishing the compact universal microarray technology over prior technologies is that all possible DNA sequence variants can be represented on DNA microarrays in a space- and cost-efficient manner, so that only a minimal number of individual DNA sequences and individual DNA spots need to be synthesized [38, 39]. Importantly, “all k-mer”-style synthetic DNA microarrays, either those with each spot representing a single k-mer or those with a compact universal design, can be applied to the study of any proteins from any genome of interest.
6 Options in Immobilizing Double-Stranded DNAs to the Slide Surface
There are a few options for the immobilization of dsDNAs to the slide surface. Generally, the DNAs either can be attached randomly by UV-crosslinking [43] or they can be end-attached, either by a reactive group at one of the DNA termini [36] or by in situ synthesis of arrays of oligonucleotides [9, 32, 33] that are subsequently double-stranded [37, 38]. In theory, end-attachment should allow the DNAs to not be kinked and to be maximally accessible for interaction with DNA binding proteins. However, gentle UV-crosslinking can work well too (Bulyk and colleagues, unpublished results). Such a UV-crosslinking protocol (i.e., millijoules setting) would need to optimize the two opposing issues of: (1) ensuring that the DNA structure is as unperturbed as possible, i.e., ideally most DNA molecules will have just one crosslink to the slide surface; (2) ensuring that most spotted DNA molecules will be attached to the slides.
All three types of dsDNA immobilization have been used successfully to create microarrays used in PBM experiments [26, 36]. In the first method, the dsDNAs were end-attached to amine-reactive slides through the use of amino-tagged universal primers, as described previously [36]. In the second method, unmodified dsDNAs can be spotted onto various other types of slides, such as polylysine slides (Bulyk and colleagues, unpublished results) or GAPS II or UltraGAPS slides (Corning), and covalently attached to the slides via UV-crosslinking in a Stratalinker (Stratagene) [42]. Finally, in situ synthesized oligonucleotide arrays can be biochemically double-stranded either by primer extension [36-38] or by self-hairpinning [29, 38].
7 DNA Microarray Quality
DNA Purification and Printing Buffer
In their published study using whole-genome yeast intergenic microarrays in PBM experiments to identify the DNA binding site specificities of the S. cerevisiae TFs Rap1, Abf1, and Mig1 [26], Bulyk and colleagues used microarrays printed with PCR products ∼60-1500 bp long, covering essentially all noncoding regions of the S. cerevisiae yeast genome [15]. Those genomic regions were amplified by PCR, and the completed PCR reactions were precipitated with ammonium acetate and isopropanol, washed with 70% ethanol, dried overnight, and resuspended in 3x SSC printing buffer at a DNA concentration of 100 to 500 ng/μl. Alternatively, the PCR products may be filtered with purification plates, such as 96-well MultiScreen® PCR Filter Plates (Millipore, Billerica, MA). The extra filtration provided by the MultiScreen® plates increases the purity of the dsDNA. Other printing buffers or additives such as Sarkosyl or betaine may aid in increasing the spot uniformity and thus improving the morphology of the printed spots. The use of different slide types can also result in different spot morphologies with given printing buffers; care should be taken to ensure that the chosen printing buffer is compatible with the chosen slide type.
Microarray Data Quality Control
Spot uniformity and good spot morphology allow more accurate quantification of spot signal intensities, and ultimately the degree of sequence-specific binding of a given DNA binding protein to each spot. Severe problems with spot morphology frequently can be attributed to the choice of printing buffer and/or post-printing processing. Obviously problematic microarrays can be identified visually (Fig. 4) [42]. More subtle differences in spot quality can be identified through analysis of the quantified signal intensity data. Care should be taken to remove from consideration in subsequent data analysis steps any spots with too low DNA concentration to permit accurate quantification of the spot signal intensities, or spots in which the DNA is spread non-uniformly throughout the pixels. Various additional filtering criteria can be applied later during data analysis to remove from consideration any remaining spots that may be noisy even after removing spots with highly variable pixel signal intensities.
Figure 4. Examples of DNA microarray spot quality.

Identical portions of yeast intergenic microarrays printed onto Corning® GAPS II slides, processed in different ways (see below) before UV-crosslinking, and then stained with SYBR Green I. Images have been false-colored as in Figure 2. Examples of microarrays with poor spot quality are shown in (a) and (b). In both of these cases, the DNA is distributed non-uniformly, with either (a) high concentrations near the centers of spots, or (b) high concentrations along spot perimeters. Both of these microarrays resulted from two separate print runs, from which microarrays were UV-crosslinked without first rehydrating and baking. An example of a good quality microarray is shown in (c). This microarray was rehydrated and then baked before being UV-crosslinked. (Reproduced from [42] with permission from The Humana Press, Inc.)
8 Determination of the DNA Binding Specificities of Proteins with Protein Binding Microarray Experiments
Protein Binding Microarray Experiments
Protocols for performing PBM experiments have been described in detail previously [26, 36, 38, 42]. Briefly, microarrays are first pre-wet and then blocked with a milk solution in order to minimize background. Milk can also be included in both the protein binding and antibody labeling reactions. Other blocking reagents may be suitable depending on the slide substrate on which the microarray was manufactured. Cover slips are typically used for the various microarray incubation steps. The use of LifterSlips™ cover slips helps to ensure a uniform distribution of the reaction mixture over the surface of the microarray. The microarrays are then incubated in a hydration chamber to prevent excessive evaporation of the reaction mixture under the cover slip.
The DNA binding protein of interest, typically at a final concentration in the range of approximately 20 nM, is initially pre-incubated with nonspecific DNA competitors. All incubations are typically performed for 1 hour at room temperature [26, 42], but can be adjusted at the discretion of the user, as can the concentration of the protein in the binding mixture. Any necessary small molecules, such as zinc when examining zinc finger proteins, should be included in all binding and subsequent reactions and washes.
Once the pre-blocking and pre-incubation steps are completed, the microarrays are washed and then the protein binding reaction mixture is applied to the microarrays. During this time, fluorophore-conjugated antibody is pre-incubated in a milk solution. As with all fluorophores, all possible care should be taken to avoid photobleaching during the course of staining the microarrays. Alexa Fluor® 488 conjugated anti-glutathione S-transferase (anti-GST) polyclonal antibody (Molecular Probes) has been used successfully [26]. Other epitope tags and/or other antibodies conjugated with other fluorophores might also be used successfully. Once the protein binding step is completed, the microarrays are washed again, and then the pre-incubated antibody mixture is applied to the microarrays. Once the antibody staining step is completed, the microarrays are washed again, and then immediately spun dry in a table-top centrifuge. The dried microarrays are then ready for scanning using an appropriate laser and filter set (for Alexa Fluor™ 488, argon ion laser (488 nm excitation) and 522 nm emission filter).
Analysis of Protein Binding Microarray Data
Quantification of the Microarray Signal Intensities and Quality Control
In order to capture signal intensities for even very low signal intensity spots, while ensuring that sub-saturation signal intensities are captured for as many spots as possible on the microarray, one can scan the microarrays at a number of different laser power (or PMT gain) settings, and then later integrate the data from these multiple scans [26, 36] using masliner software [44], as described below. The microarray TIF images can be quantified with microarray analysis software such as GenePix Pro (Axon Instruments, Inc.). After image quantification, one typically calculates the background-subtracted median intensities for use in subsequent analysis. One can then calculate the relative signal intensity data over the full series of scans taken at multiple laser power settings [26, 36]. To accomplish this task in a semi-automated fashion, one can use masliner (MicroArray Spot LINEar Regression) software, which combines the linear ranges of multiple scans from different scanner sensitivity settings onto an extended linear scale [44]. In their experiments using whole-genome yeast intergenic microarrays, Bulyk and colleagues observed that the final PBM and SYBR Green I stained microarrays frequently had post-masliner fluorescence intensities that spanned 5 to 6 orders of magnitude [26].
After masliner processing, any low quality spots, such as those with dust flecks, should be removed from further consideration [26]. Next, the data from each of the replicate microarrays are normalized according to total signal intensity, and then within each individual microarray the data are normalized sector-wise, according to their local region on the slide. The data are then normalized again so that the mean spot intensity is the same over all the sectors. After these signal intensity normalizations, a number of additional quality control filtering criteria are applied, including the removal of spots with highly variable pixel signal intensities that could result in noisy PBM data, or spots that do not have highly reproducible data over the replicate microarrays. Additional ad hoc criteria (see [26] and [42]) can also further eliminate potentially noisy data points.
Identification of the Significantly Bound Spots
Once the PBM and SYBR Green I microarray data have been quantified, normalized, and filtered to remove noisy data points, the ratio of the mean PBM signal intensity divided by the mean SYBR Green I signal intensity can be used to identify the significantly bound spots [26]. Alternatively, one could use PBM data not normalized by the amount of DNA [29]. In general, a sequence-specific DNA binding protein is expected to bind preferentially to only a relatively small fraction of possible binding sites. Likewise, the remaining sequence variants are expected to be bound nonspecifically, as all DNA binding proteins are likely to exhibit some weaker affinity for nonspecific DNA binding sites [45]. One way to calculate the significance of binding, or P-value, for a given spot is to calculate its z-score [29, 37]. However, if more than a small percentage of spots are bound sequence-specifically, then another measure of significance, such as a pseudo-z-score [26, 42], may be more appropriate.
Details on how to calculate such pseudo-z-scores have been described previously [26, 42]. Briefly, the log2 of the ratios are LOWESS-normalized and then plotted as a histogram. The resulting distribution is expected to resemble a Gaussian distribution, corresponding to spots bound only nonspecifically, with specifically bound spots localizing to the upper tail of the distribution. The Gaussian-like distribution can then be used to calculate for each spot a pseudo-z-score that represents the probability that the spot belongs to the distribution of nonspecifically bound spots. Specifically, all values less than the mode of the Gaussian-like distribution are fit to a Gaussian function using the Mathematica software package (Wolfram Research, Inc., Champaign, IL). The pseudo-z-score for each spot is then calculated based on z, the number of standard deviations that the spot’s log ratio departs from the mean of the Gaussian distribution [46].
Lastly, the pseudo-z-scores should be corrected for multiple hypothesis testing. Bulyk and colleagues previously employed the Modified Bonferroni Method [6, 47], using an initial α = 0.001. Spots meeting or exceeding this significance threshold were considered significantly ‘bound’ (Fig. 5a). Users may wish to consider spots at less stringent significance thresholds accordingly.
Figure 5. Identification of the DNA binding site motif from the significantly bound spots.
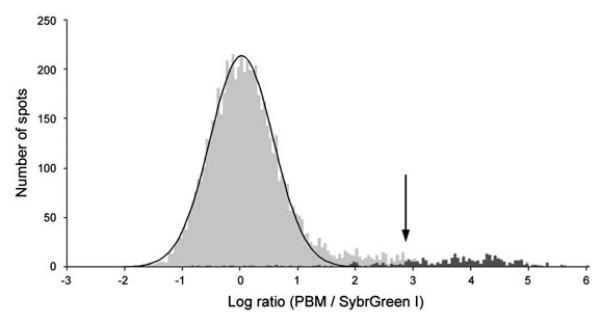
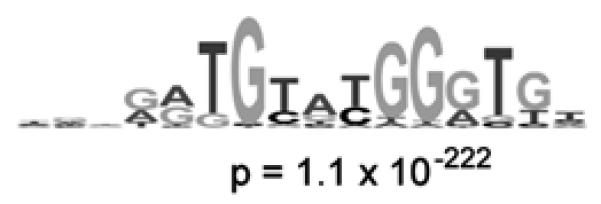
(a) Distribution of ratios of PBM data, normalized by SYBR Green I data, for the yeast TF Rap1 bound to yeast intergenic microarrays. The arrow indicates those spots passing a P-value cutoff of 0.001 after correction for multiple hypothesis testing. Indicated in dark gray are spots with an exact match to a sequence belonging to the PBM-derived binding site motif. (b) Sequence logo [53] of the PBM-derived motif for the yeast TF Rap1. (Reproduced from [26] with permission from Nature Publishing Group.)
Identification of the DNA Binding Site Motif from the Protein Binding Microarray Data
For the set of spots that are bound at the threshold significance level, one can then examine the corresponding set of DNA sequences for the likely DNA binding site motif of the given protein [26]. One might choose to search only the most significantly bound spots in order to minimize consideration of potentially false positive spots that would contribute noise to the motif finding searches. For this set of input sequences, one typically uses a motif finding algorithm, such as BioProspector [48], AlignACE [49, 50], MEME [51], or MDscan [52], in order to identify the DNA binding site motif of the protein. Since the binding site width of a TF is typically between 6 and 18 bp, the motif searches should be performed within this parameter range [26].
Once a motif has been identified by the given motif finder, one then needs to assess the likelihood of it being the DNA binding site motif of the given protein. This can be assessed statistically by calculating its group specificity score [49], which in this context indicates how specific the motif is to the set of bound spots as compared to all the spots on the microarray (for details on how to perform this calculation, and how to select the most likely TF binding site motif from the results, see [42]). In order to assess the statistical significance of the motifs resulting from this analysis, the results are compared against those resulting from analysis of a set of computational negative control sequence sets [26]. PBM-derived motifs with group specificity scores that are more significant than the group specificity scores of the corresponding computational negative control sets are considered to be good candidates for being the DNA binding site motif for the given DNA binding protein (Fig. 5b). Examples of the ranges of group specificity scores for computational negative controls and for actual PBM data for yeast TFs can be found in [26]. A graphical sequence logo [53] for each motif, such as those shown in Figure 3, is often convenient for ease of visual examination of motifs and can be generated readily [54].
9 Prediction of Functional Roles of Transcription Factors from Protein Binding Microarray Data
Cross-Species Conservation of PBM-Derived TF Binding Sites
To find evidence supporting the hypothesis that the S. cerevisiae intergenic regions bound in the in vitro PBM experiments contain functional in vivo binding sites for the given TF, one can map the PBM-derived binding sites in S. cerevisiae to the orthologous positions in the sequence alignments of the S. mikatae, S. kudriavzevii, S. bayanus, and S. paradoxus genomes, which are the four other currently available sequenced yeast genomes of the yeast sensu stricto clade [55, 56]. Significant phylogenetic conservation suggests regulatory function of the PBM-derived TF binding sites.
Functional Category Enrichment of Predicted Target Genes
Analysis of a group of genes for enrichment for a particular functional annotation previously has been used to analyze sets of yeast genes that comprise particular gene expression clusters [57]. Each of the sets of intergenic regions bound in PBMs were examined to determine whether the groups of candidate target genes, located directly downstream of the bound intergenic regions, were over-represented for particular functional groups of genes [49, 57]. The web-based tool FunSpec, with Bonferroni correction, was used for the statistical evaluation of these groups of genes, for groups of over-represented gene and protein categories with respect to existing functional category information from a number of public and published databases [58]. FunSpec uses the hypergeometric distribution to calculate a p-value for functional category enrichment [49, 57]. Among the significantly enriched categories for the target genes derived from the Rap1 PBM data, many are consistent with the known regulatory functions of Rap1 [59], including the MIPS [60] functional classification categories for ribosome biogenesis (p < 1.0 × 10-14), protein synthesis (p < 1.0 × 10-14), structural constituents of the ribosome (p < 1.0 × 10-14), and cell growth and/or maintenance (p = 3.5 × 10-12).
Analysis of Publicly Available Gene Expression Datasets to Identify Conditions in Which a Significant Fraction of PBM-Derived Target Genes are Differentially Expressed
Because different culture conditions often stimulate different cellular responses and coordinate changes in transcriptional regulation, the success of ChIP-chip experiments hinges on choosing those conditions in which the TF is expressed and actively regulating its target genes. PBMs, however, are free of this constraint and can identify TF binding site motifs and putative target genes irrespective of culture conditions. For example, Bulyk & colleagues used 643 publicly available S. cerevisiae gene expression datasets to identify conditions in which significant fractions of Abf1, Rap1, and Mig1 PBM target genes were differentially expressed. The conditions that exhibited the largest number of differentially regulated candidate target genes corresponded well with the known functions of each TF. For example, many Mig1 PBM target genes were down-regulated at least 2.5-fold in glucose and fructose, compared to other carbon sources. These results show that together with expression profiling, PBM analysis can provide insight into the functions of particular TFs and identify conditions in which they are active in vivo. Therefore, analysis of the PBM-derived predicted target genes for conditions in which these genes are co-regulated can further be used to suggest in vivo conditions for TF activity [26].
10 Applications of Protein Binding Microarrays
Two main types of studies have been performed using PBMs. In the first type, a family of closely related zinc finger proteins, including Zif268 (Egr1) and a number of artifical zinc finger proteins that arose from in vitro selections, were examined using a DNA microarray spotted with short synthetic dsDNAs specifically designed to interrogate all possible variants of a subset of the core binding site sequence (specifically, the central 3 bp of the Zif268 binding site) [36]. Because all the proteins were closely related, a DNA microarray could be designed to specifically examine the DNA binding site sequence variants expected to differ among the different proteins. A focused microarray, directed for a specific family of proteins, could be designed for other structural classes as well, as long as a consensus sequence or likely DNA binding site to use a starting point for the family is known [61]. This approach can permit one to minimize microarray manufacture costs by synthesizing only those dsDNAs thought to be most relevant for the family of proteins being examined. Similarly, if differences within a specific class of proteins are of interest, designing a focused microarray can permit one to thoroughly or near thoroughly examine all likely binding site sequence variants of interest.
In the second type of study, a more generic DNA microarray was used to probe the sequence-specific binding of TFs representing a number of different structural classes of DNA binding proteins. The DNA microarrays were spotted with PCR amplicons representing essentially all intergenic regions of the S. cerevisiae yeast genome [26]. Instead of using phage display of DNA binding domains, that study used proteins expressed with an epitope tag. Such fusion proteins can be constructed readily using available genomic clone collections currently under construction for various model organisms as well as for the human genome. Because of the longer lengths of the spotted DNAs, the DNA binding site motifs of the query TFs were identified by motif finding software [26, 42]. Since actual genomic sequences are represented on these arrays, one could also examine binding by multimeric protein complexes [30].
More recently, generic DNA microarrays spotted with short synthetics dsDNAs representing all k-mers have been described [29, 38, 39] for use in PBMs, allowing the analysis of the binding profile for all k-mers up to k=8 to 10 [29] or k=10 to 12 [38, 39] on a single 1×3 inch microarray. Such arrays have been used for the analysis of engineered polyamides [29] and for TFs [29, 38].
11 Outlook
There are predicted to be ∼1850 TFs in the human genome [62], but only a very small fraction of them have well-characterized binding specificities. Likewise, most TFs from various model organisms are of as yet undetermined DNA binding specificities and in general their regulatory functions are not well understood on a genomic scale. The challenge will be to characterize their DNA binding specificities, so that their target genes and potential combinatorial modes of transcriptional regulatory control can be discovered. Continued improvements in the synthesis of high density DNA microarrays will allow an even greater fraction of binding site sequence space to be surveyed.
In the future, PBM technology might potentially be used to derive protein-DNA binding affinities (Kd’s) for all possible DNA binding sites for a given TF. The affinities could either be interpolated from a set of reference DNAs as has been done previously [36], or they could be determined from signal intensities from microarrays probed with a range of protein concentrations, as has recently been described for peptide interactions with protein microarrays [63]. Such binding data would be important for better understanding mechanisms of transcriptional regulation, such as potential competitive binding by TFs [64], and for improved prediction of cis regulatory elements in the genome [61]
Finally, in recent years, a number of efforts have been focused on attempting to predict TF binding sites using structural information on the protein or related protein-DNA complexes. Some of these studies have attempted to determine what “recognition rules” or “recognition code” may exist that stipulate what DNA base pairs are likely to be bound by what amino acids in the context of a particular structural class of DNA binding proteins. These approaches have come from either analysis of databases of well-characterized DNA-protein interactions [65-69], computer modeling [70, 71], or from experiments employing in vitro selection from a randomized library, either of the DNA base pairs or the amino acid residues implicated in sequence-specific binding [3, 72, 73]. However, there is no obvious, simple code like the genetic code, and any recognition rules that might exist are likely to be a quite degenerate “probabilistic code” [5] and highly dependent upon the docking arrangement of the protein with its DNA binding site [74]. Such efforts will be greatly aided by the further development of high-throughput technologies for identifying TF-DNA binding site interactions, so that much larger datasets can be generated for analyses required to decipher any degenerate probabilistic codes or to be used as training sets for developing improved DNA binding site prediction algorithms. Studies like these would allow us to understand better the biophysical determinants of observed protein-DNA interactions, and perhaps to glimpse the related selective pressures that underlie observed evolutionary changes in regulatory proteins and their target DNA binding sites.
Acknowledgements
I thank Michael F. Berger and Tom Volkert for technical assistance. This work was supported in part by National Institutes of Health grants from the National Human Genome Research Institute to M.L.B. (R01 HG002966 and R01 HG003420).
Abbreviations
- PBM
protein binding microarray
- TF
transcription factor
- dsDNA
double-stranded DNA
- ChIP
chromatin immunoprecipitation
References
- 1.Wyrick J, Young R. Curr Opin Genet Dev. 2002;12:130. doi: 10.1016/s0959-437x(02)00277-0. [DOI] [PubMed] [Google Scholar]
- 2.Lockhart DJ, Winzeler EA. Nature. 2000;405:827. doi: 10.1038/35015701. [DOI] [PubMed] [Google Scholar]
- 3.Choo Y, Klug A. Proc Natl Acad Sci USA. 1994;91:11168. doi: 10.1073/pnas.91.23.11168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hallikas O, Palin K, Sinjushina N, et al. Cell. 2006;124:47. doi: 10.1016/j.cell.2005.10.042. [DOI] [PubMed] [Google Scholar]
- 5.Benos P, Bulyk M, Stormo G. Nucleic Acids Res. 2002;30:4442. doi: 10.1093/nar/gkf578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bulyk M, Johnson P, Church G. Nucleic Acids Res. 2002;30:1255. doi: 10.1093/nar/30.5.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee M-L, Bulyk M, Whitmore G, Church G. Biometrics. 2002;58:981. doi: 10.1111/j.0006-341x.2002.00981.x. [DOI] [PubMed] [Google Scholar]
- 8.Man TK, Stormo GD. Nucleic Acids Res. 2001;29:2471. doi: 10.1093/nar/29.12.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pease AC, Solas D, Sullivan EJ, et al. Proc Natl Acad Sci USA. 1994;91:5022. doi: 10.1073/pnas.91.11.5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schena M, Shalon D, Davis RW, Brown PO. Science. 1995;270:467. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 11.MacBeath G, Schreiber SL. Science. 2000;289:1760. doi: 10.1126/science.289.5485.1760. [DOI] [PubMed] [Google Scholar]
- 12.MacBeath G, Koehler AN, Schreiber SL. J. Am. Chem. Soc. 1999;121:7967. [Google Scholar]
- 13.Lieb JD, Liu X, Botstein D, Brown PO. Nat Genet. 2001;28:327. doi: 10.1038/ng569. [DOI] [PubMed] [Google Scholar]
- 14.Iyer VR, Horak CE, Scafe CS, et al. Nature. 2001;409:533. doi: 10.1038/35054095. [DOI] [PubMed] [Google Scholar]
- 15.Ren B, Robert F, Wyrick JJ, et al. Science. 2000;290:2306. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- 16.Reid JL, Iyer VR, Brown PO, Struhl K. Mol Cell. 2000;6:1297. doi: 10.1016/s1097-2765(00)00128-3. [DOI] [PubMed] [Google Scholar]
- 17.Lee T, Rinaldi N, Robert R, et al. Science. 2002;298:799. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- 18.Harbison CT, Gordon DB, Lee TI, et al. Nature. 2004;431:99. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Steensel B, Henikoff S. Nat Biotechnol. 2000;18:424. doi: 10.1038/74487. [DOI] [PubMed] [Google Scholar]
- 20.van Steensel B, Delrow J, Henikoff S. Nat Genet. 2001;27:304. doi: 10.1038/85871. [DOI] [PubMed] [Google Scholar]
- 21.Tompa R, McCallum C, Delrow J, et al. Curr Biol. 2002;12:65. doi: 10.1016/s0960-9822(01)00622-4. [DOI] [PubMed] [Google Scholar]
- 22.Oliphant A, Brandl C, Struhl K. Mol Cell Biol. 1989;9:2944. doi: 10.1128/mcb.9.7.2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amendt B, Sutherland L, Russo A. J Biol Chem. 1999;274:11635. doi: 10.1074/jbc.274.17.11635. [DOI] [PubMed] [Google Scholar]
- 24.Walter J, Dever C, Biggin M. Genes Dev. 1994;8:1678. doi: 10.1101/gad.8.14.1678. [DOI] [PubMed] [Google Scholar]
- 25.Udalova I, Mott R, Field D, Kwiatkowski D. Proc Natl Acad Sci USA. 2002;99:8167. doi: 10.1073/pnas.102674699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mukherjee S, Berger MF, Jona G, et al. Nat Genet. 2004;36:1331. doi: 10.1038/ng1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roulet E, Busso S, Camargo AA, et al. Nat Biotechnol. 2002;20:831. doi: 10.1038/nbt718. [DOI] [PubMed] [Google Scholar]
- 28.Vargo MA, Nguyen L, Colman RF. Biochemistry. 2004;43:3327. doi: 10.1021/bi030245z. [DOI] [PubMed] [Google Scholar]
- 29.Warren CL, Kratochvil NC, Hauschild KE, et al. Proc Natl Acad Sci U S A. 2006 [Google Scholar]
- 30.Doi N, Takashima H, Kinjo M, et al. Genome Res. 2002;12:487. doi: 10.1101/gr.218802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Braun P, Hu Y, Shen B, et al. Proc. Natl. Acad. Sci. U.S.A. 2002;99:2654. doi: 10.1073/pnas.042684199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hughes TR, Mao M, Jones AR, et al. Nat Biotechnol. 2001;19:342. doi: 10.1038/86730. [DOI] [PubMed] [Google Scholar]
- 33.Singh-Gasson S, Green RD, Yue Y, et al. Nat Biotechnol. 1999;17:974. doi: 10.1038/13664. [DOI] [PubMed] [Google Scholar]
- 34.Nuwaysir EF, Huang W, Albert TJ, et al. Genome Res. 2002;12:1749. doi: 10.1101/gr.362402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Albert TJ, Norton J, Ott M, et al. Nucleic Acids Res. 2003;31:e35. doi: 10.1093/nar/gng035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bulyk ML, Huang X, Choo Y, Church GM. Proc Natl Acad Sci USA. 2001;98:7158. doi: 10.1073/pnas.111163698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bulyk ML, Gentalen E, Lockhart DJ, Church GM. Nat. Biotechnol. 1999;17:573. doi: 10.1038/9878. [DOI] [PubMed] [Google Scholar]
- 38.Berger MF, Philippakis AA, Qureshi AM, et al. (manuscript in preparation)
- 39.Philippakis A, Qureshi A, He F, et al. (manuscript in preparation)
- 40.Odom DT, Zizlsperger N, Gordon DB, et al. Science. 2004;303:1378. doi: 10.1126/science.1089769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weinmann AS, Yan PS, Oberley MJ, et al. Genes Dev. 2002;16:235. doi: 10.1101/gad.943102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berger MF, Bulyk ML. In: Gene mapping, discovery, and expression (Methods in Molecular Biology) Bina M, editor. The Humana Press, Inc.; Totowa, New Jersey: 2006. [Google Scholar]
- 43.DeRisi J, Penland L, Brown PO, et al. Nat Genet. 1996;14:457. doi: 10.1038/ng1296-457. [DOI] [PubMed] [Google Scholar]
- 44.Dudley A, Aach J, Steffen M, Church G. Proc Natl Acad Sci USA. 2002;99:7554. doi: 10.1073/pnas.112683499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berg OG, Winter RB, von Hippel PH. Biochemistry. 1981;20:6929. doi: 10.1021/bi00527a028. [DOI] [PubMed] [Google Scholar]
- 46.Taylor J. An Introduction to Error Analysis. University Science Books; Sausalito, CA: 1997. [Google Scholar]
- 47.Sokal R, Rohlf R. Biometry: The Principles and Practice of Statistics in Biological Research. W. H. Freeman and Company; New York: 1995. [Google Scholar]
- 48.Liu X, Brutlag D, Liu J. Pac Symp Biocomput. 2001:127. [PubMed] [Google Scholar]
- 49.Hughes JD, Estep PW, Tavazoie S, Church GM. J Mol Biol. 2000;296:1205. doi: 10.1006/jmbi.2000.3519. [DOI] [PubMed] [Google Scholar]
- 50.Roth FP, Hughes JD, Estep PW, Church GM. Nat Biotechnol. 1998;16:939. doi: 10.1038/nbt1098-939. [DOI] [PubMed] [Google Scholar]
- 51.Bailey T, Elkan C. Proc Int Conf Intell Syst Mol Biol. 1995;3:21. [PubMed] [Google Scholar]
- 52.Liu X, Brutlag D, Liu J. Nat Biotechnol. 2002;20:835. doi: 10.1038/nbt717. [DOI] [PubMed] [Google Scholar]
- 53.Schneider TD, Stephens RM. Nucleic Acids Res. 1990;18:6097. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Crooks GE, Hon G, Chandonia JM, Brenner SE. Genome Res. 2004;14:1188. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kellis M, Patterson N, Endrizzi M, et al. Nature. 2003;423:241. doi: 10.1038/nature01644. [DOI] [PubMed] [Google Scholar]
- 56.Cliften P, Sudarsanam P, Desikan A, et al. Science. 2003;301:71. doi: 10.1126/science.1084337. [DOI] [PubMed] [Google Scholar]
- 57.Tavazoie S, Hughes J, Campbell M, et al. Nat Genet. 1999;22:281. doi: 10.1038/10343. [DOI] [PubMed] [Google Scholar]
- 58.Robinson M, Grigull J, Mohammad N, Hughes T. BMC Bioinformatics. 2002;3:35. doi: 10.1186/1471-2105-3-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Planta RJ. Yeast. 1997;13:1505. doi: 10.1002/(SICI)1097-0061(199712)13:16<1505::AID-YEA229>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 60.Mewes H, Frishman D, Guldener U, et al. Nucleic Acids Res. 2002;30:31. doi: 10.1093/nar/30.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Michelson AM, Bulyk ML. Molecular Systems Biology. 2006 doi: 10.1038/msb4100062. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Venter JC, Adams MD, Myers EW, et al. Science. 2001;291:1304. [Google Scholar]
- 63.Jones RB, Gordus A, Krall JA, MacBeath G. Nature. 2006;439:168. doi: 10.1038/nature04177. [DOI] [PubMed] [Google Scholar]
- 64.Pierce M, Benjamin KR, Montano SP, et al. Mol Cell Biol. 2003;23:4814. doi: 10.1128/MCB.23.14.4814-4825.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Desjarlais JR, Berg JM. Proc Natl Acad Sci USA. 1992;89:7345. doi: 10.1073/pnas.89.16.7345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Desjarlais JR, Berg JM. Proteins. 1992;12:101. doi: 10.1002/prot.340120202. [DOI] [PubMed] [Google Scholar]
- 67.Jacobs G. EMBO J. 1992;11:4507. doi: 10.1002/j.1460-2075.1992.tb05552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Suzuki M, Yagi N. Proc Natl Acad Sci USA. 1994;91:12357. doi: 10.1073/pnas.91.26.12357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mandel-Gutfreund Y, Baron A, Margalit H. Pac Symp Biocomput. 2001:139. doi: 10.1142/9789814447362_0015. [DOI] [PubMed] [Google Scholar]
- 70.Pomerantz JL, Sharp PA, Pabo CO. Science. 1995;267:93. doi: 10.1126/science.7809612. [DOI] [PubMed] [Google Scholar]
- 71.Pomerantz JL, Pabo CO, Sharp PA. Proc Natl Acad Sci USA. 1995;92:9752. doi: 10.1073/pnas.92.21.9752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Choo Y, Klug A. Proc Natl Acad Sci USA. 1994;91:11163. doi: 10.1073/pnas.91.23.11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rebar EJ, Pabo CO. Science. 1994;263:671. doi: 10.1126/science.8303274. [DOI] [PubMed] [Google Scholar]
- 74.Pabo CO, Nekludova L. J Mol Biol. 2000;301:597. doi: 10.1006/jmbi.2000.3918. [DOI] [PubMed] [Google Scholar]