

SUMMARY
Vertebrates harbor abundant lipopolysaccharide (LPS) or endotoxin in their gut microbiota. Here we demonstrate that the brush border enzyme intestinal alkaline phosphatase (Iap), which dephosphorylates LPS, is induced during establishment of the microbiota and plays a crucial role in promoting mucosal tolerance to gut bacteria in zebrafish. We demonstrate that Iap deficient animals are hypersensitive to LPS toxicity through a mechanism mediated by Myd88 and Tumor Necrosis Factor Receptor (Tnfr). We further show that the endogenous microbiota establish the normal homeostatic level of neutrophils in the intestine through a process involving Myd88 and Tnfr. Iap deficient animals exhibit excessive intestinal neutrophil influx, similar to wild type animals exposed to LPS. When reared germ-free, however, the intestines of Iap deficient animals are devoid of neutrophils, demonstrating that Iap functions to prevent inflammatory responses to resident gut bacteria.
INTRODUCTION
LPS, the major constituent of the outer membrane of all Gram-negative bacteria, both pathogens and mutualists, was independently discovered as a bacterial-associated substance called endotoxin that elicits septic shock in animals (Beutler and Rietschel, 2003). We now know that LPS acts as a toxin by over-stimulating Toll-like receptor (TLR) innate immune signaling, which induces pathogenic inflammatory responses. Mice deficient for TLR4, the TLR specific for LPS, or MyD88, a common adaptor downstream of TLRs, exhibit increased resistance to LPS toxicity (Kawai et al., 1999; Poltorak et al.,1998). TLR signaling through MyD88 promotes nuclear translocation of NFκB and transcription of proinflammatory cytokines such as Tumor Necrosis Factor (TNF). TNF is an important mediator of septic shock, as demonstrated by the endotoxin resistance observed in mice treated with TNF blocking antibody (Beutler et al., 1985). However, TNF is not the only mediator of LPS toxicity, as indicated by the fact that mice deficient for TNFα (Pasparakis et al., 1996), its major receptor TNFRp55 (Pfeffer et al., 1993), or both its 75 and 55 kD receptors (Rothe et al., 1993) are as sensitive as wild type mice to interperitoneal injection with high doses of LPS, although they are more resistant to low doses of LPS administered along with the hepatocyte toxin D-galactosamine. TNF and other proinflammatory cytokines induce vascular permeability, blood flow, and neutrophil recruitment to the LPS source as well as systemic responses such as fever, and in the extreme case of septic shock, disseminated intravascular coagulation, hypotension and ultimately organ dysfunction (Kasper and Harrison, 2005).
One of the most abundant sources of LPS encountered by vertebrates is their resident gut microbiota. Since the discovery of innate immune signaling pathways that recognize molecular signatures present on both pathogens and resident beneficial microbes, an unanswered question has been why gut microbial communities do not elicit pathological inflammation in their hosts. Indeed, inappropriate inflammatory responses to the microbiota manifest themselves in patients with inflammatory bowel diseases (Sartor, 2006). In healthy individuals, suggested protective mechanisms include sequestration of IgA coated gut microbes from the intestinal epithelium (Macpherson et al., 2005), barrier functions of the intestinal epithelium, and restricted expression of innate immune receptors in intestinal epithelial cells (Cario and Podolsky, 2005).
In addition to restricting innate immune signaling by sequestering proinflammatory ligands and their receptors, host cells can actively modulate the inflammatory response by modifying the proinflammatory microbial molecules themselves. Such a mechanism has been shown in the case of an acyloxyacyl hydrolase that cleaves acyl chains from the lipid A portion of LPS (Feulner et al., 2004). In mice lacking this enzyme, acylated LPS persists for longer periods of time after infection with Gram-negative pathogens and elicits increased B cell proliferation and antibody production (Lu et al., 2005). Alkaline phosphatases (AP) have also been shown to modify LPS by dephosphorylating its lipid A moiety (Beumer et al., 2003; Koyama et al., 2002; Tuin et al., 2006; van Veen et al., 2005). Lipid A, which accounts for the toxicity of LPS, contains two phosphate groups coupled to glucosamines; removal of one of the phosphate groups generates a monophosphoryl lipid A that is a 100-fold less toxic than the unmodified lipid A (Schromm et al., 1998).
In vertebrates, APs are broadly distributed throughout different organs, but for the most part their physiological substrates are unknown. Mice deficient for the ubiquitously expressed tissue-nonspecific AP (TNAP) die from seizures due to a defect in the metabolism of pyridoxal phosphate (Waymire et al., 1995), arguing that vitamin B-6 is a natural of substrate of TNAP in mice. The intestinal specific isozyme, intestinal alkaline phosphatase (IAP) has been used traditionally as a marker of enterocyte maturation, but its physiological function in the intestine is unclear. The protein is localized to the apical brush border and is enriched in surfactant-like particles that are secreted towards the intestinal lumen (Alpers et al., 1995). A sharp increase in activity of this enzyme occurs during the post-embryonic development of mammals and fish, the period during which the gut microbiota is established (Bates et al., 2006; Henning, 1985; Zambonino Infante and Cahu, 2001). IAP has been thought to play a role in digestion and absorption of casein (Li-Chan and Nakai, 1989). However IAP deficient mice have no apparent digestion deficits and in fact exhibit accelerated transport of fat droplets through the intestinal wall, resulting in obesity when fed a high fat diet (Nakano et al., 2007; Narisawa et al., 2003).
We hypothesized that Iap functions to dephosophorylate LPS associated with gut bacteria, thereby modulating intestinal inflammation in response to the resident microbiota. We tested this idea using a zebrafish model because of the ease with which we could manipulate both bacterial associations, using gnotobiology, and host gene expression, using morpholino antisense oligonucleotides (MOs). The development and physiology of the teleost and mammalian digestive tracts are very similar (Wallace et al., 2005), and both share conserved responses to their resident microbiota (Cheesman and Guillemin, 2007). Here we show that zebrafish, like mammals, respond to LPS through a mechanism that involves Myd88 and Tnfr. We report that upon colonization, gut microbes elicit low-level intestinal inflammation by a similar mechanism as exogenously administered LPS. Finally, we demonstrate that Iap function is required to detoxify LPS and to prevent intestinal inflammation in response to the resident microbiota.
RESULTS
Identification of the zebrafish intestinal alkaline phosphatase gene
In previous work we showed that brush border AP activity in the zebrafish intestine increases during the period when this organ is colonized by microorganisms, between 5 and 8 days post fertilization (dpf), but fails to increase in animals reared GF (Bates et al., 2006). We found that by inoculating 5 dpf GF zebrafish with microbiota from conventionally reared (CV) controls, we could restore normal Iap levels by 8 dpf. Furthermore we demonstrated that LPS exposure was sufficient to induce Iap activity in GF animals. To investigate the mechanism and functional significance of LPS regulation of Iap, we sought to identify zebrafish iap genes. APs in humans are encoded by four loci: TNAP on chromosome 1 and the tissue-specific isozymes IAP, placental AP, and germ-cell AP, which are clustered at chromosomal position 2q37 and appear to have arisen from tandem duplications (Millán, 2006). To isolate iap genes in zebrafish, we compared the murine Akp3 sequence that encodes IAP (accession: M61705.1) to the zebrafish genome, which yielded two alkaline phosphatase genes that mapped to chromosomes 11 and 22. Similar to humans, these ap genes were highly related but distinguished by their expression patterns (Le Du and Millan, 2002). In situ hybridization with the chromosome 11 ap transcript (accession: NM_201007), which was annotated as alkaline phosphatase (alp), exhibited low level, ubiquitous expression at 5 dpf (data not shown). In contrast, the unannotated chromosome 22 ap gene (accession: NM_001014353), which shared 75% sequence identity to murine Akp3, was expressed specifically in the intestinal epithelium of 5 dpf larvae (Figure 1A,B). RT-PCR analysis of dissected guts from 8 dpf larvae showed intestinal specific expression of the chromosome 22 ap gene, whereas the chromosome 11 ap transcript was present at low levels in the intestine as well as in the carcasses from which the intestines were removed (Figure 1C).
Figure 1.
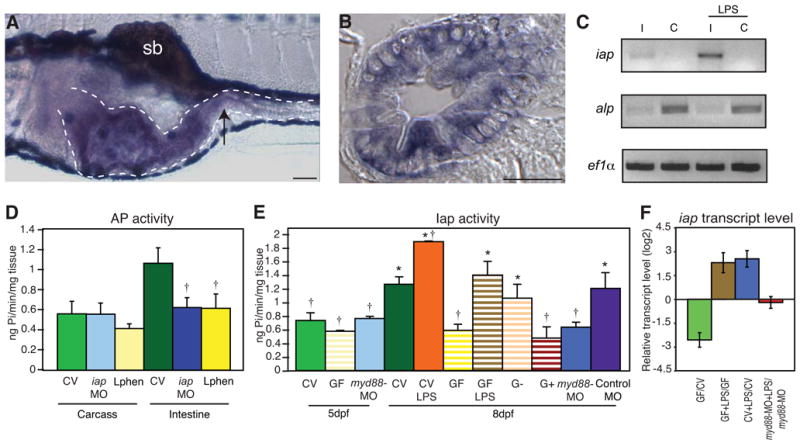
Iap activity and iap transcription are regulated by LPS. In situ hybridization of iap transcript at 5 days post fertilization (dpf) in (A) a whole mount larva, and (B) a transverse section through the mid intestine (at the point indicated by the arrow in A). The iap specific purple stain is present in the intestinal epithelium (outlined with the dotted line in A) and is distinct from the black pigment cells above and below the digestive tract and the swim bladder (sb) in A. Scale bar in panel A = 100 μm, scale bar in panel B = 5 μm. (C) Semi-quantitative RT-PCR analysis showing iap and alp expression in dissected intestines (I) and carcasses from which intestines were removed (C) of 8 dpf larvae untreated or exposed to 50 μg/ml LPS for 24h. Levels of the housekeeping gene ef1α are shown as an amplification and loading control. (D) AP activity in the carcasses and intestines of 8 dpf untreated CV WT larvae, iap-MO injected larvae or larvae exposure to 10mM L-phen from 5 dpf. † Indicates values that differ significantly as compared to the CV levels of each group (carcass or intestine), P<0.01. (E) AP activity in 5 and 8 dpf intestines from larvae reared CV (solid bars) left untreated, exposed at 5 dpf to 30 μg/ml LPS, or injected at the 1 cell stage with myd88-MO or the control galT-MO, or larvae reared GF (striped bars) left untreated, exposed at 5 dpf to 3 μg/ml LPS, or mono-associated at 5 dpf with a Gram-negative Aeromonas species (G−) or a Gram-positive Streptococcus species (G+). † Indicates values that differ significantly from CV at 8dpf, * indicates values that differ significantly from GF at 8 dpf, P<0.01. For D and E, n=10 dissected intestines/treatment for each trial, with at least 2 trials per treatment. Error bars represent standard deviation. (F) iap transcript levels, measured by qRTPCR, were reduced in 8 dpf GF versus CV animals, and elevated in 8 dpf CV and GF animals exposed for 24h to 30 μg/ml LPS, but not in 8 dpf myd88-MO injected animals reared CV and exposed for 24h to 50 μg/ml LPS. Data are representative of two repeated trials, in which all samples were run in triplicate. Error bars indicate standard deviation. All animals were WT unless otherwise indicated.
We next tested whether this gene encoded the AP activity in the zebrafish intestine by blocking expression of the gene with a splice-blocking MO specific to the gene (Figure S1A). When reared to 8 dpf, the intestines of iap-MO injected animals displayed significantly reduced AP activity as compared to wild type (WT) intestines (Figure 1D). The iap-MO specifically inhibited AP activity in the intestine and did not alter AP activity in the carcass lacking intestines (Figure 1D). We observed a similar intestinal specific inhibition of AP activity in 8 dpf fish that had been immersed from 5 dpf in a solution of 10mM L-phenylalanine (L-phen), a specific inhibitor of IAP isozymes (Fishman et al., 1963) (Figure 1D). Treatment with a control MO against an α,3galactosyl transferase (galT) gene did not alter Iap activity (Figure 1E). Based on these results, we have designated the chromosome 22 ap gene the zebrafish intestinal alkaline phosphatase (iap). Intestines from iap-MO injected animals retained some AP activity, likely due to incomplete inhibition by the iap-MO at 8 dpf, indicated by the presence of some WT iap transcript in these animals (Figure S1A). In addition, some AP activity in the intestine is likely to be encoded by the ubiquitously expressed alp gene, because the iap-MO had no inhibitory effect on the total AP activity in 5 dpf intestine but reduced AP activity in 8 dpf intestines (Figure S1B), despite being more efficient at blocking iap transcript splicing at earlier time points (data not shown).
Bacterial LPS regulates IAP transcription in zebrafish
We previously reported that purified LPS, at a concentration of 3 μg/ml, was sufficient to induce Iap activity in GF larvae to levels of CV controls (Bates et al., 2006). Here we tested whether Iap activity could be further induced by additional LPS. We observed that the Iap activity of CV larvae exposed to LPS at 30 μg/ml was significantly higher than untreated levels (Figure 1E). As we had shown previously, mono-association of GF larvae with Gram-negative bacterial isolates from the zebrafish intestine (Aeromonas and Pseudomonas species) was sufficient to induce Iap activity to CV levels, however when we mono-associated GF larvae with several Gram-positive isolates (Streptococcus and Staphylococcus species), which lack LPS in their cell walls, we observed no induction of Iap activity (Figure 1E and data not shown). These results indicated that LPS was sufficient and necessary to induce Iap activity in zebrafish.
In order to understand the mechanism of Iap induction, we examined iap transcript abundance in CV and GF larvae. Semi-quantitative RT-PCR revealed that iap transcript levels increased in the intestine upon exposure to 50 μg/ml LPS for 24h, while transcription of alp remained invariant throughout the whole organism with this treatment (Figure 1C). Using quantitative reverse transcription PCR (qRT-PCR) we found that, similar to of the regulation of Iap enzyme activity, iap transcript levels were significantly elevated in the presence of bacteria (Figure 1F). Furthermore, exposure to exogenous LPS (at 30 μg/ml for 24 hours) was sufficient to increase iap transcript levels in GF larvae and elevate iap above normal levels in CV larvae (Figure 1F).
Bacterial regulation of Iap activity is Myd88 dependent
The induction of iap in response to LPS suggested the possibility that this gene was regulated by Tlr signaling. Several orthologues of Tlr4 and a single gene encoding Myd88 have been identified in the zebrafish genome (Jault et al., 2004; Meijer et al., 2004). To test whether Myd88 was important for LPS induction of iap, we used a splice-blocking MO to zebrafish myd88 (Figure S1C). The myd88-MO injected fish exhibited no gross morphological defects (data not shown) similar to reports of zebrafish treated with a different myd88-MO (van der Sar et al., 2006) and Myd88−/− mice (Iiyama et al., 2003). To test whether Myd88 was required for the larval up-regulation of Iap activity, we dissected intestines from myd88-MO injected larvae and assayed the tissue for AP activity. Whereas the AP activity from intestines of myd88-MO injected animals was indistinguishable between CV or GF WT animals at 5 dpf, at the onset of bacterial colonization of the gut, intestinal AP activity in myd88-MO injected larvae failed to increase by 8 dpf, similar to GF WT animals (Figure 1E), suggesting that microbiota-dependent induction of Iap in CV animals was Myd88 dependent. Next we investigated whether iap transcript abundance was regulated by exogenous LPS in a Myd88 dependent manner. Using qRT-PCR we found that myd88-MO injected larvae treated with 50 μg/ml purified LPS for 24 hours fail to increase iap (Figure 1F), indicating that Myd88 was required for LPS mediated induction of iap.
LPS is toxic to zebrafish
Ingestion or intraperitoneal injection of high doses of LPS is toxic to mammals (Van Amersfoort et al., 2003). Vertebrate species exhibit a wide range of sensitivities to LPS toxicity: calves are extremely sensitive compared to rats and mice, whereas fish and frogs are more resistant (Berczi et al., 1966). We found that soaking 6 dpf zebrafish larvae in water containing high concentrations of LPS resulted in lethality, with percent survival and the kinetics of killing being proportional to the LPS dose. Larvae at 5 dpf were more resistant to LPS killing, possibly due to reduced expression of Tlr signaling pathway genes, which have been shown in some cases to have dynamic developmental expression profiles (Jault et al., 2004). At 6 dpf, a dose of 30 μg/ml LPS failed to cause death, a 50 μg/ml dose resulted in 100% mortality by 48h, a 150 μg/ml dose resulted in 100% mortality by 4.5h and a 250 μg/ml dose caused death in 100% of larvae in 2h (Figure 2A). Similar to mammals, zebrafish exposed to LPS exhibited organ failure, observed as decreased heart beating and lethargy, as well as edema and pooling of blood that was presumably a consequence of severe bradycardia (data not shown).
Figure 2.

LPS toxicity in zebrafish. (A) Dose-dependent killing of wild-type animals exposed to LPS at 6 dpf. Analysis of survival curves show they are significantly different (Logrank test, P<0.0001). (B–C) H&E stained liver sections of untreated (UT) 8 dpf larvae or exposed to 100μg/ml LPS for 24h. (B) Hepatocytes in B show typical organization in cords (dashed line) with distinct nuclei (arrow). (C) LPS treatment resulted in disorganized tissue morphology, with cell boundaries that are difficult to distinguish and swollen hepatocyte nuclei (arrowhead), in contrast to the normal-sized nuclei of red blood cells (asterisks). Scale bar in panel B,C = 5 μm. (D) tnfa and tnfb transcript levels, assayed by qRTPCR, in WT and myd88-MO injected 7 dpf larvae exposed to 50 μg/ml LPS for 4 or 8 h, or WT exposed to 50 μ/ml CIAP treated LPS for 4 h. Data are representative of two repeated trials, in which all samples were run in triplicate. Error bars indicate standard deviation. (E–F) Mpo stained transverse sections of UT 8 dpf larvae or larvae exposed to 150 μg/ml LPS for 2h at the esophageal-intestinal junction (eij). Mpo-positive cells (dark brown) are present in the liver (1) of the LPS exposed animal in F. Scale bar in panel E,F = 10 μm. (G–H) Survival curves of myd88-MO or tnfr1-MO injected 7 dpf larvae exposed to 150 or 250 μg/ml LPS. Survival curves are significantly different (Logrank test, P<0.0001). n = at least 30 total animals for each sample treatment in at least 2 independent trials. All animals were exposed to LPS at 7 dpf except those in panel A, which all began treatment at 6 dpf to allow for 48 h time period to observe toxic effects of low doses of LPS (30–50 μg/ml) prior to termination of all experiments at 8 dpf.
LPS toxicity has a characteristic histopathology in mammals including lesions in the liver, heart and intestine, as well as edema and neutrophil infiltration in affected tissues. Histological analysis of livers from zebrafish larvae exposed to 100 μg/ml LPS for 24h and sacrificed prior to death revealed swollen, disorganized hepatocytes (Figure 2C) and infiltrates of Myeloid peroxidase (Mpo) positive neutrophils (Figure 2F), similar to pathology reported in LPS-exposed mice (Inoue et al., 2005). As in LPS intoxicated mammals, we also observed intestinal infiltration of Mpo positve neutrophils in LPS treated larvae, as discussed below.
A characteristic of LPS toxicity in mammals is the induction of high levels of transcription of the proinflammatory cytokine Tnf. qRT-PCR analysis of 7 dpf larvae treated with 50 μg/ml LPS exhibited a dramatic transient induction of both zebrafish tnf orthologues, with high levels of induction at 4h after exposure that declined significantly by 8h of treatment (Figure 2D). A similar transient Tnf induction was observed in a LPS induced rat model of bacteremia (Xuan et al., 2001) and in zebrafish embryos in a bacteremia model of infection with the Gram-negative fish pathogen Edwardsiella tarda (Pressley et al., 2005).
Another characteristic of LPS toxicity in mammals is the involvement of TLR and TNF signaling. We found that larvae injected with myd88-MO expressed lower levels of tnf transcripts upon LPS exposure (Figure 2D). In addition, myd88-MO injected larvae were significantly more resistant to LPS killing at two different LPS concentrations (Figure 2G, Logrank test, P < 0.0001). To test for a possible role for Tnfr in responses to LPS we used a splice-blocking MO to the zebrafish tnfr1 (Figure S1D). tnfr1-MO injected fish were also significantly more resistant to LPS than WT fish at two different LPS concentrations (Figure 2H, Logrank test, P < 0.0001). Both the myd88- and tnfr1-MO injected larvae did succumb to the LPS treatment, although with slower kinetics, probably due to Myd88 and Tnfr independent mechanisms of LPS toxicity respectively, as well as incomplete inhibition by the MOs (Figure S1C,D).
Iap protects against LPS toxicity
We hypothesized that a function of endogenous Iap was to detoxify LPS encountered by the intestinal epithelium. First we tested whether dephosphorylated LPS was less proinflammatory and less toxic to zebrafish, as it is to mammalian cells (Koyama et al., 2002), by exposing 7 dpf larvae to 250 μg/ml LPS that had been pre-treated with purified calf intestinal alkaline phosphatase (CIAP). Dephosphorylated LPS failed to induce tnf or iap expression (Figure 2D, and data not shown), and was completely non-toxic to zebrafish, whereas mock treated LPS at the same dose caused 100% lethality by 2h (Figure 3A).
Figure 3.

Iap functions to detoxify LPS. (A) LPS pretreated with CIAP was non-toxic to zebrafish at 250 μg/ml LPS, in contrast to mock treated LPS. Inhibition of IAP activity using (B) L-phen, or (C) with iap-MO or by rearing larvae GF, significantly increased susceptibility of larvae to LPS killing. Survival curves are significantly different (except WT untreated and WT CIAP-LPS in A, WT untreated, 10mM L-phen and WT 30 μg/ml LPS in B, and iap-MO 150 μg/ml LPS and GF 150 μg/ml LPS in C, Logrank test, P < 0.0001). All animals were administered LPS at 7 dpf except in panel B, where animals exposed to 30 μg/ml LPS began treatment at 6 dpf. All animals were reared CV, unless otherwise indicated. n = at least 30 total animals for each sample group, in at least two independent trials.
Next we tested whether LPS sensitivity was increased in zebrafish in which Iap activity was depleted. Inhibition of Iap, both by immersion of 5 dpf larvae in a solution of 10mM L-phen or injection with the iap-MO resulted in significantly increased sensitivity to LPS (Figure 3B,C). A dose of 30 μg/ml LPS administered to 6 dpf WT larvae caused insignificant mortality in a 48h period, whereas larvae pretreated with 10mM L-phen at 4 dpf, followed by LPS exposure at 6 dpf, exhibited 100% mortality within the following 48h (Figure 3B, Logrank test, P < 0.0001). Control larvae treated with L-phen alone showed no lethality over the same period (Figure 3B). L-phen treated larvae were also markedly more sensitive than WT larvae when exposed to 150 μg/ml LPS at 7 dpf (Figure 3B, Logrank, P < 0.0001). Similarly, iap-MO injected larvae were more sensitive to LPS treatment with approximately 90% mortality by 2.5h when given a 150 μg/ml dose at 7 dpf, whereas control animals experienced more prolonged survival (Figure 3C, Logrank test, P < 0.0001). Finally, we observed that GF animals, which had reduced Iap activity (see Figure 1E), also exhibited increased sensitivity to 150 μg/ml LPS (Figure 3C, Logrank test, P < 0.0001). Together these data indicate that endogenous Iap conferred protection against a biologically relevant range of concentrations of exogenously administered LPS.
The microbiota regulate homeostatic numbers of intestinal neutrophils
We next asked whether Iap played a role in modulating the inflammatory response of the intestine to LPS associated with resident bacteria. We found no change in the level of tnf transcripts between GF and CV 8 dpf larvae, but this was not surprising given the transient nature of expression of these genes in response to proinflammatory stimuli (Figure 2D) or bacteremia challenge (Pressley et al., 2005). We characterized neutrophil cell homeostasis in the intestine in response to microbiota colonization. Mpo has been used as a marker of zebrafish granulocytes or neutrophils, which have been shown to infiltrate wound sites similar to mammalian neutrophils (Bennett et al., 2001; Lieschke et al., 2001; Mathias et al., 2006; Renshaw et al., 2006). mpo transcript levels are elevated in the intestines of CV relative to GF zebrafish 6 dpf larvae (Rawls et al., 2004). We counted Mpo positive neutrophils in the distal 140 μm of larval intestines, where we observed the preponderance of these cells to be distributed. We found that the average number of Mpo positive neutrophils in the intestine increased during microbial colonization of this organ, with an average of 2 neutrophils in the distal intestine of 6 dpf CV larvae, increasing to an average of 7 neutrophils by 8 dpf (Figure 4Q,R). In contrast, GF intestines were entirely devoid of Mpo positive neutrophils at 6 and 8 dpf, indicating that neutrophils were recruited into the intestine in response to the microbiota (Figure 4Q,R).
Figure 4.

Iap functions to prevent intestinal neutrophil infiltration in response to the microbiota. Whole mount larvae at 6 dpf (A–D) and transverse sections through distal zebrafish intestines at 6 dpf (E–H) and 8 dpf (I–P) with Mpo positive neutrophils visualized in dark brown in the intestinal epithelium (arrowheads) and posterior cardinal vein (arrows); (black cells surrounding the intestinal epithelium are pigment cells.) The WT intestine contained low numbers of neutrophils at 6 (A) and 8 dpf (I), whereas GF intestines were devoid of all neutrophils (B,J). Neutrophil numbers increased significantly upon exposure to 150 μg/ml LPS for 2h (C) or with inhibition of endogenous Iap with iap-MO (D,E) or L-phen (O). Neutrophil infiltration was inhibited in myd88-MO (K) or tnfr1-MO injected larvae (L), even upon exposure to 150 μg/ml LPS for 2h (M–N) or co-injection with iap-MO (F–G). In the absence of microbiota, Iap inhibition did not induce neutrophil influx (H,P). All animals were reared CV unless otherwise indicated. Scale bar in panel D (A–D) = 50 μm, scale bar in panel P (E–P) = 5 μm. (Q–R) Quantification of neutrophils per 140 μm of distal intestine, n = at least 13 animals per treatment; bar indicates average value for each group. † indicates values that differ significantly from WT, * indicates values differ significantly from WT LPS treated, P < 0.01. One-way analysis of variance (ANOVA) show treatments differ significantly, (Q) F = 99.24, P < 0.0001 (R) F = 142.7, P < 0.0001.
We next investigated the mechanism by which intestinal neutrophil recruitment occurs. We showed that the proinflammatory stimulus of 150 μg/ml LPS exposure for 2h resulted in a marked increased in Mpo positive intestinal neutophils (17 in 6 dpf and 22 in 8 dpf CV larvae, Figure 4D,E,Q,R). In contrast, exposure to 150 μg/ml CIAP-treated LPS for 2h failed to elicit any increase in neutrophils in 8 dpf larvae (Figure 4R). Based on our results showing the importance of Myd88 and Tnfr1 in other responses to LPS, we tested whether the recruitment of intestinal neutrophils could be inhibited with the myd88-MO or the tnfr1-MO. We found that myd88-MO injected larvae were devoid of Mpo positive intestinal neutrophils when reared CV and had significantly reduced numbers of these cells upon exposure to LPS (Figure 4K,M,R). Similarly, tnfr1-MO injected larvae lacked Mpo positive neutrophils in the gut epithelium when reared CV and upon exposure to LPS (Figure 4L,N,R). Our results suggest that both Tlr and Tnf signaling play a role in neutrophil influx into the intestine in response to proinflammatory stimuli as well as for the homeostatic level of intestinal neutrophils established by the resident microbiota.
Iap prevents inflammatory responses to the resident microbiota
We next examined the role of Iap in modulating the intestinal inflammatory response to the microbiota. In these experiments we either inhibited Iap at 6 dpf with the iap-MO (when splice blocking was more effective, data not shown), or at 8 dpf with L-phen. We found that the number of Mpo positive intestinal neutrophils in iap-MO injected larvae at 6 dpf or L-phen treated larvae at 8 dpf was significantly greater than in WT controls, and was similar to the number seen in LPS exposed larvae (Figure 4C,F,O,Q,R). We next tested whether this neutrophil influx was mediated by the same Myd88 and Tnfr1 based mechanisms as in WT animals exposed to LPS or microbiota. In larvae doubly injected with either a combination of iap- and myd88-MOs (Figure 4G,Q) or iap- and tnfr1-MOs (Figure 4H,Q), intestinal Mpo positive neutrophil numbers were greatly reduced (Figure 4Q), indicating that Iap functions upstream of Myd88 and Tnfr1. Finally, to demonstrate that Iap function was required upstream of proinflammatory compounds associated with the resident microbiota, we examined neutrophil numbers in Iap deficient larvae reared in the absence of microbes. When reared under GF conditions, both iap-MO injected 6 dpf larvae and L-phen treated 8 dpf larvae had very low numbers of Mpo positive neutrophils in their gut epithelia (Figure 4H,P,Q,R). Wild type GF intestines were capable of neutrophil recruitment, as demonstrated by a robust neutrophil influx of these animals in response to LPS exposure (Figure 4R). These results argue that GF Iap deficient larvae were devoid of intestinal neutrophils because they lacked the proinflammatory substrate of Iap, consistent with endogenous Iap functioning to detoxify LPS associated with resident gut bacteria.
DISSCUSION
Using a GF zebrafish model, we showed that LPS induces Iap activity during bacterial colonization of the gut (Bates et al., 2006). Although mammalian IAP is well-characterized as a marker of enterocyte cytodifferentation, its endogenous substrates were unknown. IAP is capable of dephosphorylating a number of different LPS serotypes (Tuin et al., 2006), a reaction that produces a non-toxic form of the molecule. Here we tested the hypothesis that zebrafish Iap functions to detoxify LPS associated with resident gut bacteria.
LPS and the downstream products of its signaling are known to be toxic in mammals, but this response has not been previously described in zebrafish. We report that LPS treatment in zebrafish results in dose dependent death, organ failure, characteristic histopathology including swollen hepatocytes and neutrophil infiltration, and dynamic transcriptional induction of the inflammatory cytokines, tnfa and tnfb. It is difficult to compare the lethal dose of LPS between zebrafish and mice because we used a water bourn route of administration that would lead to LPS ingestion as well as dermal and gill exposure. Within the 8 dpf larval zebrafish intestine we estimate there to be approximately 4 × 105 bacteria, most of which are Gram-negative, within a volume of approximately 1 to 4 nl, resulting in a bacterial concentration of approximately 1011 cells/ml, similar to the bacterial concentration reported for the human colon (Savage, 1977). Based on measurements of total and shed LPS from Salmonella (Freudenberg et al., 1991), this bacterial density would put the concentration of total LPS in the zebrafish intestine on the order of mg/ml and shed LPS on the order of tens of μg/ml, within the range of concentrations used in this study. In mouse models of septic shock, LPS is typically administered intraperitoneally at doses of approximately 50 mg/kg body weight to achieve 100% lethality and is approximately 10 fold less toxic when administered orally (Youngner, 1972). We showed that in zebrafish, as in mammals, the toxicity of LPS is ameliorated by inhibition of Myd88 and Tnfr. In addition, pretreatment of LPS with purified CIAP rendered it completely non-toxic to zebrafish. Finally we demonstrated that inhibition of endogenous Iap, either pharmacologically or with a gene-specific MO, resulted in hypersensitivity to LPS toxicity.
To test the role of this enzyme in preventing inflammation in response to the microbiota, we investigated the regulation of neutrophil infiltration into the intestine. We showed that the homeostatic numbers of Mpo positive neutrophils in the intestinal epithelium are established by the microbiota, with GF larvae lacking all such cells. We did not find evidence for macrophage involvement in the inflammatory response to gut microbes or exogenous LPS. Whereas RNA isolated from dissected guts of GF, CV, and LPS exposed fish demonstrated significant differences in mpo levels correlated with intestinal neutrophil numbers, we observed no differences in transcript levels of the macrophage specific gene, colony stimulating factor 1 receptor between these samples (unpublished results). We showed that establishment of intestinal immune cell homeostasis upon microbiota colonization uses the same proteins, Myd88 and Tnfr1, that promote intestinal inflammation in response to exogenously administered LPS. These results suggest the importance of innate immune signaling in the maturation of mucosal immunity, similar to subtle developmental defects observed in the gut associated lymphoid tissue of Myd88 deficient mice (Iiyama et al., 2003). In mice, MyD88 function has been shown to be required exclusively in myeloid cells and not the intestinal epithelium for microbiota-dependent responses to intestinal injury (Pull et al., 2005). It will be of interest to determine the cellular distribution and requirements for Myd88 function in the zebrafish intestine.
The microbiota’s capacity to elicit intestinal inflammation through a similar mechanism as endotoxin highlights the importance of host strategies both to select for an appropriate microbial community (a process that may go awry in some inflammatory bowel diseases (Eckburg and Relman, 2007)) and to limit the inflammatory impact of this community. Indeed, it is possible that the inhibition of innate immune responses, such as those mediated by Myd88, could alter the intestinal microbial community and thus indirectly result in a decrease in intestinal inflammation. However, when we cultured the microbiota of myd88-MO injected and control larvae reared together in the same water, we observed no significant differences in bacterial load or colony morphology between the two groups (unpublished results), arguing against a change in microbiota being responsible for the absence of Mpo positive neutrophils in the Myd88 deficient intestines.
By characterizing neutrophil infiltration in Iap deficient fish reared in the presence and absence of microbes, we show that Iap acts upstream of a proinflammatory signal associated with the microbiota. Together our results suggest the model that Iap, which is upregulated by the microbiota during colonization, functions to detoxify microbiota-associated LPS, thereby establishing a homeostatic negative feedback loop that prevents excessive Tlr and Tnf signaling and intestinal inflammation (Figure 5). Thus Iap plays an important role in limiting the proinflammatory potential of resident gut microbes and promoting mucosal tolerance to the Gram-negative bacteria that constitute a significant proportion of the zebrafish microbiota (Bates et al., 2006; Rawls et al., 2006).
Figure 5.

A model of Iap function in the intestinal epithelium. Iap is induced by microbiota-associated LPS and dephosphorylates this LPS, thereby establishing a homeostatic negative feedback loop that reduces signaling through Tlrs and Tnf and prevents excessive intestinal inflammation.
Importantly, Iap enzymatic activity is highly localized to the apical intestinal epithelium (Bates et al., 2006), and thus its LPS detoxifying activity would not be expected to impair the host’s detection of Gram-negative pathogens that have penetrated the intestinal epithelial barrier. In addition to its brush border localization, mammalian IAP has also been shown to be secreted by enterocytes (Alpers et al., 1995), raising the possibility that this enzyme may have extra-intestinal activities, as has been suggested by studies in which rats injected intravenously with purified calf IAP followed by LPS exhibited reduced hepatocyte damage compared to control rats injected with LPS alone (Tuin et al., 2006). Just as the immune system is exquisitely specialized in different tissues, the localized modification of microbial products may represent an underappreciated aspect of microbial-host interactions. Localized expression of a host LPS-detoxifying acyloxacyl hydrolase (AOAH) to the renal cortex of the kidney and secretion of the enzyme into urine may help protect the urinary tract from excessive inflammatory responses to Gram-negative bacteria (Munford, 2005). Notably, no Aoah homologues have been found in fish genomes (Munford and Varley, 2006), suggesting that other LPS-detoxifying mechanisms, such as those involving APs, may play more important roles in these species. Another host enzyme expressed in lung epithelial cells, the mammalian lactonase, Paraoxonase-2 (PON2), has been shown to degrade the bacterial quorum sensing homoserine lactone autoinducer and interfere with signaling mechanisms necessary for Pseudomonas aeruginosa tracheal infection (Stoltz et al., 2007). Here we have demonstrated that Iap is another of what may prove to be a large number of host enzymes devoted to modifying bacterial signals encountered by specific tissues. Experiments to test whether IAP’s function in preventing inflammatory responses to the microbiota is conserved in mammals are underway.
Humans exhibit a wide range of LPS responsiveness. Possible mechanisms for this phenotypic diversity include extensive polymorphism in the TLR4 gene in human populations, with TLR4 alleles that are less responsive to LPS being correlated with greater incidence of Gram-negative bacterial infection but reduced risk for diseases associated with chronic inflammation (Miller et al., 2005). A considerable degree of genetic polymorphism also exists in mammalian AP genes (Millán, and allelic variants in Akp2, encoding TNAP, have been shown to regulate serum AP activity in mice (Foreman et al., suggesting the possibility of variation in IAP genes contributing to LPS responsiveness. Developmental variation in IAP activity may also have important implications for human health. For example, the premature intestine of infants afflicted with necrotising enterocolitis may be prone to severe inflammatory responses due to a paucity of IAP activity.
Bacterial-animal mutualisms are characterized by careful negotiations between partners in order to optimize their shared environment. Reciprocal signaling exists in these mutualisms with microbial signals modulating host processes that then directly or indirectly influence the composition or activity of the microbiota. Microbial gut residents have been shown to actively suppress inflammation by inhibiting multiple steps in the κ pathway (Ismail and Hooper, 2005) Here we show that in zebrafish a signal common to many resident gut bacteria, LPS, up-regulates Iap, which functions to prevent excessive intestinal inflammation, a response that would be detrimental to microbiota and host alike.
EXPERIMENTAL PROCEDURES
Gnotobiotic zebrafish husbandry
All experiments with zebrafish were performed using protocols approved by the University of Oregon Institutional Animal Care and Use Committee and following standard protocols (Westerfield, 1993). CV, GF and mono-associated larvae were generated and sterility of GF embryos was assessed as previously described (Bates et al., 2006). To generate GF iap-MO injected animals, embryos were produced by in vitro fertilization following the standard procedure (Westerfield, 1993), except for the use of antibiotic embryo medium as described previously (Bates et al., 2006) to fertilize the eggs, and then were injected with iap-MO as described below. The injected embryos were then derived GF as previously described (Bates et al., 2006), except the duration of soaking in 0.003% sodium hypochloride was reduced from 30 to 20 min and the embryos were additionally soaked in a solution of 0.1 % poly(vinylpyrrolidone)-iodine for 2 min. Bacteria used for mono-associations were zebrafish isolates Aeromonas veronii biovar sobria, Pseudomonas fluorescens (Bates et al., 2006), Streptococcus and Staphylococcus strains (unpublished data) and a Streptococcus isolate from mouse shown to colonize zebrafish (Rawls et al., 2006). Mono-associated animals were generated by injecting bacterial cultures into flasks of GF 5 dpf larvae to a final concentration of 106 colony forming units (CFU)/ml, which achieved a final bacterial load in 8 dpf larvae comparable to that of 8 dpf CV larvae ((Bates et al., 2006) and unpublished data).
LPS and L-phenylalanine treatments
Filter-sterilized solutions of LPS (E. coli serotype 0111:B4, Fluka) were injected into flasks of GF and CV larvae to a final concentration of 3–250 μg/ml at 7 dpf unless otherwise indicated. Of note, LPS purchased from Sigma produced variable levels of killing, whereas four different lots of LPS purchased from Fluka gave very reproducible results. Commercial sources of LPS have been shown to contain trace contaminants of peptidoglycan (PGN) and lipoproteins (Hirschfeld et al., 2000). To test whether the LPS toxicity could be due to PGN contaminants, we exposed 7 dpf zebrafish larvae to solutions of purified PGN (Staphylococcus aureus 77140, Fluka) up to 1 mg/ml, but did not observe and killing over 24 hours with these treatments. To test whether the LPS toxicity could be due to lipoprotein contaminants, we exposed 7 dpf zebrafish to solutions of synthetic lipopeptide, Pam3Cys-Ser-(Lys)4, (ALX-165-066, Axxora), known to be an immune adjuvant (Erhard et al., 2000), up to 500 μg/ml, and also did not observe killing over 24 hours with these treatments. To generate dephosphorylated LPS, LPS was incubated with 0.148 U/μl CIAP purified from bovine intestinal mucosa (ALPI12G, Biozyme laboratories) for 4h at 37°C, then incubated at 80°C for 10 min to destroy CIAP activity. Mock treated LPS was generated by incubating LPS under identical conditions without the addition of CIAP. To inhibit IAP, larvae were incubated in a solution of 10mM L-phen for the duration of the experiment, starting at 5 dpf unless otherwise indicated. Some residual Iap activity was observed in the L-phen treated larvae, however higher doses of the compound were not readily soluble and appeared to be toxic to the fish.
Alkaline phosphatase
AP activity was quantified as previously described (Bates et al., 2006). Briefly, dissected intestines from 10 larvae, or the carcasses from which the intestines had been removed, were pooled, weighed, homogenized and incubated in p-nitrophenylphosphate liquid substrate system (Sigma) for 30 minutes and absorbance was measured at 405 nm.
Morpholino injections
Splice-blocking morpholinos (MOs) were obtained from Gene Tools (Corvalis, OR), except for MyD88e2i2, which was a generous gift from Dr. Lalita Ramakrishnan (University of Washington). All MOs were injected into embryos at the one cell stage at the indicated final amounts: IAPe2i2 (3 pmoles), TRlv1/TRlv2 (1.2 pmoles and 6 pmoles, respectively), MyD88e2i2 (25 pmoles), galTe2i2 (5.9 pmoles). MO oligo sequences are as follows: IAPe2i2: 5′-TGTAAAGTCGTCTTCATCACTCACC-3′, MyD88 e2i2: 5′-GTTAAACACTGACCCTGTGGATCAT-3′, TRlv1: 5′-TACGTCCTTGTGCATTGCTGGCATC-3′, TRlv2: 5′-CTGCATTGTGACTTACTTATCGCAC-3′. galTe2i2: 5′AAATCATTATGCACTCACCTGATGG-3′. Splice-blocking was observed by RT-PCR analysis of cDNA from injected larvae using specific primers designed to span the splice site. Primer sequences are as follows: IAPe1F: 5′-TCAGAGGCTCGGGATGTGTTTG-3′, IAPe5R: 5′-GACTTTCCTTGTGCTTTGGCG-3′; MyD88elF: 5′-TCTTGACGGACTGGGAAACTCG-3′, MyD88e5R: 5′-GATTTGTAGACGACAGGGATTAGCC-3′; TR1F: 5′-GCATGGATCCATATCAGGACTTGGTGGA-3′, TR1R: 5′TCGAGAATTCTTACGAAACGCTTGTGTT-3′. The myd88-MO showed near complete splice-blocking and the tnfr1-MO and iap-MO partially blocked splicing of the target transcripts at 8 dpf (Figure S1)
Semi-quantitative RT-PCR analysis
Dissected intestines or the remaining carcasses of 20 control animals or 20 animals treated with 150 μg/ml LPS for 2h, were pooled and RNA was harvested by homogenizing and extracting with Trizol reagent (Invitrogen). The RNA was further purified of contaminating genomic DNA using Turbo DNA-free kit (Ambion). The RNAs were used as templates for generating cDNAs with Superscript III reverse transcriptase and random primers (Invitrogen) following the manufacturer’s instructions. cDNA amplification by PCR was subsequently carried out for 25 cycles, with each cycle consisting of denaturation at 94° for 15s, primer annealing at 59° for 30s, and extension at 72° for 1 min. Assays were performed on 200ng cDNA using Taq DNA polymerase (Roche) and 20 pmol of the gene-specific primers. Primer sequences are as follows: IAPF: 5′-ATGGGAGTGTCCACGGTTTCAG-3′ IAPR: 5′-CGATGCCAACAGACTTTCCTTG-3′, ALPF: 5′-GAAGGTCGTACAACTGCTTATCC-3′, ALPR: 5′-GATTCCCACTGATTTGCCTGC-3′, EF1AF: 5′-CGTCTGCCACTTCAGCATGTG-3′, EF1AR: 5′ACTTGCAGGCGATGTGAGCAG-3′. Assays were performed using a TProfessional thermocycler (Biometra).
SYBR-Green Real-Time Quantitative (q)RT-PCR analysis
RNA was harvested from GF, CV, or MO injected whole larvae at 8dpf and cDNAs were generated as above. qRT-PCR assays were performed in 25 μl reactions with cDNA corresponding to 600 ng of total RNA, and 300nM gene-specific or control primers. Gene-specific primers were designed using Primer Express 2.0 software (Applied Biosystems). Primer sequences are as follows; IAP151: 5′-GCCCTCACACTGCCTCTCA-3′, IAP233: 5′-GAAACCGTGGACACTCCCATT-3′, TNF1F: 5′-TCCTCAGACCACGGAAAAGTG-3′, TNF1R: 5′-CAACCCATTTCAGCGATTGTC-3′, TNF2F: 5′-GCTGGATCTTCAAAGTCGGGTGTA-3′, TNF2R: 5′-TGTGAGTCTCAGCACACTTCCATC-3′, EF1AF: 5′-CGTCTGCCACTTCAGCATGTG-3′, EF1AR: 5′-ACTTGCAGGCGATGTGAGCAG-3′. Assays were performed in triplicate using an ABI Prism 7900HT Sequence Detection System (Applied Biosystems). Data were normalized to EF1A (ΔΔ Ct analysis, as described in the ABI User Bulletin #2, http://docs.appliedbiosystems.com/pebiodocs/04303859.pdf).
Histology
Unless otherwise specified, zebrafish larvae were fixed in 4% paraformaldehyde overnight, embedded in paraffin, and 7 μm thick sections were cut and mounted on glass slides. Samples were imaged on a Nikon TE2000 inverted microscope with Nomarski and fluorescence optics. Digital images were obtained with a CoolSNAP camera (Photometrics) or a QImage Micropublisher camera (QImaging Corp). Images were collected and analyzed using Metamorph and processed in Adobe Photoshop. Hematoxylin and eosin (H&E) stained 7 μm transverse sections were analyzed for histopathology related to LPS treatment.
In situ hybridization
5 dpf larvae were fixed in 4% PFA overnight and washed in 1XPBST. Whole-mount larvae were equilibrated into HYB+ buffer (Westerfield, 1993) and prehybridized at 65°C for 4h. RNA probes were prepared according to Boehringer instructions (Cat. #1175025) and 100 ng of RNA probe in HYB+ was heated at 65°C for 60 minutes. Larvae were hybridized with RNA probes for 36h. Probes were removed by soaking larvae for 30 minutes at 70°C in 50% formamide in 2XSSC, followed by rinsing for 30 minutes at 70°C in 2XSSC and rinsing 2 times for 30 minutes at 70°C in 0.2XSSC. Probes were detected according to the standard protocol (Westerfield, 1993) and larvae were sectioned as described previously for imaging.
Myeloid peroxidase
Larvae were fixed in 4% PFA overnight and washed in 1XPBS. Whole-mount larvae were stained with Myeloperoxidase kit (Sigma) for histochemical identification of neutrophils. Mpo activity has been used as a marker of zebrafish neutrophils, although weak staining has also been observed in eosinophils and erythrocytes (Lieschke et al., 2001), therefore care was taken not to overdevelop the Mpo stain. Following staining, larvae were washed in 1XPBS 0.1 % Tween, embedded in paraffin and 7 μm transverse sections were mounted on glass slides. Cells with strong Mpo staining in the intestinal epithelium were quantified in 20 serial sections rostral to the anus, corresponding to the distal 140 μm of the intestine.
Microbiota enumeration and analysis
To compare the microbiota of myd88-MO injected and WT siblings, these embryos were placed in polycarbonate cylinders with mesh bottoms and reared in the same tank in a shared microbiological aquatic environment. At 8 dpf, animals were euthanized with tricaine methane sulfonate (MS222, Sigma) and rinsed 3 times in sterile water. 10 WT or myd88-MO injected larvae were each placed in 100 μl sterile water, homogenized, diluted, and cultured on tryptic soy agar to count CFU and examine colony morphologies. To estimate the concentration of bacteria in the larval zebrafish intestine, from which we inferred LPS concentration, we measured the average number of 16S rRNA genes amplified from a known cell number of Aeromonas veronii biovar sobria, a dominant member of the zebrafish microbiota, to be approximately 4 copies/CFU. Based on our previous measurements of 16S rRNA copies amplified per zebrafish (Bates et al., 2006), we calculated the bacterial load per 8 dpf larva to be approximately 4 × 105. Based on measurements of histological sections and empirical observations from microinjecting known volumes of liquid into the larval gut, we estimate the volume of this organ to be approximately 1 to 4 nl.
Statistical analysis
AP activity assays were repeated with at least two trials per treatment and data were analyzed using two-sample t-Tests assuming unequal variances in Excel (Microsoft Office). Survival curves were performed at least twice, with a combined total of at least 30 fish per treatment. Data for survival curves were graphed and analyzed using GraphPad Prism (Kaplan-Meier analysis, GraphPad Software, http://www.graphpad.com). Neutrophil cell counts were also graphed and analyzed using GraphPad Prism (Scatter plot using one grouping variable for one-way ANOVA), and were also analyzed using two-sample t-Tests assuming unequal variances in Excel (Microsoft Office).
Supplementary Material
Acknowledgments
We thank Rose Gaudreau and the staff of the University of Oregon Zebrafish Facility for fish husbandry, Poh Kheng Loi for histology services, Mimi Shirasu-Hiza for assistance with statistical analysis, Tamara Pozos and Lalita Ramakrishnan for the gift of the myd88-MO, Hilary Clay for sharing unpublished information on the tnfr1-MO, members of the Guillemin lab for insightful discussions, and Sarah Cheesman, Hilary Clay, Tory Herman, Robin Lesley and Lalita Ramakrishnan for critical reading of the manuscript. This research was supported by NIH grants R21 DK067065-01 and R01 DK075549-01 and a Burroughs Wellcome Fund Career Award in the Biomedical Sciences (to K.G.). NIH grant HD22486 provided support for the Oregon Zebrafish Facility.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alpers DH, Zhang Y, Ahnen DJ. Synthesis and parallel secretion of rat intestinal alkaline phosphatase and a surfactant-like particle protein. The American journal of physiology. 1995;268:E1205–1214. doi: 10.1152/ajpendo.1995.268.6.E1205. [DOI] [PubMed] [Google Scholar]
- Bates JM, Mittge E, Kuhlman J, Baden KN, Cheesman SE, Guillemin K. Distinct signals from the microbiota promote different aspects of zebrafish gut differentiation. Developmental biology. 2006;297:374–386. doi: 10.1016/j.ydbio.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Bennett CM, Kanki JP, Rhodes J, Liu TX, Paw BH, Kieran MW, Langenau DM, Delahaye-Brown A, Zon LI, Fleming MD, et al. Myelopoiesis in the zebrafish, Danio rerio. Blood. 2001;98:643–651. doi: 10.1182/blood.v98.3.643. [DOI] [PubMed] [Google Scholar]
- Berczi I, Bertok L, Bereznai T. Comparative studies on the toxicity of Escherichia coli lipopolysaccharide endotoxin in various animal species. Canadian journal of microbiology. 1966;12:1070–1071. doi: 10.1139/m66-143. [DOI] [PubMed] [Google Scholar]
- Beumer C, Wulferink M, Raaben W, Fiechter D, Brands R, Seinen W. Calf intestinal alkaline phosphatase, a novel therapeutic drug for lipopolysaccharide (LPS)-mediated diseases, attenuates LPS toxicity in mice and piglets. The Journal of pharmacology and experimental therapeutics. 2003;307:737–744. doi: 10.1124/jpet.103.056606. [DOI] [PubMed] [Google Scholar]
- Beutler B, Milsark IW, Cerami AC. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science. 1985;229:869–871. doi: 10.1126/science.3895437. [DOI] [PubMed] [Google Scholar]
- Beutler B, Rietschel ET. Innate immune sensing and its roots: the story of endotoxin. Nature reviews. 2003;3:169–176. doi: 10.1038/nri1004. [DOI] [PubMed] [Google Scholar]
- Cario E, Podolsky DK. Intestinal epithelial TOLLerance versus inTOLLerance of commensals. Molecular immunology. 2005;42:887–893. doi: 10.1016/j.molimm.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Cheesman SE, Guillemin K. We know you are in there: Conversing with the indigenous gut microbiota. Research in microbiology. 2007;158:2–9. doi: 10.1016/j.resmic.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Eckburg PB, Relman DA. The role of microbes in Crohn’s disease. Clin Infect Dis. 2007;44:256–262. doi: 10.1086/510385. [DOI] [PubMed] [Google Scholar]
- Erhard MH, Schmidt P, Zinsmeister P, Hofmann A, Munster U, Kaspers B, Wiesmuller KH, Bessler WG, Stangassinger M. Adjuvant effects of various lipopeptides and interferon-gamma on the humoral immune response of chickens. Poultry science. 2000;79:1264–1270. doi: 10.1093/ps/79.9.1264. [DOI] [PubMed] [Google Scholar]
- Feulner JA, Lu M, Shelton JM, Zhang M, Richardson JA, Munford RS. Identification of acyloxyacyl hydrolase, a lipopolysaccharide-detoxifying enzyme, in the murine urinary tract. Infection and immunity. 2004;72:3171–3178. doi: 10.1128/IAI.72.6.3171-3178.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman WH, Green S, Inglis NI. L-phenylalanine: an organ specific, stereospecific inhibitor of human intestinal alkaline phosphatase. Nature. 1963;198:685–686. doi: 10.1038/198685b0. [DOI] [PubMed] [Google Scholar]
- Foreman JE, Blizard DA, Gerhard G, Mack HA, Lang DH, Van Nimwegen KL, Vogler GP, Stout JT, Shihabi ZK, Griffith JW, et al. Serum alkaline phosphatase activity is regulated by a chromosomal region containing the alkaline phosphatase 2 gene (Akp2) in C57BL/6J and DBA/2J mice. Physiological genomics. 2005;23:295–303. doi: 10.1152/physiolgenomics.00062.2005. [DOI] [PubMed] [Google Scholar]
- Freudenberg MA, Meier-Dieter U, Staehelin T, Galanos C. Analysis of LPS released from Salmonella abortus equi in human serum. Microb Pathog. 1991;10:93–104. doi: 10.1016/0882-4010(91)90070-q. [DOI] [PubMed] [Google Scholar]
- Henning SJ. Ontogeny of enzymes in the small intestine. Annual review of physiology. 1985;47:231–245. doi: 10.1146/annurev.ph.47.030185.001311. [DOI] [PubMed] [Google Scholar]
- Hirschfeld M, Ma Y, Weis JH, Vogel SN, Weis JJ. Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine toll-like receptor 2. J Immunol. 2000;165:618–622. doi: 10.4049/jimmunol.165.2.618. [DOI] [PubMed] [Google Scholar]
- Iiyama R, Kanai T, Uraushihara K, Ishikura T, Makita S, Totsuka T, Yamazaki M, Nakamura T, Miyata T, Yoshida H, et al. Normal development of the gut-associated lymphoid tissue except Peyer’s patch in MyD88-deficient mice. Scandinavian journal of immunology. 2003;58:620–627. doi: 10.1111/j.1365-3083.2003.01346.x. [DOI] [PubMed] [Google Scholar]
- Inoue K, Takano H, Shimada A, Yanagisawa R, Sakurai M, Yoshino S, Sato H, Yoshikawa T. Urinary trypsin inhibitor protects against systemic inflammation induced by lipopolysaccharide. Molecular pharmacology. 2005;67:673–680. doi: 10.1124/mol.104.005967. [DOI] [PubMed] [Google Scholar]
- Ismail AS, Hooper LV. Epithelial cells and their neighbors. IV. Bacterial contributions to intestinal epithelial barrier integrity. American journal of physiology. 2005;289:G779–784. doi: 10.1152/ajpgi.00203.2005. [DOI] [PubMed] [Google Scholar]
- Jault C, Pichon L, Chluba J. Toll-like receptor gene family and TIR-domain adapters in Danio rerio. Molecular immunology. 2004;40:759–771. doi: 10.1016/j.molimm.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Kasper DL, Harrison TR. Harrison’s principles of internal medicine. 16. New York: McGraw-Hill Medical Pub. Division; 2005. [Google Scholar]
- Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–122. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- Koyama I, Matsunaga T, Harada T, Hokari S, Komoda T. Alkaline phosphatases reduce toxicity of lipopolysaccharides in vivo and in vitro through dephosphorylation. Clinical biochemistry. 2002;35:455–461. doi: 10.1016/s0009-9120(02)00330-2. [DOI] [PubMed] [Google Scholar]
- Le Du MH, Millan JL. Structural evidence of functional divergence in human alkaline phosphatases. The Journal of biological chemistry. 2002;277:49808–49814. doi: 10.1074/jbc.M207394200. [DOI] [PubMed] [Google Scholar]
- Li-Chan E, Nakai S. Enzymic dephosphorylation of bovine casein to improve acid clotting properties and digestibility for infant formula. The Journal of dairy research. 1989;56:381–390. doi: 10.1017/s0022029900028843. [DOI] [PubMed] [Google Scholar]
- Lieschke GJ, Oates AC, Crowhurst MO, Ward AC, Layton JE. Morphologic and functional characterization of granulocytes and macrophages in embryonic and adult zebrafish. Blood. 2001;98:3087–3096. doi: 10.1182/blood.v98.10.3087. [DOI] [PubMed] [Google Scholar]
- Lu M, Zhang M, Takashima A, Weiss J, Apicella MA, Li XH, Yuan D, Munford RS. Lipopolysaccharide deacylation by an endogenous lipase controls innate antibody responses to Gram-negative bacteria. Nature immunology. 2005;6:989–994. doi: 10.1038/ni1246. [DOI] [PubMed] [Google Scholar]
- Macpherson AJ, Geuking MB, McCoy KD. Immune responses that adapt the intestinal mucosa to commensal intestinal bacteria. Immunology. 2005;115:153–162. doi: 10.1111/j.1365-2567.2005.02159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias JR, Perrin BJ, Liu TX, Kanki J, Look AT, Huttenlocher A. Resolution of inflammation by retrograde chemotaxis of neutrophils in transgenic zebrafish. J Leukoc Biol. 2006 doi: 10.1189/jlb.0506346. [DOI] [PubMed] [Google Scholar]
- Meijer AH, Gabby Krens SF, Medina Rodriguez IA, He S, Bitter W, Ewa Snaar-Jagalska B, Spaink HP. Expression analysis of the Toll-like receptor and TIR domain adaptor families of zebrafish. Molecular immunology. 2004;40:773–783. doi: 10.1016/j.molimm.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Millán JL. Mammalian alkaline phosphatases: from biology to applications in medicine and biotechnology. Weinheim: Chichester, Wiley-VCH; 2006. [Google Scholar]
- Miller SI, Ernst RK, Bader MW. LPS, TLR4 and infectious disease diversity. Nat Rev Microbiol. 2005;3:36–46. doi: 10.1038/nrmicro1068. [DOI] [PubMed] [Google Scholar]
- Munford RS. Detoxifying endotoxin: time, place and person. Journal of endotoxin research. 2005;11:69–84. doi: 10.1179/096805105X35161. [DOI] [PubMed] [Google Scholar]
- Munford RS, Varley AW. Shield as signal: lipopolysaccharides and the evolution of immunity to gram-negative bacteria. PLoS Pathog. 2006;2:e67. doi: 10.1371/journal.ppat.0020067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano T, Inoue I, Koyama I, Kanazawa K, Nakamura K, Narisawa S, Tanaka K, Akita M, Masuyama T, Seo M, et al. Disruption of the murine intestinal alkaline phosphatase gene Akp3 impairs lipid transcytosis and induces visceral fat accumulation and hepatic steatosis. American journal of physiology. 2007;292:G1439–1449. doi: 10.1152/ajpgi.00331.2006. [DOI] [PubMed] [Google Scholar]
- Narisawa S, Huang L, Iwasaki A, Hasegawa H, Alpers DH, Millan JL. Accelerated fat absorption in intestinal alkaline phosphatase knockout mice. Molecular and cellular biology. 2003;23:7525–7530. doi: 10.1128/MCB.23.21.7525-7530.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasparakis M, Alexopoulou L, Episkopou V, Kollias G. Immune and inflammatory responses in TNF alpha-deficient mice: a critical requirement for TNF alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. The Journal of experimental medicine. 1996;184:1397–1411. doi: 10.1084/jem.184.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer K, Matsuyama T, Kundig TM, Wakeham A, Kishihara K, Shahinian A, Wiegmann K, Ohashi PS, Kronke M, Mak TW. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell. 1993;73:457–467. doi: 10.1016/0092-8674(93)90134-c. [DOI] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Pressley ME, Phelan PE, 3rd, Witten PE, Mellon MT, Kim CH. Pathogenesis and inflammatory response to Edwardsiella tarda infection in the zebrafish. Developmental and comparative immunology. 2005;29:501–513. doi: 10.1016/j.dci.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Pull SL, Doherty JM, Mills JC, Gordon JI, Stappenbeck TS. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:99–104. doi: 10.1073/pnas.0405979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawls JF, Mahowald MA, Ley RE, Gordon JI. Reciprocal gut microbiota transplants from zebrafish and mice to germ-free recipients reveal host habitat selection. Cell. 2006;127:423–433. doi: 10.1016/j.cell.2006.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawls JF, Samuel BS, Gordon JI. Gnotobiotic zebrafish reveal evolutionarily conserved responses to the gut microbiota. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:4596–4601. doi: 10.1073/pnas.0400706101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renshaw SA, Loynes CA, Trushell DM, Elworthy S, Ingham PW, Whyte MK. A transgenic zebrafish model of neutrophilic inflammation. Blood. 2006;108:3976–3978. doi: 10.1182/blood-2006-05-024075. [DOI] [PubMed] [Google Scholar]
- Rothe J, Lesslauer W, Lotscher H, Lang Y, Koebel P, Kontgen F, Althage A, Zinkernagel R, Steinmetz M, Bluethmann H. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature. 1993;364:798–802. doi: 10.1038/364798a0. [DOI] [PubMed] [Google Scholar]
- Sartor RB. Mechanisms of disease: pathogenesis of Crohn’s disease and ulcerative colitis. Nature clinical practice. 2006;3:390–407. doi: 10.1038/ncpgasthep0528. [DOI] [PubMed] [Google Scholar]
- Savage DC. Microbial ecology of the gastrointestinal tract. Annu Rev Microbiol. 1977;31:107–133. doi: 10.1146/annurev.mi.31.100177.000543. [DOI] [PubMed] [Google Scholar]
- Schromm AB, Brandenburg K, Loppnow H, Zahringer U, Rietschel ET, Carroll SF, Koch MH, Kusumoto S, Seydel U. The charge of endotoxin molecules influences their conformation and IL-6-inducing capacity. J Immunol. 1998;161:5464–5471. [PubMed] [Google Scholar]
- Stoltz DA, Ozer EA, Ng CJ, Yu JM, Reddy ST, Lusis AJ, Bourquard N, Parsek MR, Zabner J, Shih DM. Paraoxonase-2 deficiency enhances Pseudomonas aeruginosa quorum sensing in murine tracheal epithelia. Am J Physiol Lung Cell Mol Physiol. 2007;292:L852–860. doi: 10.1152/ajplung.00370.2006. [DOI] [PubMed] [Google Scholar]
- Tuin A, Huizinga-Van der Vlag A, van Loenen-Weemaes AM, Meijer DK, Poelstra K. On the role and fate of LPS-dephosphorylating activity in the rat liver. American journal of physiology. 2006;290:G377–385. doi: 10.1152/ajpgi.00147.2005. [DOI] [PubMed] [Google Scholar]
- Van Amersfoort ES, Van Berkel TJ, Kuiper J. Receptors, mediators, and mechanisms involved in bacterial sepsis and septic shock. Clinical microbiology reviews. 2003;16:379–414. doi: 10.1128/CMR.16.3.379-414.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Sar AM, Stockhammer OW, van der Laan C, Spaink HP, Bitter W, Meijer AH. MyD88 innate immune function in a zebrafish embryo infection model. Infection and immunity. 2006;74:2436–2441. doi: 10.1128/IAI.74.4.2436-2441.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Veen SQ, van Vliet AK, Wulferink M, Brands R, Boermeester MA, van Gulik TM. Bovine intestinal alkaline phosphatase attenuates the inflammatory response in secondary peritonitis in mice. Infection and immunity. 2005;73:4309–4314. doi: 10.1128/IAI.73.7.4309-4314.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace KN, Akhter S, Smith EM, Lorent K, Pack M. Intestinal growth and differentiation in zebrafish. Mech Dev. 2005;122:157–173. doi: 10.1016/j.mod.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Waymire KG, Mahuren JD, Jaje JM, Guilarte TR, Coburn SP, MacGregor GR. Mice lacking tissue non-specific alkaline phosphatase die from seizures due to defective metabolism of vitamin B-6. Nature genetics. 1995;11:45–51. doi: 10.1038/ng0995-45. [DOI] [PubMed] [Google Scholar]
- Westerfield M. The zebrafish book a guide for the laboratory use of zebrafish Danio (Brachydanio) rerio. Eugene, OR: Institute of Neuroscience University of Oregon; 1993. [Google Scholar]
- Xuan D, Nicolau DP, Nightingale CH, Quintiliani R. Circulating tumor necrosis factor-alpha production during the progression of rat endotoxic sepsis. Chemotherapy. 2001;47:194–202. doi: 10.1159/000063221. [DOI] [PubMed] [Google Scholar]
- Youngner JS. Bacterial lipopolysaccharide: oral route for interferon production in mice. Infection and immunity. 1972;6:646–647. doi: 10.1128/iai.6.4.646-647.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambonino Infante JL, Cahu CL. Ontogeny of the gastrointestinal tract of marine fish larvae. Comp Biochem Physiol C Toxicol Pharmacol. 2001;130:477–487. doi: 10.1016/s1532-0456(01)00274-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.