

Summary
During neurogenesis in Xenopus, apicobasally polarised superficial and non-polar deep cells take up different fates: deep cells become primary neurons while superficial cells stay as progenitors. It is not known whether the proteins that affect cell polarity also affect cell fate and how membrane polarity information may be transmitted to the nucleus. Here, we examine the role of the polarity components, apically enriched aPKC and basolateral Lgl2, in primary neurogenesis. We report that a membrane-tethered form of aPKC (aPKC-CAAX) suppresses primary neurogenesis and promotes cell proliferation. Unexpectedly, both endogenous aPKC and aPKC-CAAX show some nuclear localisation. A constitutively active aPKC fused to a nuclear localisation signal has the same phenotypic effect as aPKC-CAAX in that it suppresses neurogenesis and enhances proliferation. Conversely, inhibiting endogenous aPKC with a dominant-negative form that is restricted to the nucleus enhances primary neurogenesis. These observations suggest that aPKC has a function in the nucleus that is important for cell fate specification during primary neurogenesis. In a complementary experiment, overexpressing basolateral Lgl2 causes depolarisation and internalisation of superficial cells, which form ectopic neurons when supplemented with a proneural factor. These findings suggest that both aPKC and Lgl2 affect cell fate, but that aPKC is a nuclear determinant itself that might shuttle from the membrane to the nucleus to control cell proliferation and fate; loss of epithelial cell polarity by Lgl2 overexpression changes the position of the cells and is permissive for a change in cell fate.
Keywords: aPKC, Lgl, Polarity, Primary neurons, Xenopus
INTRODUCTION
Apicobasal polarity is important for the function of specialised epithelial cells, including the formation of a permeability barrier, differential secretion and adhesion (Muller, 2001; Shin et al., 2006). Polarity may also be important for generating cell diversity by directly influencing the cell fate choice of polarised versus non-polar cells (Betschinger and Knoblich, 2004; Gotz and Huttner, 2005). Epithelial polarised cells consist of molecularly distinct apical and basolateral membrane domains separated by tight junctions, with specific protein complexes localised to these domains. Notable among these are the apical aPKC-Par3-Par6 complex and the basolateral Lgl-Dlg-Scribb complex. Atypical protein kinase C (aPKC) is a serine/threonine kinase that is recruited to the cell membrane via its interaction with the scaffolding PDZ-domain proteins Par3 and Par6 and is activated by the Rho-GTPase Cdc42 (Joberty et al., 2000; Lin et al., 2000). Lethal giant larvae (Lgl) is a basolaterally localised cytoskeletal protein that contains an Lgl domain of unknown function and many WD40 domains for multiple protein interactions (Vasioukhin, 2006). Apical and basolateral complexes show antagonistic interactions, have been conserved during evolution from Drosophila to vertebrates, and are functionally important for establishing and maintaining the polarity of cells and their specialised junctions (Muller and Bossinger, 2003; Suzuki and Ohno, 2006).
Although the molecules that control epithelial polarity and their interactions are widely conserved, comparatively little is known about their actual influence on cell fate. The influence of polarity on fate is best understood in Drosophila neuroblasts, a population of polarised progenitor cells that delaminate from an epithelium, inheriting some features of apicobasal epithelial polarity. In a Drosophila neuroblast, apically localised protein complexes direct the segregation of cell fate determinants to an apolar basal cell upon asymmetric division (reviewed by Knoblich, 2008). The basal cell then becomes a ganglion mother cell (GMC), which has a limited proliferative capacity, while the sibling polar cell remains as a neuroblast and undergoes repeated cell divisions. Many basal determinants have been identified, but a direct role of the apical side in fate determination of the neuroblast, if any, has remained unclear. The main role of the apical aPKC-Par complex is thought to be in limiting the activity of the basolateral complex and direct fate determinants to the basolateral side, where they are differentially inherited by the daughter cell (Betschinger and Knoblich, 2004). Thus, so far, the emphasis for direct cell fate determination has been on molecules localised to the basal side. Only recently has a role for apical determinants been considered in reports showing that cortical aPKC is required and sufficient to promote neuroblast self-renewal (Lee et al., 2006; Rolls et al., 2003). However, it is totally unclear how the apical polarity information is transmitted from the apical side to the nucleus of the polarised cell.
The Drosophila neuroblast paradigm has been extended to vertebrate neurogenesis, in which asymmetric divisions generate cells of different proliferative capacity (reviewed by Knoblich, 2008; Gotz and Huttner, 2005). In vertebrates, the role of the aPKC-Par complex in neural cell fate diversification has been somewhat controversial. In the mouse cortex and zebrafish retina, loss of aPKC has been reported to affect tissue architecture and cause loss of adherens junctions but not to affect cell fate (Cui et al., 2007; Imai et al., 2006). In the chicken neural tube, overexpression of membrane-tethered aPKC disrupts adherens junctions and enhances proliferation, but does not affect neuronal differentiation (Ghosh et al., 2008; Henrique and Schweisguth, 2003). In the mouse cortex, a conditional deletion of Cdc42, an activator of aPKC, changes cell position and affects proliferation (Cappello et al., 2006), whereas loss of Par3 (Pard3) and gain of Par3/Par6 (Pard6) function affects cell proliferation (Costa et al., 2008). The exact timing of the knockout, molecular redundancy and changes in cell position are some of the factors that have complicated the interpretation of functional experiments in many vertebrates. Furthermore, it is not known how any changes that occur at the cell membrane are communicated to the nucleus.
The frog neural plate consists of two layers of cells - apicobasally polarised superficial cells and non-polar inner cells - that interdigitate during neurulation (Davidson and Keller, 1999; Hartenstein, 1989). By lineage-labelling individual cells, it has been shown that the deep layer clones contain primary neurons and secondary precursors, although never as part of the same clone, whereas the superficial layer clones predominantly contain secondary precursors. Furthermore, whereas most deep-cell-derived primary neuronal progenitors divide only once, generating two neurons, secondary progenitors continue to proliferate at least until the end of embryogenesis [stage (st.) 35/36]. Both types of progenitors divide symmetrically up to the stage of analysis (st.35/36), although it is possible that secondary progenitors might switch to an asymmetric mode of division later on (Hartenstein, 1989). We have subsequently shown that superficial cells are in fact incompetent to undergo early or primary neurogenesis, as they do not form early neurons even when confronted with the same signals or transcription factors that readily induce neurogenesis in the apolar deep cells (Chalmers et al., 2002). We have shown that superficial and deep cells are generated by repeated oriented division of apicobasally polarised epithelial cells at blastula stages and we suggested that during these divisions, only the inner layer cells become competent to form primary neurons (Chalmers et al., 2003). Superficial cells are polarised and, as in other embryonic polarised cells, aPKC localises to the apical membrane, whereas Lgl2 is localised to the basolateral side (Chalmers et al., 2005). In deep apolar cells, there is no cortical aPKC, and Lgl2 is localised throughout the membrane. Based on these findings we have proposed that the frog early ectoderm represents an excellent model system in which to study the influence of polarity on cell fate. Differences in fate and competence are observed throughout the Xenopus ectoderm, both neural and non-neural (epidermal) (Chalmers et al., 2002) (and references therein). Previous studies have shown that PAR1 specifies inner cell fate (ciliated cells) downstream of aPKC in the non-neural ectoderm (Ossipova et al., 2007).
Here, we have used the experimentally tractable model system of Xenopus ectoderm to investigate whether polarity affects cell fate and, in particular, primary neurogenesis. Since post-mitotic primary neurons are not normally derived from polarised superficial cells, we specifically asked three questions: first, whether expressing aPKC in the deep layer is sufficient to suppress primary neurogenesis and, conversely, whether inhibiting aPKC increases primary neurogenesis; third, whether overexpression of Lgl2, which depolarises superficial cells (Chalmers et al., 2005), increases primary neurogenesis.
Our findings show that overexpression of a constitutively active membrane-targeted aPKC (aPKC-CAAX) suppresses primary neurogenesis in deep cells, enhances superficial gene expression and promotes cell proliferation. Interestingly, both endogenous aPKC and expressed aPKC-CAAX are detected in the nucleus, and the effects of aPKC-CAAX on neurogenesis can be fully phenocopied by a nuclear version of activated aPKC. Blocking endogenous aPKC with a dominant-negative form, which is exclusively targeted to the nucleus, shows the opposite phenotype in that it enhances primary neurogenesis. These results suggest that the nuclear fraction of aPKC is functionally important in mediating the fate of the cells. Overexpression of Lgl2 results in depolarisation of superficial cells, which are then internalised and form additional primary neurons when an excess of the proneural factor, X-ngnr-1, is also supplied.
We conclude that aPKC itself acts as a nuclear determinant, possibly transmitting polarity information from the membrane to the nucleus. The nuclear localisation of aPKC places it in a position from which it is possible to act on nuclear targets directly, affecting neurogenesis and proliferation. The loss of membrane cell polarity by Lgl2 overexpression releases cells from their intrinsic inability to undergo primary neurogenesis, but is not sufficient to promote a primary neuronal fate.
MATERIALS AND METHODS
DNA constructs
Constructs Xt-aPKCλ, Xt-aPKCλ-CT, Xt-Lgl and GFP-Xt-Lgl have been described previously (Chalmers et al., 2005). Plasmids pCS2-Rat-aPKCζ-CAAX and pCS2-GAPGFP were kind gifts from S. Sokol and J. Smith (Moriyoshi et al., 1996). The remainder of the Xt-aPKC constructs were generated by PCR-based cloning using the Xt-aPKCλ plasmid as a template and primer sets with introduced restriction sites (details of primers are available on request). The PCR products were cloned into the pCS2 vector.
Overexpression in whole embryos
For unilateral injections, Xenopus embryos were processed as described (Bourguignon et al., 1998). The following amounts of mRNA were injected: Xt-aPKCλ 1 ng, Xt-aPKCλ-CT 0.5 ng, NLS-Xt-aPKCλ 1 ng, NLS-Xt-aPKC-CT 0.25-0.5 ng, Rat-aPKCζ-CAAX 0.25-0.5 ng, Xt-aPKC-NT-CAAX 0.25-0.5 ng, NLS-Xt-aPKC-NT 1-2 ng, Xt-Lgl 1-2 ng, GFP-Xt-Lgl 1-2 ng; 0.5 ng GFP or lacZ mRNA was co-injected as lineage tracer. Embryos were cultured until the desired stage and fixed in MEMFA (0.1 M MOPS pH 7.4, 2 mM EGTA, 1 mM MgSO4, 3.7% formaldehyde) (Chalmers et al., 2002).
X-Gal staining and whole-mount in situ hybridisation followed by sectioning
Antisense probes for N-tubulin, Uroplakin 1B and Prothymosin alpha RNAs were made as previously described (Chalmers et al., 2006). X-Gal staining, in situ hybridisation and sectioning were carried out as described (Bourguignon et al., 1998).
Time-lapse video microscopy
Time-lapse images of injected embryos were generated using a Leica MZFL111 microscope, a 3CCD colour video camera (Sony) and Northern Eclipse software (Empix Imaging). Movies were generated using Windows Movie Maker software (Microsoft).
Cell culture and transfections
HeLa cells were maintained in DMEM supplemented with 10% FCS and antibiotics (Sabherwal et al., 2004). Individual plasmid DNA constructs were transfected into the cells by lipofection (using Lipofectamine 2000, Invitrogen), following the manufacturer's protocol.
Whole cell lysates, subcellular fractionation and western blot analysis
Cells grown to ∼50-60% confluency in 60-mm dishes were transfected with the desired constructs. Twenty-four hours post-transfection, PBS-washed cells were scraped using 0.5 ml IMP buffer (50 mM Tris-acetate pH 7.5, 300 mM NaCl, 1 mg/ml BSA, 2% NP40, 1 mM EDTA, protease inhibitors) and rotated at 4°C for 10-15 minutes, followed by centrifugation at 13,000 rpm (16,100 g) using a table-top centrifuge. Supernatant was collected as cell lysates, 10-20 μl of which were run on a gel for further analysis.
Nuclear and cytoplasmic fractions from HeLa cells were prepared as described (Sabherwal et al., 2004). For preparing cytoplasmic and nuclear fractions from embryos, 20-25 embryos were gently lysed in 500 μl cytoplasmic extraction buffer A (10 mM HEPES pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 1 mM DTT) using a glass pipette, incubated on ice for 10 minutes, then centrifuged at 400 g for 10 minutes at 4°C. Supernatant was retained as the cytoplasmic fraction. The pellet was dissolved again in buffer A to lyse any remaining cells and then centrifuged at 15,000 g for 10 minutes at 4°C. Supernatant was discarded and the nuclear pellet was finally dissolved in 1× sample buffer containing urea (50 mM Tris pH 6.8, 2% SDS, 0.1% Bromophenol Blue, 10% glycerol, 100 mM DTT, 8 M urea) by incubating at 37°C for 30-60 minutes to provide the total nuclear fraction.
Protein detection by western blot was performed as described (Sabherwal et al., 2004). The following primary antibodies were used: rat anti-HA-HRP (1:1000, Roche), rabbit anti-aPKC (1:200, Santa Cruz), mouse anti-tubulin as cytoplasmic control (1:10,000, Sigma T9026), rabbit anti-NBS/P95 as nuclear control for HeLa cells (1:1000, Cell Signaling) (Regad et al., 2007), and rabbit anti-lamin B1 as nuclear control for embryos (1:1000, ab16048, Abcam).
BrdU incorporation, labelling and detection
To label dividing cells in S phase, gastrula stage embryos (st.10.5) were injected into their blastocoel twice with 4.2 nl of undiluted stock solution of 5-bromo-2′-deoxyuridine (BrdU) as provided by the BrdU Labelling Kit (Roche, No. 11296736001), followed by soaking in the same solution (diluted 1:50) for 2 hours at 18°C. Cells with incorporated BrdU were detected on sections following the manufacturer's protocol.
Single-cell Micro-Ruby labelling and internalisation assay
Single cells in control and experimental embryos were injected at blastula stage with the lysine-fixable cell tracer dye Micro-Ruby (1 nl of 2% aqueous solution) (dextran, tetramethylrhodamine biotin; Molecular Probes, No. D-7162). Embryos were grown until the desired stage, MEMFA fixed, sectioned and immunostained as required. For the internalisation assay, cells on sections were counted to estimate the fraction of Micro-Ruby-positive (red) deep cells relative to the total number of Micro-Ruby-positive cells. Statistical analysis for Student's t-test was carried out using SigmaStat version 3.00.
Generation of rabbit polyclonal antibody against X-MyT1
Rabbit polyclonal antibody against the neuronal zinc-finger protein X-MyT1 (Bellefroid et al., 1996) was generated against the purified fusion protein (MBP-MyT1 N-terminus) containing the first 311 amino acids from full-length X-MyT1 (Eurogentec). The serum was affinity purified using Affigel 15 (BioRad) coupled to the GST-MyT1 N-terminal domain.
Cryosectioning and antibody staining
For immunohistochemistry, embryos fixed in MEMFA were sectioned on a cryostat after embedding in 15% fish gelatin/15% sucrose and stained following a standard protocol (Chalmers et al., 2003; Regad et al., 2007).
Immunocytochemistry on HeLa cells grown on coverslips was performed as described (Sabherwal et al., 2004). The following primary antibodies were used for both immunocyto- and immunohistochemistry: rabbit anti-aPKC (1:200, Santa Cruz), rat anti-HA epitope (1:200, Roche), mouse anti-GFP (1:500, Roche), mouse anti-PH3 (1:500, Upstate), mouse anti-BrdU (1:100, Roche) and rabbit anti-X-MyT1 (1:1000).
RESULTS
Overexpression of membrane-tethered aPKC (aPKC-CAAX) inhibits primary neurogenesis
To find out whether polarity components affect cell fate, we first asked whether aPKC overexpression in deep cells affects primary neurogenesis in Xenopus embryos. Overexpression of full-length aPKC (Fig. 1A) did not significantly alter primary neurogenesis (Fig. 1B) (16% of embryos showed a minor reduction in N-tubulin expression on the injected side, n=111) although the protein was easily detected by immunohistochemistry on sections of late neurula stage embryos (data not shown). Removal of the N-terminal regulatory domain of aPKC results in a constitutively active form (Fig. 1A) (aPKC-CT), which expands the apical domain of superficial cells at the expense of the basolateral domain, resulting in hyper-apicalised superficial cells (Chalmers et al., 2005). However, aPKC-CT (0.5 ng) had no significant effect on N-tubulin expression (Fig. 1B) (11% of embryos showed a minor reduction, n=98).
Fig. 1.

The effect of overexpressing aPKC on primary neurogenesis in Xenopus. (A) Schematic representation of the various aPKC constructs used in this work. SV40NLS, nuclear localisation signal from SV40; CAAX, prenylation signal. (B) mRNAs for aPKC (1 ng), aPKC-CT (0.5 ng), aPKC-CAAX (0.25-0.5 ng), NLS-aPKC (1 ng) and NLS-aPKC-CT (0.25-0.5 ng) were injected unilaterally together with lacZ mRNA (0.25 ng) and embryos were analysed by in situ hybridisation for N-tubulin expression. Only aPKC-CAAX and NLS-aPKC-CT showed significant inhibitory effects on primary neurogenesis (arrows). Scale bar: 250 μm.
Given these results, we searched for other ways to activate aPKC in the deep cells. A membrane-tethered version of aPKC (aPKC-CAAX, in which aPKC is linked to a prenylation signal) has previously been shown to function as a constitutively active form in Drosophila and Xenopus (Lee et al., 2006; Ossipova et al., 2007; Ghosh et al., 2008; Sotillos et al., 2004). Overexpressing aPKC-CAAX (0.25-0.5 ng) reduced primary neurogenesis in 47% of injected embryos (n=111) (Fig. 1B). These results led us to conclude that cortical localisation of aPKC activates it in the deep cells and leads to a reduction in primary neurons, which are normally produced by these cells.
Endogenous aPKC and aPKC-CAAX localise to the nucleus in HeLa cells and Xenopus embryos
aPKC-CAAX overexpression suppressed the expression of N-tubulin, but it was not clear how this effect was mediated. We reasoned that information must be transmitted from the cell cortex to the nucleus and considered that either a signal transduction pathway downstream of aPKC affects gene expression via a number of intermediates, or that aPKC is transported to the nucleus, functioning as a nuclear determinant itself.
To gain a better understanding of the correlation between subcellular localisation and function, we first looked at the subcellular localisation of various aPKC constructs in HeLa cells. Full-length aPKC was found to be mainly cytoplasmic, with some cortical and little nuclear staining (Fig. 2B), whereas aPKC-CT was mainly nuclear and cytoplasmic with very little or no cortical staining (Fig. 2B). As expected, aPKC-CAAX was found to localise to the cortex in HeLa cells, co-localising with GAP-GFP (a marker for plasma membrane) (Fig. 2B). Interestingly, aPKC-CAAX also showed diffuse nuclear staining (Fig. 2B). Some intra-nuclear aggregates were also observed (Fig. 2B), which might represent nuclear membranes as the presence of prenylation signal(s) in nuclear proteins may result in their accumulation along the inner nuclear membrane (Prufert et al., 2004; Ralle et al., 2004; Wright and Philips, 2006).
Fig. 2.

Subcellular localisation and expression of aPKC constructs. (A) Immunohistochemistry using anti-aPKC antibody (green) on sections showing ectoderm of st.10.5 Xenopus embryos (apical side up) from non-injected control (left) or injection with aPKC-CAAX or NLS-aPKC-CT mRNAs. Arrows indicate nuclear staining. (B) Immunocytochemistry on HeLa cells transfected with the constructs shown and assayed with anti-aPKC (red) and anti-GFP (green) antibodies. Nuclei are blue (DAPI staining). (C) Western blot analysis with nuclear lysates from HeLa cells transfected with HA-tagged constructs as shown and assayed with anti-aPKC and anti-HA-antibodies. Endogenous aPKC shows some nuclear localisation, as shown by the anti-aPKC antibody, but HA-tagged aPKC-CAAX shows greater nuclear accumulation than HA-aPKC, as shown by the anti-HA antibody. P95 and tubulin were used as nuclear and cytoplasmic controls, respectively. (D) Western blot analysis with nuclear lysates from st.10.5 Xenopus embryos injected with HA-tagged constructs as shown and blotted with anti-HA and anti-aPKC antibodies. Both HA-tagged aPKC and aPKC-CAAX show nuclear accumulation. Lamin and tubulin were used as nuclear and cytoplasmic controls, respectively. (E) Expression analysis using whole cell lysates from HeLa cells overexpressing various aPKC constructs as shown. GFP, control for transfection and loading. Scale bars: 50 μm in A; 250 μm in B.
To confirm the nuclear localisation of aPKC biochemically, we performed western blot analysis on nuclear fractions obtained from HeLa cells (Fig. 2C). Interestingly, endogenous aPKC localised to the nucleus in non-transfected cells, and overexpressed full-length aPKC also showed nuclear localisation. Surprisingly, the membrane-tethered form, aPKC-CAAX, showed even higher levels of nuclear localisation. Together with its higher biological activity in the embryo (Fig. 2C), this finding suggested that aPKC might function as a nuclear determinant itself.
To confirm whether a similar observation holds true for embryos, we performed immunohistochemistry on gastrula stage Xenopus embryos. Although nuclear aPKC-CAAX staining was weak compared with the very strong cortical staining, it was easily detected (Fig. 1A). We also observed the nuclear localisation of endogenous aPKC (Fig. 1A). In western blots of nuclear and cytoplasmic fractions obtained from the embryos at three different stages (pre-gastrula st.8, gastrula st.10.5 and neurula st.16), we detected both overexpressed aPKC-CAAX and endogenous aPKC in the nuclear fraction (Fig. 2D, st.10.5; st.8 and st.16 not shown).
The nuclear version of aPKC-CT (NLS-aPKC-CT) phenocopies aPKC-CAAX in reducing N-tubulin expression
To check our hypothesis that aPKC functions as a nuclear determinant, we cloned the SV40 T-antigen nuclear localisation signal (NLS) (Gorlich and Kutay, 1999) at the N-terminus of both aPKC and aPKC-CT. It is known that aPKC contains an endogenous NLS but this might be subject to regulation (Perander et al., 2001), whereas the SV40 NLS would be expected to be constitutively active in the context of aPKC. Immunostaining using both HeLa cells and sections from Xenopus embryos overexpressing NLS-aPKC and NLS-aPKC-CT clearly demonstrated that both proteins show increased nuclear accumulation in comparison to aPKC and aPKC-CT (Fig. 2B; Fig. 1A). Overexpression of the nuclear version of full-length aPKC (NLS-aPKC, 1 ng) in embryos had a negligible effect on the expression of N-tubulin, but NLS-aPKC-CT (0.25-0.5 ng) strongly suppressed primary neurogenesis (Fig. 1B) (59% of injected embryos showed a significant reduction in N-tubulin expression, n=53). Since aPKC-CAAX localises to the nucleus and NLS-aPKC-CT phenocopies aPKC-CAAX in suppressing neurogenesis, we suggest that the likely mechanism of aPKC-CAAX action involves the translocation of the activated aPKC from the membrane to the nucleus. The apparent paradox lies in the observation that although aPKC-CT also localises to the nucleus, possibly owing to its small size (∼42 kDa) (Fig. 2B), it does not affect neurogenesis significantly; however, this might be explained by the inherently lower expression level of this construct (Fig. 2E), which could be due to lower translation and/or stability.
Overexpression of either a membrane-targeted or nuclear version of dominant-negative aPKC enhances primary neurogenesis
Our data so far suggest that a nuclear kinase activity of aPKC might be responsible for the suppression of primary neurogenesis. This interpretation is based on overexpression experiments and although it is possible that aPKC acts in the nucleus, it is not clear whether it normally does so. Furthermore, the nuclear-targeted construct NLS-aPKC-CT (Fig. 2A) shows some apparent membrane localisation and therefore it is difficult to distinguish with certainty whether the important kinase activity is at the plasma membrane or in the nucleus. Loss-of-function experiments with aPKC morpholinos or chemical inhibitors are not appropriate for addressing this question because they result in widespread cell death, precluding further analysis (Chalmers et al., 2005), and do not allow selective inhibition of a subcellular fraction of aPKC. Previous studies have shown that an N-terminal regulatory domain (Fig. 1A) acts as a dominant-negative form of aPKC (aPKC-NT) (Chalmers et al., 2005). To inhibit endogenous aPKC specifically in the membrane or the nucleus, we fused the human influenza hemagglutinin derived epitope (HA)-tagged dominant-negative form of aPKC (consisting of the entire regulatory domain with the first 200 amino acids of aPKC) with either a prenylation (CAAX) signal at its C-terminus or an SV40 NLS at its N-terminus (Fig. 3A), and asked which of the two, if any, would give rise to a phenotype opposite to that of aPKC overexpression. Interestingly, both constructs enhanced primary neurogenesis (Fig. 3C) [45% (n=31) and 50% (n=22) of embryos injected with HA-aPKC-NT-CAAX and NLS-aPKC-NT-HA, respectively, showed a significant increase in N-tubulin expression]. As expected, whereas HA-aPKC-NT-CAAX exhibited predominantly cortical localisation with weak nuclear staining (preventing a distinction between membrane and nuclear function), NLS-aPKC-NT-HA showed exclusively nuclear localisation (Fig. 3B). Embryos injected with the nuclear dominant-negative construct showed enhanced N-tubulin expression, strongly supporting the hypothesis that the nuclear fraction of aPKC normally restricts primary neurogenesis. With both injections, supernumerary N-tubulin-positive cells were observed in the deep layer of the ectoderm (Fig. 3C), although some were found in an intermediate position between the superficial and deep layers (data not shown) and some were also found in the superficial layer (Fig. 3C, middle image and inset). Unlike aPKC-overexpressing constructs, dominant-negative aPKC did not affect neural plate architecture in any obvious way.
Fig. 3.

Effects of overexpressing dominant-negative aPKC contructs on primary neurogenesis. (A) The two dominant-negative forms of aPKC used. (B,C) Xenopus embryos unilaterally injected with either HA-aPKC-NT-CAAX or NLS-aPKC-NT-HA mRNA were processed for (B) immunohistochemistry on sectioned ectoderm using anti-HA (green) and anti-aPKC (red) antibodies, or (C) for in situ hybridisation at st.16 for N-tubulin expression. Whereas HA-aPKC-NT-CAAX showed predominantly cortical staining with weak nuclear localisation, NLS-aPKC-NT-HA showed exclusively nuclear localisation (B). Overexpression of both dominant-negatives enhanced neurogenesis (C). Arrows point to nuclear staining (B), ectopic neurons (C, upper row) and ectopic neurons in superficial neuroectoderm (C, lower row, middle panel inset). Dotted lines (C, lower row) indicate the boundary between ectoderm and mesoderm. N, notochord. Scale bars: 50 μm in B; 1 mm in C, upper row; 500 μm in C, lower row.
Overexpression of either aPKC-CAAX or NLS-aPKC-CT enhances cell proliferation in ectoderm
Embryos overexpressing either NLS-aPKC-CT or aPKC-CAAX showed a remarkable thickening of both neural and non-neural ectoderm (Fig. 4A,B; Fig. 5A). To find out whether this was restricted to the deep or superficial layer, we used Uroplakin 1B, a marker expressed in the superficial layers of both neural and epidermal ectoderm (Chalmers et al., 2006), and Prothymosin alpha, which is normally expressed in both neural and epidermal deep ectoderm (Chalmers et al., 2006). There was a strong enhancement in Uroplakin 1B staining, as seen in several layers in embryos (with some patchy areas of missing staining) upon overexpression of either aPKC-CAAX or NLS-aPKC-CT, both in neural and epidermal ectoderm (Fig. 4A). There was no compensatory reduction in Prothymosin alpha staining, which even occasionally increased, suggesting that both layers are thickened upon overexpression of aPKC.
Fig. 4.

Effects of overexpressing aPKC constructs on expression of superficial and deep layer markers. Whole-mounts and sections of st.16 Xenopus embryos showing in situ hybridisation for (A) the superficial ectodermal marker Uroplakin 1B and (B) the deep ectodermal marker Prothymosin alpha. The dotted line marks the boundary between ectoderm (above) and mesendoderm (below). Both ectodermal layers are thickened following injection of either aPKC-CAAX or NLS-aPKC-CT mRNA. n, total number for embryos analysed. Scale bars: 250 μm.
Fig. 5.

Effect of overexpressing aPKC constructs on cell proliferation. (A) Neurula stage Xenopus embryos overexpressing aPKC-CAAX and NLS-aPKC-CT showed suppression of N-tubulin expression and a thickened ectoderm (arrows). (B) Quantitation of phospho-histone H3 (PH3)-positive nuclei relative to the total number of nuclei as obtained from at least three embryos in each set and a minimum of five sections per embryo. (C) Sections of st.10.5 embryos injected with aPKC-CAAX and NLS-aPKC-CT and co-immunostained with anti-PH3 (black in DIC image, top row, and red in bottom row) and anti-aPKC (green, in middle and bottom rows) antibodies. Sections show multilayered, thickened ectoderm and increased PH3 staining in both layers of the ectoderm. The bottom row shows the middle row at higher magnification, with anti-aPKC and anti-PH3. Scale bars: 250 μm in A; 50 μm in C.
To find out whether this ectodermal thickening is due to an increase in cell proliferation, we stained the sections from embryos overexpressing either aPKC-CAAX or NLS-aPKC-CT with antibodies against aPKC and either the M-phase marker phospho-histone 3 (PH3) or BrdU (after embryos had been BrdU saturated at st.10.5). We found no increase in BrdU staining in the thickened ectoderm at neurula stages (see Fig. S1 in the supplementary material). However, at gastrula stage there was an increase in the total number of cells and an increase in the absolute number of PH3-positive cells, resulting in a net increase in the number of PH3-positive over total nuclei in the ectoderm (1.48-fold for aPKC-CAAX and 1.54-fold for NLS-aPKC-CT) (Fig. 5B,C). The ectoderm over the blastocoel roof was thickened compared with that in the control sections (Fig. 5C), and cells in mitosis were observed in both the superficial and deep layers of the ectoderm (Fig. 5C). We cannot exclude the possibility that additional effects of aPKC on radial intercalation contribute to the thickening of the ectoderm. Thus, together with the suppression of primary neurogenesis, aPKC overexpression increases proliferation throughout the ectoderm at the gastrula stage.
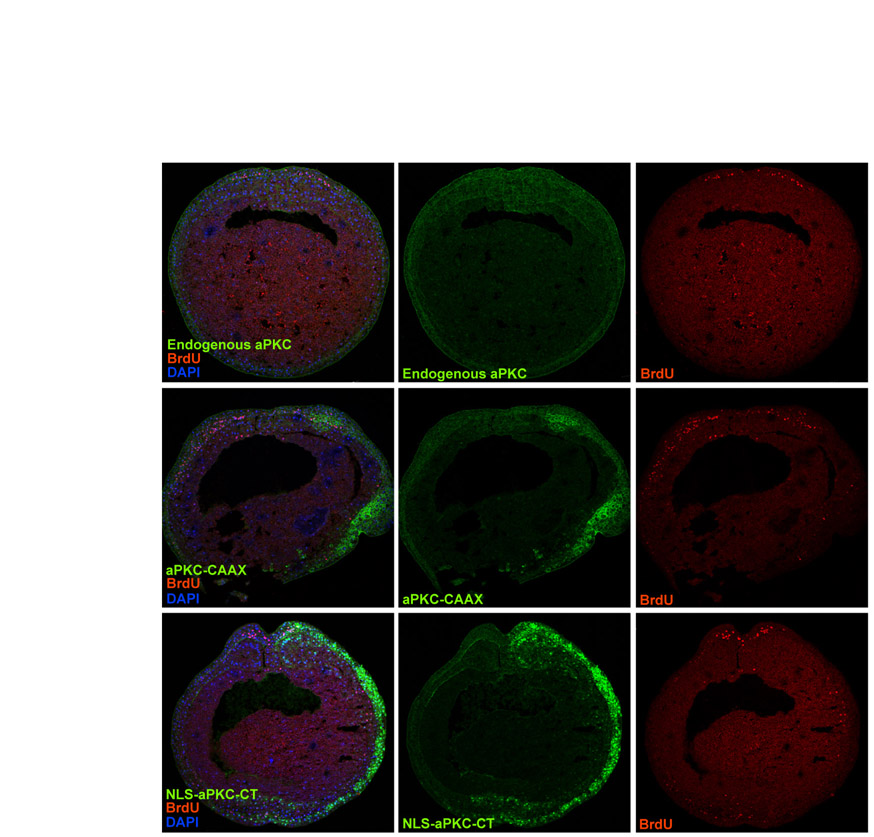
To show that events taking place at the gastrula stage are relevant to neurogenesis, we asked how many of the primary neurons are generated by the gastrula stage, and how this is affected by overexpression of aPKC constructs. Gastrula stage (st.10.5) embryos were injected with BrdU and cultured to tadpole stage (st.25), at which point we analysed them by double staining for BrdU and X-Myt1, a marker for post-mitotic neurons (Bellefroid et al., 1996). In the neural tube of tailbud stage embryos, all ventricular zone progenitors were positive for BrdU as expected. In the control non-injected neural tubes, many X-Myt1+ cells were also BrdU+, representing the neuronal population that was born at or after BrdU injection (st.10.5). However, a small proportion (∼10%) of X-Myt1+ cells were BrdU-, suggesting that they were born (replicated for the last time) before st.10.5. This is consistent with previous reports showing that the majority of primary neurons are born between the late gastrula and early neurula stages (Lamborghini, 1980; Hartenstein, 1989), with st.10.5 being the first at which post-mitotic neurons can be detected. In experimental embryos overexpressing either aPKC-CAAX or NLS-aPKC-CT, the total number of BrdU+ cells was increased on the injected side, and the number of early born neurons (X-Myt1+, BrdU-) was reduced (Fig. 6A,B).
Fig. 6.

Effect of overexpressing aPKC-CAAX or NLS-aPKC-CT on the neuronal population born during gastrulation. Xenopus embryos were unilaterally injected at the 2-cell stage with either aPKC-CAAX or NLS-aPKC-CT, BrdU saturated at st.10.5 and immunostained for BrdU and for the neuronal differentiation marker X-MyT1 on sections of tailbud at st.25. (A) Top row shows anti-aPKC staining (green), with nuclei in blue (DAPI), to distinguish between injected and non-injected sides for aPKC-CAAX and NLS-aPKC-CT injections. Bottom row shows adjacent sections co-stained for BrdU (red) and X-MyT1 (green). Neural progenitors are positive for BrdU only and appear red, neurons born after BrdU labelling are co-labelled and appear orange (arrow), and neurons born before st.10.5 (BrdU saturation) appear green. The number of neurons born before st.10.5 (green) is reduced after overexpression of either aPKC construct. The dashed lines in circle delineate the neural tube and the straight dashed lines mark the midline of the neural tube. (B) Quantification of the results from A.
Co-expression of Lgl2 with Neurogenin 1 (X-ngnr-1) causes ectopic neurogenesis outside the deep layer of neuroectoderm
Apicobasal polarity of epithelial cells is maintained by the antagonistic interactions of apically enriched aPKC and basally localised Lgl2 (reviewed by Suzuki and Ohno, 2006; Knoblich, 2008). In the frog, overexpression of Lgl2 leads to the loss of apical aPKC localisation, loss of apical pigment and depolarisation of outer cells (Chalmers et al., 2005). To understand further how polarity affects cell fate, we asked how the complete depolarisation of superficial cells by Lgl2 overexpression would affect primary neurogenesis. Embryos overexpressing Lgl2 (1 ng) showed no change in N-tubulin expression (n=35) (Fig. 7D-F) by comparison with control embryos injected with lacZ mRNA (0.25 ng) alone (n=32) (Fig. 7A-C). This result led us to hypothesise that the loss of cell polarity is not sufficient for the superficial cells to generate primary neurons and that they might additionally need some proneural factor to do so. Neurogenin 1 (X-ngnr-1) has been shown to be a very potent proneural factor, and overexpressing X-ngnr-1 alone (0.25 ng) resulted in ectopic neurons, but only in the deep layer of ectoderm as the polarised superficial cells are not competent to respond (n=41) (Fig. 7G-I) (Chalmers et al., 2002). Co-expression of X-ngnr-1 with Lgl2 also did not result in ectopic neurogenesis in the superficial layer. However, we observed ectopic N-tubulin staining in an intermediate position between the deep and superficial layers (n=26) (Fig. 7J-L).
Fig. 7.

Effect of Lgl2 overexpression on primary neurogenesis. Overexpression of Lgl2 (1 ng) had no effect on expression of the primary neuron marker N-tubulin (n=35) (D-F) by comparison with control embryos injected with lacZ mRNA only (0.25 ng, n=32) (A-C). But when Lgl2 was co-expressed with the proneurogenic factor X-ngnr-1 (0.25 ng, n=26) (J-L), ectopic primary neurons were found in a position intermediate between the superficial and deep layers (L, arrow), whereas embryos injected with X-ngnr-1 alone (n=41) (G-I) showed ectopic neurogenesis only in the deep layer of neuroectoderm (I, arrow). Also note the absence of lacZ staining in the superficial layer cells of embryos injected with Lgl2 and X-ngnr-1 (L, arrowhead), as compared with lacZ (C) or X-ngnr-1 (I) injected embryos, indicating the possibility of internalisation of superficial cells due to Lgl2 overexpression. Scale bars: 250 μm.
Depolarised superficial cells resulting from Lgl2 overexpression become internalised and form neurons when supplemented with a proneural factor
Ectopic neurons in an intermediate position could be the result of internalisation of depolarised superficial cells. To address this possibility, we followed the fate of GFPLgl2-expressing cells by injecting single depolarised cells with Micro-Ruby at blastula stage (Fig. 8A) and determined the location of cells that were positive for both GFPLgl2 and Micro-Ruby at st.16-17. Control embryos showed labelled cells that were approximately evenly distributed between the inner and outer layers (Fig. 8A). This was expected because inner layer cells are generated by oriented divisions of outer layer cells during blastula stages. By contrast, embryos overexpressing GFPLgl2 had a considerably higher fraction of deep layer labelled cells (∼88%) (Fig. 8A).
Fig. 8.

Lgl2-expressing cells become internalised and contribute to primary neurogenesis. (A) The distribution of GFPLgl2-expressing cells in the inner versus outer ectodermal layer of neurula stage Xenopus embryos was assessed by an internalisation assay (see Materials and methods for details) that involved lineage-labelling individual GFPLgl2-expressing outer cells by Micro-Ruby injection at the blastula stage (a,b). The fraction of Micro-Ruby-labelled cells in the inner layer over the total number of labelled cells was increased in GFPLgl2-injected embryos as compared with control embryos (c,d). (B) Time-lapse images of an embryo overexpressing GFPLgl2, showing early loss of apicobasal cell polarity in superficial cells (apical de-pigmentation, arrow in a) and subsequent internalisation of these cells at mid-gastrula stage (g-j). (C) To determine whether internalised cells resulting from GFPLgl2 overexpression contribute to increased neurogenesis, the Micro-Ruby-based internalisation assay was combined with in situ hybridisation for N-tubulin, as analysed in sections of st.16 neurulae from embryos that had been injected at the 2-cell stage with either X-ngnr-1 alone or in combination with GFPLgl2 (a-f). Bar charts show that the percentage of Micro-Ruby-labelled deep layer cells relative to the total number of labelled cells was increased when GFPLgl2 was co-injected and that ∼76% of these cells, on average, were also N-tubulin positive. Scale bars: 500 μm in B; 50 μm in C.
To confirm that this was due to internalisation, we followed the fate of GFPLgl2-overexpressing cells by time-lapse microscopy. At the onset of the appearance of GFPLgl2, the superficial cells started to lose their polarity (as shown by loss of pigmentation), with the patch of depolarised cells expanding gradually. At around the mid-gastrulation stage, this patch was rapidly internalised (Fig. 8B).
To show that the internalised, GFPLgl2-injected cells give rise to neurons, we combined the internalisation assay with in situ hybridisation for N-tubulin (Fig. 8C). As before, X-ngnr-1-injected embryos had Micro-Ruby-labelled cells almost equally distributed between the inner and outer layers (similar to non-injected embryos) (Fig. 8A,C). By contrast, when X-ngnr-1 and GFPLgl2 were co-injected, 82% of the Micro-Ruby-injected cells were internal and the majority (76%) of these were also positive for N-tubulin (Fig. 8C) (compared with 52% N-tubulin-positive when embryos were injected with X-nngnr-1 alone), demonstrating that these internalised cells give rise to neurons. Thus, inhibition of apical aPKC results in the internalisation of superficial cells, which generate ectopic neurons in an intermediate position when supplemented with X-ngnr-1.
DISCUSSION
The Xenopus ectoderm consists of a superficial, outer, epithelial polarised cell layer overlying a layer of inner, apolar cells; these two cell layers follow different fates during development (Asashima and Grunz, 1983; Bradley et al., 1996; Chalmers et al., 2002; Hartenstein, 1989; Jones and Woodland, 1986; Muller and Hausen, 1995). This difference in fate applies to the entire ectoderm, but in the neural ectoderm it is manifested as a difference in the competence to generate primary neurons; these are normally generated by the inner layer of cells only. We have previously shown that aPKC and Lgl2 are functionally involved in determining the apical and basolateral membrane domain, respectively, acting in a mutually antagonistic manner (Chalmers et al., 2005). Here, we have asked whether the same polarity molecules are also important for influencing the fate of cells in this vertebrate model system.
Our results show that localisation of aPKC to the membrane of the deep cells of the neural plate, which are normally apolar and do not have aPKC localised to their membrane, inhibits these cells from becoming primary neurons. Surprisingly, membrane localisation of aPKC also increases its nuclear fraction and this is likely to be important because blocking aPKC activity with a nuclear dominant-negative enhances primary neurogenesis. Overexpression of Lgl2 leads to internalisation of outer cells and allows them to become competent for primary neurogenesis.
Thus, both of the polarity proteins tested, apical aPKC and basolateral Lgl2, have the ability to affect cell fate. However, our results place the emphasis on aPKC, which, through a nuclear function, seems to be instructive for suppressing neurogenesis and enhancing proliferation.
Apical proteins in cell fate determination
Work over many years in Drosophila neuroblasts has led to a model in which the role of the apically localised proteins in fate determination is restricted to targeting fate determinants to the basal side of the cell, leading to their subsequent inheritance by the basal apolar cells. This previous work has placed the emphasis of direct fate determination on the basolateral side of the cell (Knoblich, 2008). Our work puts emphasis on fate determination by the apical side, by showing that aPKC is sufficient and necessary to influence the fate of the cells in which it is expressed. This is more in line with reports from mammalian neurogenesis which suggested that it is the apical side of polarised neuroepithelial cells that determines their fate, even though it was still not clear what the apical determinants might be or whether the mechanism was direct (Gotz and Huttner, 2005; Kosodo et al., 2004). Recent reports have identified Par3, Par6 and Cdc42 as apical determinants of proliferation in mouse cortical neurogenesis, although the precise mechanism by which they influence cell fate is not yet clear (Cappello et al., 2006; Costa et al., 2008). It is possible that these recently identified apical determinants exert their effects on cell proliferation directly or indirectly via aPKC, which, in the present work, has been identified as a nuclear determinant of cell proliferation and fate. Interestingly, a recent study found that aPKC is asymmetrically inherited in neural stem cell division (Marthiens and ffrench-Constant, 2009).
aPKC as a nuclear determinant of cell fate
We have three lines of evidence to support the idea that the nuclear fraction of aPKC is important for determination of cell fate. First, the aPKC-CAAX phenotype can be entirely phenocopied by a nuclear-targeted form of the catalytic domain of aPKC (NLS-aPKC-CT). Second, aPKC-CAAX shows increased nuclear accumulation, alongside its strong cortical localisation. Third, a dominant-negative form of aPKC, which is exclusively nuclear, shows a phenotype opposite to that of aPKC-CAAX, suggesting that the nuclear fraction is functionally important.
In non-polarised cells (HeLa and PC12 cells), it has been shown that aPKC contains both a bipartite NLS and a nuclear export signal and can shuttle between the cytoplasm and the nucleus (Perander et al., 2001; Zhou et al., 1997). Whereas overexpressed wild-type aPKC and the endogenous aPKC are both detected in the nucleus, the membrane-targeted aPKC (aPKC-CAAX) shows increased nuclear accumulation and increased activity in our developmental fate assays. We interpret these findings to mean that there might be communication between membrane and nuclear aPKC and that activation of aPKC in the membrane increases its nuclear localisation and activity. Indeed, a construct with the SV40 NLS fused to full-length aPKC (NLS-aPKC) is inactive in this fate assay, suggesting that nuclear accumulation of aPKC alone is not sufficient for its activity; an activation step, most likely taking place at the membrane, is also important. The mechanism of nuclear accumulation after activation at the membrane is currently unknown, but protein modifications taking place at the membrane could alter intra-molecular interactions, releasing the pseudo-substrate inhibition on the kinase domain and also enhancing nuclear localisation (Perander et al., 2001).
Once in the nucleus, aPKC could affect cell proliferation and fate by influencing, directly or indirectly, the activity of transcription factors. Although the targets of aPKC in our system are not yet known, we note that several nuclear targets of aPKC have been reported previously, including nucleolin, nuclear heterogenous ribonucleoprotein A1 (hnRNP A1) and the general transcription factor SP1 (Municio et al., 1995; Pal et al., 1998; Zhou et al., 1997). It is important to emphasise that our findings do not show that the membrane fraction of aPKC is not functionally important. Indeed, the membrane-tethered dominant-negative aPKC shows a change in cell fate along with depolarisation (de-pigmentation) of the cells that is not observed with the constitutively nuclear dominant-negative aPKC. Further experimentation is needed to distinguish a cytoskeletal versus nuclear role and to understand exactly how they might be related.
aPKC affects cell proliferation
In normal development, the inner layer of the neural ectoderm contains a mixture of proliferating and post-mitotic cells, whereas the outer layer contains only proliferative cells (Chalmers et al., 2002; Hartenstein, 1989). We found that aPKC increased cell proliferation in both epidermal and neural ectoderm at an early stage in development, leading to an increase in the total number of cells and to a thickening of the ectoderm. In contrast to our observations, a previous report did not find an increase in cell proliferation upon aPKC-CAAX overexpression in Xenopus (Ossipova et al., 2007). This is most likely due to differences in the stage of analysis; in spite of the thickened ectoderm, we also did not find any increase in the number of proliferative cells at neurula stage; however, we did find that the number of proliferative cells, as well as the total number of cells and the mitotic index, were increased earlier, at the gastrula stage. Since some post-mitotic neurons are generated at this early stage in development, perturbations in cell polarity at the gastrula stage affect the number of primary neurons at the tadpole stage. The role of aPKC in promoting proliferation is in line with previous reports from Drosophila (Lee et al., 2006; Rolls et al., 2003) and chicken (Ghosh et al., 2008) and thus it is likely to be a conserved feature of aPKC function. Our data demonstrate that it is the nuclear kinase activity of aPKC that is responsible for keeping the cells in a proliferating state, as the effect on cell proliferation of overexpressing NLS-aPKC-CT is similar to that of aPKC-CAAX.
Permissive versus instructive role of fate determinants
Lgl2 and aPKC are thought to act antagonistically (Suzuki and Ohno, 2006; Atwood and Prehoda, 2009) and, indeed, in our system Lgl2 overexpression delocalises aPKC from the apical membrane (Chalmers et al., 2005). Thus, the Lgl2 overexpression phenotype would be expected to share at least some features with the dominant-negative aPKC phenotypes. Indeed, the dominant-negative aPKC constructs and Lgl2 overexpression cause an increase in N-tubulin; however, the dominant-negative nuclear aPKC does not appear to depolarise cells and does not cause the massive internalisation that is observed with Lgl2 overexpression. In addition, dominant-negative nuclear aPKC is sufficient to increase primary neurogenesis, whereas overexpression of Lgl2 only increases the population that is competent for primary neurogenesis. Although further experimentation is needed to understand Lgl2 function in neurogenesis, one interpretation of these results is that blocking nuclear aPKC function has a direct and instructive effect on cell fate, whereas Lgl2 might interfere primarily with the cytoskeletal function of aPKC and in consequence has a milder, permissive effect on cell fate. This is compatible with a model in which Lgl2 overexpression removes an inhibitory influence for primary neurogenesis, but additional signals/nuclear factors are needed for neurogenesis to occur.
In conclusion, our results have revealed a role for polarity molecules in cell fate, particularly with respect to their proliferative potential. The diversification of fate between inner apolar cells and outer polarised cells is a conserved phenomenon in early vertebrate development, although the precise fate that inner and outer cells follow is different in each case (e.g. inner cell mass versus trophectoderm in the mouse, enveloping layer versus embryo in the zebrafish, outer versus inner ectoderm cells in Xenopus). We expect that the mechanisms uncovered in this study would be conserved in other systems.
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/cgi/content/full/136/16/2767/DC1
Supplementary Material
We thank Jeremy Green and Sergei Sokol for discussions. We thank the Wellcome Trust (grant 057819/Z/05) and BBSRC (grant BB/E003044/1) for funding. N.P. is a Wellcome Trust Senior Research Fellow. A.D.C. is a Medical Research Council Career Development Fellow. Deposited in PMC for release after 6 months.
References
- Asashima, M. and Grunz, H. (1983). Effects of inducers on inner and outer gastrula ectoderm layers of Xenopus laevis. Differentiation 23, 206-212. [DOI] [PubMed] [Google Scholar]
- Atwood, S. X. and Prehoda, K. E. (2009). aPKC phosphorylates miranda to polarize fate determinants during neuroblast asymmetric cell division. Curr. Biol. 19, 723-729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellefroid, E. J., Bourguignon, C., Hollemann, T., Ma, Q., Anderson, D. J., Kintner, C. and Pieler, T. (1996). X-MyT1, a Xenopus C2HC-type zinc finger protein with a regulatory function in neuronal differentiation. Cell 87, 1191-1202. [DOI] [PubMed] [Google Scholar]
- Betschinger, J. and Knoblich, J. A. (2004). Dare to be different: asymmetric cell division in Drosophila, C. elegans and vertebrates. Curr. Biol. 14, R674-R685. [DOI] [PubMed] [Google Scholar]
- Bourguignon, C., Li, J. and Papalopulu, N. (1998). XBF-1, a winged helix transcription factor with dual activity, has a role in positioning neurogenesis in Xenopus competent ectoderm. Development 125, 4889-4900. [DOI] [PubMed] [Google Scholar]
- Bradley, L., Wainstock, D. and Sive, H. (1996). Positive and negative signals modulate formation of the Xenopus cement gland. Development 122, 2739-2750. [DOI] [PubMed] [Google Scholar]
- Cappello, S., Attardo, A., Wu, X., Iwasato, T., Itohara, S., Wilsch-Brauninger, M., Eilken, H. M., Rieger, M. A., Schroeder, T. T., Huttner, W. B. et al. (2006). The Rho-GTPase cdc42 regulates neural progenitor fate at the apical surface. Nat. Neurosci. 9, 1099-1107. [DOI] [PubMed] [Google Scholar]
- Chalmers, A. D., Welchman, D. and Papalopulu, N. (2002). Intrinsic differences between the superficial and deep layers of the Xenopus ectoderm control primary neuronal differentiation. Dev. Cell 2, 171-182. [DOI] [PubMed] [Google Scholar]
- Chalmers, A. D., Strauss, B. and Papalopulu, N. (2003). Oriented cell divisions asymmetrically segregate aPKC and generate cell fate diversity in the early Xenopus embryo. Development 130, 2657-2668. [DOI] [PubMed] [Google Scholar]
- Chalmers, A. D., Pambos, M., Mason, J., Lang, S., Wylie, C. and Papalopulu, N. (2005). aPKC, Crumbs3 and Lgl2 control apicobasal polarity in early vertebrate development. Development 132, 977-986. [DOI] [PubMed] [Google Scholar]
- Chalmers, A. D., Lachani, K., Shin, Y., Sherwood, V., Cho, K. W. and Papalopulu, N. (2006). Grainyhead-like 3, a transcription factor identified in a microarray screen, promotes the specification of the superficial layer of the embryonic epidermis. Mech. Dev. 123, 702-718. [DOI] [PubMed] [Google Scholar]
- Costa, M. R., Wen, G., Lepier, A., Schroeder, T. and Gotz, M. (2008). Par-complex proteins promote proliferative progenitor divisions in the developing mouse cerebral cortex. Development 135, 11-22. [DOI] [PubMed] [Google Scholar]
- Cui, S., Otten, C., Rohr, S., Abdelilah-Seyfried, S. and Link, B. A. (2007). Analysis of aPKClambda and aPKCzeta reveals multiple and redundant functions during vertebrate retinogenesis. Mol. Cell. Neurosci. 34, 431-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson, L. A. and Keller, R. E. (1999). Neural tube closure in Xenopus laevis involves medial migration, directed protrusive activity, cell intercalation and convergent extension. Development 126, 4547-4556. [DOI] [PubMed] [Google Scholar]
- Ghosh, S., Marquardt, T., Thaler, J. P., Carter, N., Andrews, S. E., Pfaff, S. L. and Hunter, T. (2008). Instructive role of aPKCzeta subcellular localization in the assembly of adherens junctions in neural progenitors. Proc. Natl. Acad. Sci. USA 105, 335-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlich, D. and Kutay, U. (1999). Transport between the cell nucleus and the cytoplasm. Annu. Rev. Cell Dev. Biol. 15, 607-660. [DOI] [PubMed] [Google Scholar]
- Gotz, M. and Huttner, W. B. (2005). The cell biology of neurogenesis. Nat. Rev. Mol. Cell Biol. 6, 777-788. [DOI] [PubMed] [Google Scholar]
- Hartenstein, V. (1989). Early neurogenesis in Xenopus: the spatio-temporal pattern of proliferation and cell lineages in the embryonic spinal cord. Neuron 3, 399-411. [DOI] [PubMed] [Google Scholar]
- Henrique, D. and Schweisguth, F. (2003). Cell polarity: the ups and downs of the Par6/aPKC complex. Curr. Opin. Genet. Dev. 13, 341-350. [DOI] [PubMed] [Google Scholar]
- Imai, F., Hirai, S., Akimoto, K., Koyama, H., Miyata, T., Ogawa, M., Noguchi, S., Sasaoka, T., Noda, T. and Ohno, S. (2006). Inactivation of aPKClambda results in the loss of adherens junctions in neuroepithelial cells without affecting neurogenesis in mouse neocortex. Development 133, 1735-1744. [DOI] [PubMed] [Google Scholar]
- Joberty, G., Petersen, C., Gao, L. and Macara, I. G. (2000). The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat. Cell Biol. 2, 531-539. [DOI] [PubMed] [Google Scholar]
- Jones, E. A. and Woodland, H. R. (1986). Development of the ectoderm in Xenopus: tissue specification and the role of cell association and division. Cell 44, 345-355. [DOI] [PubMed] [Google Scholar]
- Knoblich, J. A. (2008). Mechanisms of asymmetric stem cell division. Cell 132, 583-597. [DOI] [PubMed] [Google Scholar]
- Kosodo, Y., Roper, K., Haubensak, W., Marzesco, A. M., Corbeil, D. and Huttner, W. B. (2004). Asymmetric distribution of the apical plasma membrane during neurogenic divisions of mammalian neuroepithelial cells. EMBO J. 23, 2314-2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamborghini, J. E. (1980). Rohon-beard cells and other large neurons in Xenopus embryos originate during gastrulation. J. Comp. Neurol. 189, 323-333. [DOI] [PubMed] [Google Scholar]
- Lee, C. Y., Robinson, K. J. and Doe, C. Q. (2006). Lgl, Pins and aPKC regulate neuroblast self-renewal versus differentiation. Nature 439, 594-598. [DOI] [PubMed] [Google Scholar]
- Lin, D., Edwards, A. S., Fawcett, J. P., Mbamalu, G., Scott, J. D. and Pawson, T. (2000). A mammalian PAR-3-PAR-6 complex implicated in Cdc42/Rac1 and aPKC signalling and cell polarity. Nat. Cell Biol. 2, 540-547. [DOI] [PubMed] [Google Scholar]
- Marthiens, V. and ffrench-Constant, C. (2009). Adherens junction domains are split by asymmetric division of embryonic neural stem cells. EMBO Rep. 10, 515-520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyoshi, K., Richards, L. J., Akazawa, C., O'Leary, D. D. and Nakanishi, S. (1996). Labeling neural cells using adenoviral gene transfer of membrane-targeted GFP. Neuron 16, 255-260. [DOI] [PubMed] [Google Scholar]
- Muller, H. A. (2001). Of mice, frogs and flies: generation of membrane asymmetries in early development. Dev. Growth Differ. 43, 327-342. [DOI] [PubMed] [Google Scholar]
- Muller, H. A. and Hausen, P. (1995). Epithelial cell polarity in early Xenopus development. Dev. Dyn. 202, 405-420. [DOI] [PubMed] [Google Scholar]
- Muller, H. A. and Bossinger, O. (2003). Molecular networks controlling epithelial cell polarity in development. Mech. Dev. 120, 1231-1256. [DOI] [PubMed] [Google Scholar]
- Municio, M. M., Lozano, J., Sanchez, P., Moscat, J. and Diaz-Meco, M. T. (1995). Identification of heterogeneous ribonucleoprotein A1 as a novel substrate for protein kinase C zeta. J. Biol. Chem. 270, 15884-15891. [DOI] [PubMed] [Google Scholar]
- Ossipova, O., Tabler, J., Green, J. B. and Sokol, S. Y. (2007). PAR1 specifies ciliated cells in vertebrate ectoderm downstream of aPKC. Development 134, 4297-4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal, S., Claffey, K. P., Cohen, H. T. and Mukhopadhyay, D. (1998). Activation of Sp1-mediated vascular permeability factor/vascular endothelial growth factor transcription requires specific interaction with protein kinase C zeta. J. Biol. Chem. 273, 26277-26280. [DOI] [PubMed] [Google Scholar]
- Perander, M., Bjorkoy, G. and Johansen, T. (2001). Nuclear import and export signals enable rapid nucleocytoplasmic shuttling of the atypical protein kinase C lambda. J. Biol. Chem. 276, 13015-13024. [DOI] [PubMed] [Google Scholar]
- Prufert, K., Vogel, A. and Krohne, G. (2004). The lamin CxxM motif promotes nuclear membrane growth. J. Cell Sci. 117, 6105-6116. [DOI] [PubMed] [Google Scholar]
- Ralle, T., Grund, C., Franke, W. W. and Stick, R. (2004). Intranuclear membrane structure formations by CaaX-containing nuclear proteins. J. Cell Sci. 117, 6095-6104. [DOI] [PubMed] [Google Scholar]
- Regad, T., Roth, M., Bredenkamp, N., Illing, N. and Papalopulu, N. (2007). The neural progenitor-specifying activity of FoxG1 is antagonistically regulated by CKI and FGF. Nat. Cell Biol. 9, 531-540. [DOI] [PubMed] [Google Scholar]
- Rolls, M. M., Albertson, R., Shih, H. P., Lee, C. Y. and Doe, C. Q. (2003). Drosophila aPKC regulates cell polarity and cell proliferation in neuroblasts and epithelia. J. Cell Biol. 163, 1089-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabherwal, N., Schneider, K. U., Blaschke, R. J., Marchini, A. and Rappold, G. (2004). Impairment of SHOX nuclear localization as a cause for Leri-Weill syndrome. J. Cell Sci. 117, 3041-3048. [DOI] [PubMed] [Google Scholar]
- Shin, K., Fogg, V. C. and Margolis, B. (2006). Tight junctions and cell polarity. Annu. Rev. Cell Dev. Biol. 22, 207-235. [DOI] [PubMed] [Google Scholar]
- Sotillos, S., Diaz-Meco, M. T., Caminero, E., Moscat, J. and Campuzano, S. (2004). DaPKC-dependent phosphorylation of Crumbs is required for epithelial cell polarity in Drosophila. J. Cell Biol. 166, 549-557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, A. and Ohno, S. (2006). The PAR-aPKC system: lessons in polarity. J. Cell Sci. 119, 979-987. [DOI] [PubMed] [Google Scholar]
- Vasioukhin, V. (2006). Lethal giant puzzle of Lgl. Dev. Neurosci. 28, 13-24. [DOI] [PubMed] [Google Scholar]
- Wright, L. P. and Philips, M. R. (2006). Thematic review series: lipid posttranslational modifications. CAAX modification and membrane targeting of Ras. J. Lipid Res. 47, 883-891. [DOI] [PubMed] [Google Scholar]
- Zhou, G., Seibenhener, M. L. and Wooten, M. W. (1997). Nucleolin is a protein kinase C-zeta substrate. Connection between cell surface signaling and nucleus in PC12 cells. J. Biol. Chem. 272, 31130-31137. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}