

Abstract
The distribution of the haemoglobin receptor gene (hmbR) was investigated in disease and carried Neisseria meningitidis isolates revealing that the gene occurred at a significantly higher frequency in disease isolates compared to those obtained from carriage. Where hmbR was absent, the locus was occupied by the cassettes exl2 or exl3, or with a “pseudo hmbR” gene designated exl4. The hmbR locus in published N. meningitidis genomes, as well as N. gonorrhoeae and N. lactamica ST-640, exhibited characteristics of a pathogenicity island. These data are consistent with a role for the hmbR gene in meningococcal disease.
Keywords: haemoglobin receptor, meningococcal disease, virulence, pathogenicity island
INTRODUCTION
The documented absence of the haemoglobin receptor gene among a number of Neisseria meningitidis isolates led to the discovery that the gene was located on a genetic island containing multiple cassettes including the putative proteins Exl2, Exl3 followed by other unidentified open reading frames (ORF) [1, 2]. Lack of the hmbR gene among some meningococcal isolates, combined with the association of the gene with mobile genetic elements, indicates that it may form part of a DNA virulence island recently acquired by meningococci [3, 4].
Here the distribution of the hmbR gene among over 700 disease and carriage-associated meningococci from three separate isolate collections was investigated, revealing a statistically significant association between disease and the presence of the hmbR gene. In silico analysis of the hmbR genetic locus among the genomes belonging to N. meningitidis isolates Z2491, FAM18, MC58, NMC053442, N. gonorrhoeae FA1090 and N. lactamica ST-640 indicated that the gene was located on a putative pathogenicity island. DNA arrays comparing the genomes of isolates belonging to the ST-41/44 clonal complex showed a higher likelihood of the hmbR gene being found among isolates from cases of invasive disease.
METHODS
Meningococcal isolates
Three isolate collections were examined. The first comprised 96 mainly disease-associated isolates assembled globally from 1937 to 1996 and described by Maiden et al [5]. The second contained 203 carried and invasive isolates forming part of a collection assembled in 1993 in the Czech Republic [6]. The third collection included 462 isolates obtained in England and Wales between 1999 and 2000, of which 265 were carried and 197 were disease-associated. These isolates were from the Meningococcal Reference Unit and the UK carriage study.
PCR amplification and nucleotide sequence determination
The complete hmbR gene was amplified using primers PN1 5′-CCGGAAGGAATGATGCCGCACAGG-3′ and NE3 5′-TTAAAACTTCCATTCCAGCG-3′. Isolates hmbR negative were further screened by PCR using primers hemO 5′-GAATGATGCCGCAGAG-3′ and col2 5′-GCATTCAGGTCAAAATCC-3′, which are located in the hemO and col genes on either side of the genetic island thus amplifying the whole region. Following this procedure, amplicons were sequenced using the primers hemO, exl2-R 5′-CCGTTCAGGGATTTACCGGC-3′, exl3-F 5′-GCCTGCAAATACGCCGAAGCC-3′, exl3-R 5′-GCCTGCAAATACGCCGAAGCCG-3′, exl3-seqF 5′-GGCGGAGTGCAAAATGATGC-3′ and exl3-seqR 5′-GCCATCTTTTAATTTAGCCGC-3′ revealing the exchangeable cassette replacing the hmbR gene. Nucleotide accession numbers can be found in the Appendix A1.
Whole genome comparisons of the ST41/44 clonal complex meningococci by DNA array
Techniques and data analysis were as described previously [7, 8] with data deposited in the Array express database www.ebi.ac.uk/arrayexpress/query/entry with accession number E-MEXP-343.
Statistical analyses
These were conducted using Intercooled STATA version 10 software for windows. Multivariable logistic regression models were used to estimate the strength of association between hmbR status and whether the isolate originated from a healthy carrier or a clinical case.
RESULTS
Distribution of the hmbR gene and association with disease
A total of 761 N. meningitidis isolates were screened for the presence of the hmbR gene. A minority of disease isolates lacking the gene were present with 11 (14%), 1 (7%) and 5 (3%) hmbR negative isolates present in all three collections respectively, indicative of a significant association of the gene with virulence (Table 1). The odds ratio for the association of the hmbR gene with all disease isolates and for each isolate collection provided further evidence for the predisposition of disease isolates to contain the hmbR gene. The over-representation of the gene among disease N. meningitidis isolates was apparent with the hyper-invasive lineages ST-4, ST-5, ST-8, ST-11, ST-18 and ST-32 exclusively hmbR positive (Appendix A2).
Table 1.
Distribution of the hmbR gene in all collections
| Isolate collections | hmbR positive (%) | hmbR negative (%) | Disease association OR [95% confidence interval] | Total |
|---|---|---|---|---|
| 107 MLST Disease isolates | 68 (86) | 11 (14) | 3.63 [0.98 to 13.35] | 79 |
| 107 MLST Carriage isolates | 10 (59) | 7 (41) | 17 | |
| Czech 1993 Disease isolates | 37 (97) | 1 (3) | 3.51 [1.19 to 10.40] | 38 |
| Czech 1993 Carriage isolates | 125 (76) | 40 (24) | 165 | |
| UK 1999 Disease isolates | 192 (97) | 5 (3) | 10.30 [4.033 to 26.26] | 197 |
| UK 1999 Carriage isolates | 210 (79) | 55 (21) | 265 | |
| All Disease isolates | 297 (95) | 17 (5) | 4.42 [2.72 to 7.16] | 314 |
| All Carriage isolates | 345 (77) | 102 (23) | 447 | |
| Total | 642 | 119 | 761 |
Results are significant (P < 0.0001). Relative risk = 1.26 [95% confidence interval: 1.18 to 1.34].
Association of exl2 and exl3 with carried clonal complexes
Isolates lacking the hmbR gene were found among clonal complexes ST-1, ST-22, ST-23, ST-41/44, ST-60, ST-106, ST-198, ST-549 in addition to many isolates currently unassigned to a clonal complex (Appendix A2). The exl2 exchangeable cassette was principally associated with serogroup Y isolates of the ST-23 clonal complex (59%).The exl3 genetic island was present in clonal complexes ST-1, ST-106, ST-549, ST-22 and ST-60.
Characterisation of a “pseudo hmbR” gene designated exl4
A total of 28 isolates from the clonal complexes ST-41/44 and ST-198 lacked both exl genetic islands and the hmbR gene (Appendix A2). BLAST searches of the sequence replacing exl2, exl3 and hmbR in these isolates revealed 97% sequence identity with a “pseudo hmbR gene” identified in the sequenced genome of isolate NMC053442, a Chinese ST-4821 serogroup C isolate [9]. Further analysis of the 2, 477 bp long “pseudo hmbR gene” belonging to isolate NMC053442 revealed 62% sequence identity over a length of 977 bp fragments with published hmbR sequences indicating that this may not be an hmbR gene but may instead contain fragments of the hmbR gene. Furthermore, the “pseudo hmbR gene” identified in the ST-41/44 and ST-198 isolates, as well as isolate NMC053442, was found to contain a polydeoxyguanosine phosphate tract occurring at 663 bp, much earlier than that of published hmbR genes, which are known to occur at 1159 bp. Consequently, this novel ORF replacing hmbR was designated exl4.
Clonal Complex ST-41/44 and hmbR
DNA arrays were used to compare the genomes of isolates belonging to recognised invasive sequence types (e.g. ST-41) with isolates belonging to sequence types not associated with disease (e.g. ST-44, ST-110 and ST-111). Within the ST-41/44 clonal complex, the association of hmbR with disease N. meningitidis isolates was significant (P< 0.0001) with 65 (98%) of the 66 disease-associated isolates containing the hmbR gene compared to 41 (69%) out of 59 carriage isolates. Genome comparisons between ST-41 and ST-44 isolates did not show any other major differences.
Genetic arrangement of the hmbR and exl loci in Neisseria
In silico analysis of the hmbR locus among the sequenced genomes from N. meningitidis, N. gonorrhoeae and N. lactamica identified seven different cassettes (A-G). Region I contained hemO, a gene involved in the catabolism of heme. Region II included the collagenase gene homologue, col, bordered by a 27 bp inverted repeat. Region III contained the hmbR gene, or the exchangeable islands exl2, exl3 and exl4 (Figure 1). The previously documented hmbR locus belonging to N. meningitidis GA0929 containing the exl2 exchangeable cassette was also included in the alignment [2]. Multiple unidentified ORFs, Correia elements [10] and inverted repeats were identified - all key characteristics of pathogenicity islands.
Figure 1. Genetic arrangement of the hmbR locus among Neisseria.
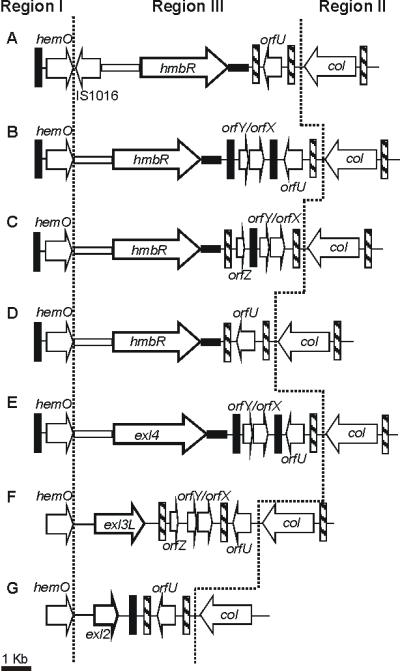
A. N. gonorrhoeae FA1090 [accession number AE004969]; B. N. meningitidis FAM18 [accession number AM421808]; C. N. meningitidis MC58 [accession number AE002098]; D. N. meningitidis Z2491 [accession number AL157959]; E. N. meningitidis NMC053442 [accession number CP000381]; F. N. lactamica ST-640 and G. N. meningitidis isolate GA0929 [accession number AF319532]. Annotation was derived from the published genome data available at TIGR, the Sanger institute and data published by Kahler et al [2]. Permission for use of the N. lactamica genome data was kindly given by Julian Parkhill at the Sanger Institute and genome data is available at http://www.sanger.ac.uk/Projects/N_lactamica//.
Each ORF is represented by an arrow the direction of which indicating the theoretical direction of transcription. Thin black vertical bars indicate Correia elements, hatched vertical bars represent inverted repeats. Open horizontal blocks denote highly conserved 174 bp regions located at the 5′ end of the hmbR and exl4 ORFs. Black horizontal blocks represent 112 bp conserved regions located immediately after the hmbR or exl4 ORFs. Both conserved regions were not found in N. lactamica or N. meningitidis GA0929.
DISCUSSION
Neisseria meningitidis remains a major cause of meningitis and septicaemia worldwide, however despite its pathogenic potential, N. meningitidis is also a common human commensal [11, 12]. The capsular polysaccharide, type IV pili and iron acquisition systems are all necessary pathogenesis determinants, but while each of these factors is required for invasive disease it is not sufficient, with the likelihood that many other genetic factors contribute to the ability of a meningococcus to cause invasive disease. The search for new virulence factors in this species is hampered by the lack of a suitable animal model representative of human disease. An alternative strategy is to employ a molecular epidemiological approach to establish a statistical association of a gene with disease. Here, the distribution of the haemoglobin receptor gene was examined in over 700 meningococci isolated from cases of disease and asymptomatic carriage.
The hmbR gene was first identified in 1995 and the importance of haemoglobin utilisation during invasion was demonstrated when an N. meningitidis hmbR mutant was attenuated in an infant rat model of meningococcal infection [13]. Data presented in this study described a statistically significant association of the hmbR gene with disease N. meningitidis isolates, providing epidemiological evidence for the importance of the hmbR gene in meningococcal virulence (Table 1). Combined with the documented low diversity of the gene [14], these data indicate a potential role of HmbR in future vaccines.
The absence of hmbR among N. meningitidis isolates has been reported [1] and, due to the prevalence of the hpuAB complex among those isolates, it was suggested that hmbR may have been a recent acquisition by meningococci. Examination of the genetic island among the genomes from N. meningitidis FAM18, Z2491, MC58 and NMC053442, N. gonorrhoeae FA1090 and N. lactamica ST-640 revealed that the region contained many inverted repeats, a feature of pathogenicity islands (PAIs) (Figure 1) [4]. Further PAI characteristics included an overall G+C content of 47% below the mean 51% of the Neisseria genome content, the presence of cryptic ORFs, the insertion sequence IS1060 in N. gonorrhoeae FA1090 as well as Correia elements among N. meningitidis isolates [3]. Finally, the hmbR locus was not composed of a homogeneous piece of DNA but was instead made up of several mosaic-like ORFs, another key PAI feature [3, 4, 15].
In the present study, a minority of 17 (5%) of the 314 disease N. meningitidis isolates from all three isolate collections lacked the hmbR gene indicative of the over-representation of the gene among disease isolates (Table 1). Most cases of meningococcal disease are caused by only a few clonal complexes of related sequence types (STs) referred to as hyper-invasive lineages [11]. The association of the hmbR gene with disease was apparent in isolates belonging to such lineages including ST-4, ST-5, ST-8, ST-11, ST-18 and ST-32 where, without exception, all isolates contained the gene (Appendix A2).
The importance of the hmbR gene in disease isolates was consistent among meningococci of the ST-41/44 clonal complex, members of which have been the prevalent cause of disease in parts of Northern Europe during the past decade and responsible for epidemic disease in New Zealand [16]. This complex of closely related clones is unusual in that it is centred on two sequence types, ST-41 and ST-44, rather than possessing a single central ST [5]. ST-41 related meningococci are in general isolated from cases of invasive disease, whereas ST-44 related clones are typically associated with carriage [11]. In the current study, all of the ST-41 related isolates possessed an hmbR gene. Conversely, all of those isolates lacking the gene were among ST-44 related meningococci. Using this DNA array, no other major differences were observed between the ST-41 and ST-44 related meningococci.
Alternative cassettes for the HmbR locus were the putative lipoproteins Exl2 and Exl3 [2]. The function of both Exl2 and Exl3 is unknown, however they both present 69% sequence identity with the TbpB-like protein, GNA2132, as well as TbpB, LbpB and HpuA - consistent with a putative role in iron uptake or metabolism [2]. In agreement with Kahler et al, the genetic island, exl2, was predominantly found among meningococci with a serogroup Y polysaccharide capsule belonging to the ET-508/ST-23 clonal complex (Appendix A2). The predominance of exl2 in meningococci from the ST-23 clonal complex may be a result of clonal expansion. Clonal complexes are often associated with particular phenotypic characteristics such as serogroup or subcapsular antigens [17].
The exl3 genetic island was predominantly found among meningococci belonging to the lineages ST-22, ST-106, ST-549 and ST-60 (Appendix A2). However, in agreement with previous analysis, exl3 was also found among isolates in the ST-1 clonal complex which, in contrast, primarily contained disease-causing serogroup A meningococci [2]. The absence of the hmbR gene among serogroup A meningococci has been documented, however all of the isolates contained the hpuB gene part of the HpuAB bipartite receptor for Hb and hapto-haemoglobin acquisition indicating that this was the main haemoglobin receptor for serogroup A meningococci [1]. In the present study, all of the serogroup A lineage ST-5 (N=13) and ST-7 (N=2) meningococci contained hmbR genes, suggesting that replacement of the hmbR gene with the exl3 locus was limited to serogroup A clonal complex ST-1 meningococci.
A total of 19 clonal complex ST-41/44 and nine ST-198 isolates contained neither the hmbR gene nor either of the exl genetic islands. Instead the locus was replaced with an ORF designated exl4 sharing 97 % identity with the “pseudo hmbR” gene from the recently sequenced genome of ST-4821 isolate NMC053442 [9]. The “pseudo hmbR” gene belonging to isolate NMC053442 was highly divergent from other hmbR genes with BLAST searches revealing 62% identity in a 977 bp fragment of the hmbR gene. Previous studies have documented the relatively high conservation of hmbR among N. meningitidis isolates raising the question of whether the gene identified in the Chinese isolate NMC053442 may be another exl island containing a hmbR fragment [14].
In conclusion, an epidemiological survey of the hmbR gene among both disease and carriage associated N. meningitidis isolates revealed a statistically significant association of the hmbR gene with virulent N. meningitidis isolates consistent with a role for hmbR during pathogenesis. This study highlights the power of molecular epidemiological studies in determining novel virulence determinants.
Supplementary Material
Acknowledgments
This work was funded by the National Institute for Biological Standards and Control (NIBSC), the Wellcome Trust and the University of Manchester.
Footnotes
The authors of this paper do not have any commercial or other associations that might pose a conflict of interest (e.g. pharmaceutical stock ownership, consultancy, advisory board membership, relevant patents, or research funding).
This information was presented at the 16th International Pathogenic Neisseria Conference, Rotterdam, The Netherlands. September 7-12 2008, 1st Session Epidemiology.
REFERENCES
- 1.Richardson AR, Stojiljkovic I. HmbR, a hemoglobin-binding outer membrane protein of Neisseria meningitidis, undergoes phase variation. J Bacteriol. 1999;181:2067–74. doi: 10.1128/jb.181.7.2067-2074.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kahler CM, Blum E, Miller YK, Ryan D, Popovic T, Stephens DS. exl, an exchangeable genetic island in Neisseria meningitidis. Infect Immun. 2001;69:1687–96. doi: 10.1128/IAI.69.3.1687-1696.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hacker J, Kaper JB. Pathogenicity islands and the evolution of microbes. Annu Rev Microbiol. 2000;54:641–79. doi: 10.1146/annurev.micro.54.1.641. [DOI] [PubMed] [Google Scholar]
- 4.Gal-Mor O, Finlay BB. Pathogenicity islands: a molecular toolbox for bacterial virulence. Cell Microbiol. 2006;8:1707–19. doi: 10.1111/j.1462-5822.2006.00794.x. [DOI] [PubMed] [Google Scholar]
- 5.Maiden MC, Bygraves JA, Feil E, et al. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A. 1998;95:3140–5. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jolley KA, Kalmusova J, Feil EJ, et al. Carried meningococci in the Czech Republic: a diverse recombining population. J Clin Microbiol. 2000;38:4492–8. doi: 10.1128/jcm.38.12.4492-4498.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bille E, Zahar JR, Perrin A, et al. A chromosomally integrated bacteriophage in invasive meningococci. J Exp Med. 2005;201:1905–13. doi: 10.1084/jem.20050112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perrin A, Bonacorsi S, Carbonnelle E, et al. Comparative genomics identifies the genetic islands that distinguish Neisseria meningitidis, the agent of cerebrospinal meningitis, from other Neisseria species. Infect Immun. 2002;70:7063–72. doi: 10.1128/IAI.70.12.7063-7072.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peng J, Yang L, Yang F, et al. Characterization of ST-4821 complex, a unique Neisseria meningitidis clone. Genomics. 2008;91:78–87. doi: 10.1016/j.ygeno.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 10.Correia FF, Inouye S, Inouye M. A 26-base-pair repetitive sequence specific for Neisseria gonorrhoeae and Neisseria meningitidis genomic DNA. J Bacteriol. 1986;167:1009–15. doi: 10.1128/jb.167.3.1009-1015.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yazdankhah SP, Kriz P, Tzanakaki G, et al. Distribution of serogroups and genotypes among disease-associated and carried isolates of Neisseria meningitidis from the Czech Republic, Greece, and Norway. J Clin Microbiol. 2004;42:5146–53. doi: 10.1128/JCM.42.11.5146-5153.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cartwright KA, Stuart JM, Jones DM, Noah ND. The Stonehouse survey: nasopharyngeal carriage of meningococci and Neisseria lactamica. Epidemiol Infect. 1987;99:591–601. doi: 10.1017/s0950268800066449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stojiljkovic I, Hwa V, de Saint Martin L, et al. The Neisseria meningitidis haemoglobin receptor: its role in iron utilization and virulence. Mol Microbiol. 1995;15:531–41. doi: 10.1111/j.1365-2958.1995.tb02266.x. [DOI] [PubMed] [Google Scholar]
- 14.Stojiljkovic I, Larson J, Hwa V, Anic S, So M. HmbR outer membrane receptors of pathogenic Neisseria_spp.: iron-regulated, hemoglobin-binding proteins with a high level of primary structure conservation. J Bacteriol. 1996;178:4670–8. doi: 10.1128/jb.178.15.4670-4678.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hacker J, Carniel E. Ecological fitness, genomic islands and bacterial pathogenicity. A Darwinian view of the evolution of microbes. EMBO Rep. 2001;2:376–81. doi: 10.1093/embo-reports/kve097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dyet K, Devoy A, McDowell R, Martin D. New Zealand’s epidemic of meningococcal disease described using molecular analysis: implications for vaccine delivery. Vaccine. 2005;23:2228–30. doi: 10.1016/j.vaccine.2005.01.050. [DOI] [PubMed] [Google Scholar]
- 17.Urwin R, Russell JE, Thompson EA, Holmes EC, Feavers IM, Maiden MC. Distribution of surface protein variants among hyperinvasive meningococci: implications for vaccine design. Infect Immun. 2004;72:5955–62. doi: 10.1128/IAI.72.10.5955-5962.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.