

Abstract
The tumor suppressor p53 is in equilibrium at cellular concentrations between dimers and tetramers. Oncogenic mutant p53 (mut) exerts a dominant-negative effect on co-expression of p53 wild-type (wt) and mut alleles in cancer cells. It is believed that wt and mut form hetero-tetramers of attenuated activity, via their tetramerization domains. Using electrospray mass spectrometry on isotopically labeled samples, we measured directly the composition and rates of formation of p53 complexes in the presence and absence of response element DNA. The dissociation of tetramers was unexpectedly very slow (t1/2 = 40 min) at 37 °C, matched by slow association of dimers, which is approximately four times longer than the half-life of spontaneous denaturation of wt p53. On mixing wt tetramers with the oncogenic contact mutant R273H of low DNA affinity, we observed the same slow formation of only wt4, wt2mut2, and mut4, in the ratio 1:2:1, on a cellular time scale. On mixing wt and mut with response element DNAs P21 and BAX, we observed only the complexes wt4.DNA, wt2mut2.DNA, and mut4.DNA, with relative dissociation constants 1:4:71 and 1:13:85, respectively, accounting for the dominant-negative effect by weakened affinity. p53 dimers assemble rapidly to tetramers on binding to response element DNA, initiated by the p53 DNA binding domains. The slow oligomerization of free p53, competing with spontaneous denaturation, has implications for the possible regulation of p53 by binding proteins and DNA that affect tetramerization kinetics as well as equilibria.
Keywords: mass spectrometry, protein-protein interactions, slow association, tetramerization domain
The tumor suppressor p53 is a transcription factor that is inactivated by mutation in some 50% of human cancers (1, 2). p53 consists of an unstructured N-terminal domain (residues 1–94) which points away from the rest of the protein (3), a folded core domain (residues 94–292), a linker to the tetramerization domain (residues 325–355) and an unstructured C-terminal domain (356–394). Nearly all of the mutations reside in its DNA-binding domain. Mutations can be either ‘contact,’ which remove residues that interact with DNA and lower affinity, or ‘structural,’ which destabilize the protein, sometimes with concomitant conformational changes (4). The transcriptionally active form of p53 is a homo-tetramer, linked via its tetramerization domains. Since most cancer mutations are located outside the tetramerization domain, in the core domain, the mutant (mut) proteins retain wild-type (wt) ability to form tetramers. Consequently the formation of hetero-tetramer complexes between wt and mut p53 is observed both in vivo and in vitro (5). It is thought that formation of hybrids between the wt and the mut protein is the cause of the trans-dominant effect of the mut over the wt in heterozygous cells (dominant-negative effect), either following somatic mutation or from birth as in the case of Li-Fraumeni syndrome (5, 6). In such events, the chances of initiating cancer are increased (7), and the p53 heterozygous cell may enhance invasion and migration of the tumor (8). The extent of the dominant-negative effect is mutant-specific (9). The exact natures of wt and mut homo-tetramers, as well as the dynamics of tetramer formation are only poorly understood. The equilibrium constant for the formation of the tetramers, approximately 20 nM, is such that there is an equilibrium between dimers and tetramers in the cell, and dimer-tetramerization phenomena may be important in the regulation of the activity of p53 (10–15).
It is difficult to analyze equilibria between complexes of p53 and DNA in mixtures of wt and mut proteins using standard bulk measurements. However, mass spectrometry (MS) gives another route for direct study of discrete populations of molecules. Intact protein complexes of masses up to megaDaltons can be studied using soft ionization electro-spray (ESI-MS) techniques (16–18). The kinetics of formation of homo-protein complexes and the exchange of subunits can be monitored using ESI-MS and isotopically labeled samples, and mass spectrometry can be used as a quantitative tool for characterizing protein complexes (19–21). Here, we have used ESI-MS to determine the dissociation and assembly kinetics of the subunits of the wt p53 tetramer in a framework, stabilized by four mutations in the core domain that do not perturb the structure (22, 23) that allows the protein to be studied over extended periods (22, 23). All experiments were performed on the core plus tetramerization domain construct (residues 94–356) which has the same quaternary structure as full-length protein (24). Parallel experiments were done on the R273H contact mutant (25), which has the same stability as the other construct (26).
We measured the composition and the kinetics of formation of complexes formed on mixing wt p53 with the most frequent contact mutant, R273H. We also measured directly the complexes formed between response element DNAs in the homo- and hetero-hybrids of wt and mut. We discovered that the formation and exchange of complexes are very slow in the absence of DNA, and that only a subset of the possible combinations of wt and mut hybrids were formed. We were also able to quantify the affinity for different compositions of wt and contact mut hybrids for two representative response element DNAs.
Results
Exchange Rate of p53 Subunits.
The exchange of p53CT subunits was studied using an equimolar solution of an unlabeled protein (12C–14N = “wtL”) with expected molecular weight of 120.2 kDa and a uniformly labeled protein with expected molecular weight of 127.0 kDa (13C–15N = “wtH”). The measured values were identical, and the experimental errors were less than ±0.020 kDa. The initial concentration of wtL4 and wtH4 were 30 μM each, in monomers (= 7.5 μM tetramers) as verified by several techniques. The equilibrium constant for the formation of tetramers from dimers is approximately 20 nM, so the starting species were nearly 100% tetrameric.
The spectra at different time points in Fig. 1, illustrate the subunit exchange of wt at room temperature. The natural and heavy isotopically labeled p53CT, wtL4, and wtH4 homo-tetramers, were essentially the only species present immediately after mixing and had their expected molecular weights. A new set of peaks corresponding to wtL2wtH2 emerged in the spectrum taken after 0.5 h, which increased in intensity until equilibrium was reached after approximately 10 h. No further hybrids were seen up to 10 days of incubation. To determine the relative abundance of the different species, we simulated the mass spectra with an in-house program based on LabVIEW, which deconvolutes each unresolved peak into the sum of its components (27). The relative abundances for each species were then plotted against time and fitted to a simple first-order exponential (Fig. 2A). The half time for exchange was 2.3 h. The equilibrium constants between wtL4, wtL2wtH2 and wtH4 were directly calculated from the ratios of intensities at equilibrium. The same exchange pattern and rates were observed using the R273H mutant (Fig. 2B). The same results were observed for incubation of p53 wt with p53 R273H. Again, only 2:2 hetero-tetramers were formed during co-incubation, with the same rate constants as found for the homo-tetramer. Repetition of the experiments between 4–65 μM (monomers) using different labeling regimes gave the same rate constant, indicating that data were independent of concentration in a range well above the dissociation constant between wt4 and wt2.
Fig. 1.

Exchange of p53CT subunits during incubation at 20 °C as determined by ESI-MS. Selected MS spectra of 12C-14N (blue) and 13C-15N (black) p53CT at different time points. The numbers represent the charge states of the respective peaks. No species other than wtL2wtH2 was observed for up to 10 days.
Fig. 2.

Exchange of p53CT wt-wt and mut-mut 20 °C. Both have similar kinetics and patterns. (A) wtL (12C-14N)/wtH (13C-15N) exchange, as shown in Fig. 1. The relative intensities of the species were calculated from the spectra and plotted against time. (B) mutL/mutH exchange. The relative intensities of the species were calculated from the spectra and plotted against time. For both A and B, we plotted the average of L4 and H4 (L4/H4).
The same experiments were repeated at 37 °C. After ≈2 h, the system reached equilibrium for the primary dimer exchange (Fig. 3), 5-fold faster than incubation at 20 °C with t1/2 of 40 min. But, sets of peaks corresponding to L3H1, and L1H3 were also observed. The protein was not stable for long enough to reach the theoretical equilibrium distribution of 1:4:6:4:1 for mutL4, mutL3mutH1, mutL2mutH2, mutL1mutH3, mutH4, respectively. It should be noted that wild-type protein that does not contain the four stabilizing mutations has a half life of only 9–16 min at 37 °C (28).
Fig. 3.

Exchange of p53CT R273H subunits during incubation at 37 °C. (A) MS spectra over time of 12C-14N (blue) with 13C-15N (black). Additionally 1:3, 3:1 hetero-tetramer species appear that were not observed at 20 °C. (B) The relative intensities of each species, mutL4, mutL3mutH1, mutL2mutH2, mutL1mutH3, and mutH4, were extracted from the spectra using simulation and then plotted against time. Equilibrium was not achieved because of aggregation of the protein after 6 h. The same results were found for wt-mut.
It can be shown for the reaction:
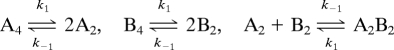 |
where, for example, A and B are different forms of p53 monomers, at concentrations well above the dissociation constant for formation of tetramers, the rate constant for exchange is k1.
Binding of DNA to p53 Homo- and Heterotetramers.
The exchange of subunits is immediately quenched by the addition of response element DNA so binding of DNA may be measured without perturbing the equilibrium. We measured the binding of an equimolar mixture of the preequilibrated wt and R273H mut tetrameric p53 to a 29-residue, double-stranded oligomer containing the P21 DNA response element sequence, which is of high affinity (29), and to a 31 residue oligomer containing the low affinity BAX sequence (29). The mixture of wt and R273H might simulate the hybrids found in vivo in heterozygous cells. Incubation was at 4 °C for 24 h to form a 1:2:1 distribution of active protein. Then, various concentrations of response element DNA were added, the binding of which equilibrated within the 5 min of sample preparation time. The center of gravity of the spectra was progressively shifted with increasing DNA concentration. First, the peaks corresponding to wt4.DNA appeared followed by wt2mut2.DNA and, finally, mut4.DNA. Sequentially, the peaks of wtL4 disappeared first, followed by wt2mut2. DNA-free mut4, was still present at the highest concentration of DNA, even for the high affinity response element P21. The mut4.DNA complex did not form until sufficient DNA had been added that all of the wt4 and wt2mut2 had been bound, leading to a lag in the binding curve.
Calculation of Absolute KDs of wt4, wt2mut2, and mut4 Complexes with DNA.
The concentrations of protein used in the MS-ESI experiments (∼1 μM in tetramers) were too far above the KDs of the wt4.P21DNA complexes for accurate determination of dissociation constants. The low concentrations of added DNA were nearly all bound to the wt4 protein and the wt2mut2 so that the concentration of unbound DNA was hard to calculate accurately (Fig. 4A). The concentration of p53 in the sample was verified by a stoichiometric titration of P21 DNA into the preincubated mixture of p53, which was about 100 times above its dissociation constant of tetramers from DNA (Fig. 5). The concentration thus measured was ∼80% of that calculated from the dilutions, which is very reasonable given that there could be handling losses and some denaturation. The fractions bound were plotted against the concentration of free protein, after subtraction of the bound protein from the total (Fig. 4 B and C), and fitted to a simple Michaelis-Menten equation to calculate the KDs.
Fig. 4.

Binding of P21 and BAX DNA response elements to preequilibrated wt p53CT (blue) and p53CT R273H (black). (A) MS spectra of wt and mut at different P21 DNA concentrations. (B and C) Fitting of intensities to simple binding isotherms: wt4.DNA (filled circles); wt2mut2.DNA; and (filled squares) mut4.DNA (filled diamonds).
Fig. 5.

Determination of active p53 from stoichiometric binding of P21 DNA. The DNA was added to a nominal concentration of 1 μM total of tetramers of wtH4 and wtL4 that had been preincubated to equilibrate. Each resultant complex could be followed independently. The concentration of total tetramers at the equivalence point was 0.785 ± 0.019 μM.
The weak binding to mut4 allowed a reasonable measurement of the binding isotherm since it was effectively titrated after all of the other two complexes were saturated with DNA and when there was excess unbound DNA over protein. The KD for the R273H tetramer bound to P21 DNA was 1.47 ± 0.17 μM. Calculation of the KDs for the other two complexes required accurate measurement of the amount of DNA bound to the protein and subtracting it from the total added DNA to give the free DNA. The values thus calculated for P21 bound to wt4 and wt2mut2 were very approximately, because of the uncertainties, 8 ± 5 nM and 57 ± 25 nM (Fig. 4B). The KDs for binding to BAX were more accurately determined for the weaker binding of BAX DNA (Fig. 4C), and were 184 ± 5 nm, 3.4 ± 0.3 μM, and 18 ± 1.5 μM for wt2, wt2mut2, and mut4, respectively.
Calculation of Relative KDs of wt4, wt2mut2, and mut4 Complexes with DNA.
We used a method for the direct calculation of the relative affinities of the different proteins for the response elements that avoided any estimation of the concentrations of unbound DNA under conditions when most was bound to protein. For a simple 1:1 binding of tetrameric p53 to a response element DNA containing four subsites: Kw = [wt4][DNA]/[wt4.DNA]; Kw/m = [wt2mut2][DNA]/[wt2mut2.DNA]; and Km = [mut4][DNA]/[mut4.DNA]. In a mixture of all of the protein components and DNA, the concentration of free DNA is the same for all equilibria. Therefore, we can calculate the ratios of all of the equilibrium constants from taking the ratios of the free and bound proteins thus: Kw/m/Kw = [wt2mut2][wt4.DNA]/[wt4][wt2mut2.DNA] and Km/Kw/m = [mut4][wt2mut2.DNA]/[wt2mut2][mut4.DNA]. Further, we can calculate the ratios of [wt4]/[wt4.DNA], [wt2mut2]/[wt2mut2.DNA], and [mut4]/[mut4.DNA] at each point quite simply. If the amplitude of, say, the signal for wt4.DNA at any DNA concentration, [D], is xD, and the amplitude at saturating DNA is x∞, then [wt4]/[wt4.DNA] = (x∞- xD)/xD. Accordingly, we do not need to know the absolute concentrations of DNA or protein to calculate the ratio of KDs. The method is most accurate for the regions where xD is significantly above zero and below x∞, that is, [DNA] is close to the KD values. The range of [DNA] used, 0, 2.8 × 10−8 to 10−5 M, was enough to span the KDs and have binding to wt4 and mut4 close to saturation. But the weak binding of DNA to the mut4 species precluded direct measurement of the saturating amplitude so we assume that it was the same as for wt4. (The amplitude of the wt2mut2 was twice that of wt4, as expected.)
For P21 DNA: Kw/m/Kw = 3.8 ± 0.3; Km/Kw/m = 18.7 ± 8; and so Km/Kw = 71 ± 31.
The values calculated from the ratios of individual KDs were in acceptable agreement to those measured directly, Kw/m/Kw = ∼7; Km/Kw/m = ∼26. For BAX DNA: Kw/m/Kw = 12.9 ± 1.9; Km/Kw/m = 6.6 ± 0.8; and so Km/Kw = 85 ± 16. The values calculated from the ratios KDs are in reasonable agreement, Kw/m/Kw = ∼18; Km/Kw/m = ∼5.
Discussion
The use of ESI-MS has allowed us to measure the association-dissociation kinetics of p53 subunits and also the affinity of mutant tetrameric protein for DNA. The rate constant for the dissociation of tetrameric protein was surprisingly and unexpectedly low. The half-life for the dissociation into dimers at 20 °C was 2.3 h and the only species of mixed hybrids formed between wild-type and mutant p53 was wt2mut2, without any of the 3:1 and 1:3 species. The half-life was 40 min at 37 °C. The dissociation constant of tetrameric p53 into dimers is approximately 20 nM, which means that at a concentration of approximately 20 nM p53 (20 nM of dimers) the half-life for association of dimers into tetramers is also 40 min. The second-order rate constant for association of dimers into tetramers is only approximately 1.4 × 104 s−1M−1, some one to two orders of magnitude lower than for most protein-protein interactions.
These experiments would not have been possible using wild-type p53 but required the superstable quadruple mutant that has been key to our structural studies (22, 24, 25, 30–32): a half-life of 40 min at 37 °C for formation of tetramers is longer than the half-life of 9–16 min for the spontaneous denaturation of wild-type p53 at 37 °C (28). The combination of the high instability of wild-type p53 and its slow oligomerization has profound implications: unless there are special factors within the cell, p53 will spontaneously denature faster than it will form active tetramers. However, there are routes for p53 to associate other than the initial dimerization of a pair of dimers via their tetramerization domains. For example, very dilute solutions of p53 that are mainly dimeric form DNA-bound tetramers within seconds when added to response element DNA (33). This reaction must occur by the rapid binding of 1 dimer to one pair of DNA binding sites, followed by the second dimer to the second pair of sites—DNA is thus a catalyst for association. The finding of slow association rate constants shows that the co-operative binding of p53 dimers to DNA is the result of a stepwise addition of dimers rather than resulting from a fast preequilibrium between dimeric and tetrameric p53.
We speculate that the slow association and dissociation rate constants for oligomerization might be part of a regulatory mechanism of p53 for processes where the presence of either dimers or tetramers is important for function or trafficking. It is possible that there may be proteins (or other DNAs) that can catalyze the association of p53. For example, we have recently found that 14–4-3γ enhances the association equilibrium of p53 by binding to its C-termini (34). So, there may be a further mode of regulation of p53 by proteins altering the rate of p53 association.
The slow rate of shuffling of subunits also has implications for the dominant negative effect of mutants where wild-type and mutant alleles are present. For contact mutants that are as stable as wild type, there will be formed just the species wt4, wt2mut2, mut4 in the ratio 1:2:1, which, given the affinities we have measured here, will lower the effective concentration of wt4 tetramers competent for binding by about a factor of 4, rather than closer to 16 expected for complete shuffling of subunits in the ratio 1:4:6:4:1. Unstable structural mutants that denature far faster than they can oligomerize with wild type will effectively lower the concentration of wild-type p53 by a factor of 2.
Although we do not know the full reasons for the high instability and slow rates of association of p53, it would seem that they are part of a complex scheme for regulation of p53 that invokes a myriad of active networks with secondary features of inherent safeguards for the spontaneous denaturation of p53. The slow rates of equilibration of p53 has to be considered in interpreting biophysical data.
Materials and Methods
Materials.
The double-stranded P21 and BAX oligonucleotides TGAGGAACATGTCCCAACATGTTGAGCTC and TGGGCTCACAAGTTAGAGACAAGCCTGGGCG, respectively, were purified by high-pressure liquid chromatography.
Protein Expression and Purification.
The p53 constructs were expressed in Escherichia coli BL21 and purified as described below. Human p53CT construct (94–356) and p53CT R273H were cloned into a pET24a-HLTV plasmid containing a 5′ extension expressing a His6-tag, a lipoamyl domain and a TEV protease cleavage site. p53CT is a superstable pseudowild-type mutant with the following mutations in the core domain: M133L/V203A/N239Y/N268D.
p53CT and p53CT R273H were grown at 37 °C in 2× TY medium with 30 mg/mL kanamycin, and 0.1 mM Zn2+. Isopropyl 1-thio-β-D-galactopyranoside (IPTG) was added to a final concentration of 1 mM to induce expression. The cells were incubated for 15 h at 25 °C before they were harvested and the proteins were purified as described (24, 30) except expression of 13C- and 15N-labeled p53 was in M9 minimal medium supplemented with a vitamin mix together with 1 g/L 15NH4Cl and 13C D-glucose (30). Protein concentration was determined spectrophotometrically using a molar extinction coefficient ε280 = 20,400 M−1cm−1. The concentration was verified by amino acid analysis (Protein and Nucleic Acid Chemistry Facility, Department of Biochemistry, University of Cambridge).
Mass Spectrometry.
The buffer was exchanged to 500 mM ammonium acetate, pH 6.9, before spectrometry, using Micro Bio-Spin 2–4 columns (Bio-Rad). Nanoflow electrospray mass spectra (nESI-MS) were recorded on a Synapt HDMS system and an LCT Premier instrument (Waters Corp.) optimized for the transmission of noncovalent complexes (35). Typically, 3 μL solution containing p53 were electrosprayed from gold-coated glass capillaries (36). To preserve the non-covalent interactions in the p53 tetramer, the MS parameters used for the Synapt were: capillary voltage, 1.3–2.0 kV; sample cone, 150 V; trap and transfer collision energy, 100 V; backing pressure 5 mbar, source pressure 6–7e−2 mbar; trap and IMS pressure 5e−2 and 5e−1 mbar, respectively; time-of-flight analyser pressure 2e−6 mbar. The following parameters were used or the LCT Premier: capillary voltage, 1.1–2.0 kV; sample cone, 200 V; ion guide 1, 150 V; aperture 1, 150 V; ion energy, 55 V; source pressure 2.8 mbar; time-of-flight analyser pressure 4.5e7 mbar. All spectra were calibrated internally using a solution of cesium iodide (100 mg/mL). Data were processed with MassLynx 4.0 software (Waters/Micromass) with minimal smoothing and without background subtraction.
Spectra Simulation and Kinetic Modeling.
The spectra were quantitatively analyzed with the LabVIEW program (27). We generated a simulated charge-state distribution for each of the component complexes, which were modeled using three parameter Gaussians. The observed m/z values of the complexes were used as midpoints and the peak width was chosen according to the observed broadness of the experimental peaks. The peak heights were scaled with a three-parameter Gaussian curve that was fitted for every complex across the tops of the corresponding peaks in the spectra. The derived simulations of the components were summed and the parameters optimized to achieve maximal agreement of the summed simulation with the experimental mass spectra (Fig. S1). The individual simulations give the peak areas for the whole charge distribution of each complex, even where peaks belonging to different species overlap. These values were used to calculate the relative abundance of the different species, which were expressed as a percentage of the total peak intensity and plotted against time.
DNA Binding Measurements by Mass Spectrometry.
The wild-type and mutant homo-tetramer samples were mixed and incubated at 4 °C rather than room temperature to prevent any loss of DNA binding as a consequence of denaturation of the core. The incubation lasted until the binomial distribution of 1:2:1 for wt4:wt2mut2:mut4 was reached, as verified by MS. DNA binding assays were then performed at equilibrium. A concentration of 55–65 μM protein was diluted 5 times in 500 mM ammonium acetate with or without DNA oligomers. The final concentration of the DNA oligomers ranged from 25 nM to 10 μM.
Relative DNA-Binding Analysis.
The relative abundance of the bound p53 forms, that is, wt4.DNA, wt2mut2.DNA, mut4.DNA, to the response elements was assessed from the sum of the associated peak intensities of all charge states for each mass spectrum. Then, this value was divided by the total peak intensity in the spectra and presented as a percentage. Each spectrum represents one concentration point, and the plot was the percentage bound species versus concentration of ligand.
Supplementary Material
Acknowledgments.
We thank Dr. A. Joerger for comments on the manuscript.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0907840106/DCSupplemental.
References
- 1.Joerger AC, Fersht AR. Structural biology of the tumor suppressor p53. Annu Rev Biochem. 2008;77:557–582. doi: 10.1146/annurev.biochem.77.060806.091238. [DOI] [PubMed] [Google Scholar]
- 2.Petitjean A, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28:622–629. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- 3.Wells M, et al. Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proc Natl Acad Sci USA. 2008;105:5762–5767. doi: 10.1073/pnas.0801353105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Joerger AC, Fersht AR. Structure-function-rescue: The diverse nature of common p53 cancer mutants. Oncogene. 2007;26:2226–2242. doi: 10.1038/sj.onc.1210291. [DOI] [PubMed] [Google Scholar]
- 5.Junk DJ, et al. Different mutant/wild-type p53 combinations cause a spectrum of increased invasive potential in nonmalignant immortalized human mammary epithelial cells. Neoplasia. 2008;10:450–461. doi: 10.1593/neo.08120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nicholls CD, McLure KG, Shields MA, Lee PW. Biogenesis of p53 involves cotranslational dimerization of monomers and posttranslational dimerization of dimers. Implications on the dominant negative effect. J Biol Chem. 2002;277:12937–12945. doi: 10.1074/jbc.M108815200. [DOI] [PubMed] [Google Scholar]
- 7.Marutani M, et al. Dominant-negative mutations of the tumor suppressor p53 relating to early onset of glioblastoma multiforme. Cancer Res. 1999;59:4765–4769. [PubMed] [Google Scholar]
- 8.Dong P, et al. p53 dominant-negative mutant R273H promotes invasion and migration of human endometrial cancer HHUA cells. Clin Exp Metastasis. 2007;24:471–483. doi: 10.1007/s10585-007-9084-8. [DOI] [PubMed] [Google Scholar]
- 9.Dearth LR, et al. Inactive full-length p53 mutants lacking dominant wild-type p53 inhibition highlight loss of heterozygosity as an important aspect of p53 status in human cancers. Carcinogenesis. 2007;28:289–298. doi: 10.1093/carcin/bgl132. [DOI] [PubMed] [Google Scholar]
- 10.van Dieck J, Fernandez-Fernandez MR, Veprintsev DB, Fersht AR. Modulation of the oligomerization state of p53 by differential binding of proteins of the S100 family to p53 monomers and tetramers. J Biol Chem. 2009;284:13804–13811. doi: 10.1074/jbc.M901351200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cai BH, et al. Functional four-base A/T gap core sequence CATTAG of P53 response elements specifically bound tetrameric P53 differently than two-base A/T gap core sequence CATG bound both dimeric and tetrameric P53. Nucleic Acids Res. 2009;37:1984–1990. doi: 10.1093/nar/gkp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fernandez-Fernandez MR, Veprintsev DB, Fersht AR. Proteins of the S100 family regulate the oligomerization of p53 tumor suppressor. Proc Natl Acad Sci USA. 2005;102:4735–4740. doi: 10.1073/pnas.0501459102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maki CG. Oligomerization is required for p53 to be efficiently ubiquitinated by MDM2. J Biol Chem. 1999;274:16531–16535. doi: 10.1074/jbc.274.23.16531. [DOI] [PubMed] [Google Scholar]
- 14.Stommel JM, et al. A leucine-rich nuclear export signal in the p53 tetramerization domain: Regulation of subcellular localization and p53 activity by NES masking. EMBO J. 1999;18:1660–1672. doi: 10.1093/emboj/18.6.1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Itahana Y, Ke H, Zhang Y. p53 oligomerization is essential for its C-terminal lysine acetylation. J Biol Chem. 2008;284:5158–5164. doi: 10.1074/jbc.M805696200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization for mass spectrometry of large biomolecules. Science. 1989;246:64–71. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 17.Sharon M, Robinson CV. The role of mass spectrometry in structure elucidation of dynamic protein complexes. Annu Rev Biochem. 2007;76:167–193. doi: 10.1146/annurev.biochem.76.061005.090816. [DOI] [PubMed] [Google Scholar]
- 18.Wendt S, et al. Quantitative evaluation of noncovalent chorismate mutase-inhibitor binding by ESI-MS. J Am Soc Mass Spectrom. 2003;14:1470–1476. doi: 10.1016/j.jasms.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 19.Keetch CA, et al. L55P transthyretin accelerates subunit exchange and leads to rapid formation of hybrid tetramers. J Biol Chem. 2005;280:41667–41674. doi: 10.1074/jbc.M508753200. [DOI] [PubMed] [Google Scholar]
- 20.Chitta RK, Rempel DL, Gross ML. Determination of affinity constants and response factors of the noncovalent dimer of gramicidin by electrospray ionization mass spectrometry and mathematical modeling. J Am Soc Mass Spectrom. 2005;16:1031–1038. doi: 10.1016/j.jasms.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 21.Sobott F, Benesch JL, Vierling E, Robinson CV. Subunit exchange of multimeric protein complexes. Real-time monitoring of subunit exchange between small heat shock proteins by using electrospray mass spectrometry. J Biol Chem. 2002;277:38921–38929. doi: 10.1074/jbc.M206060200. [DOI] [PubMed] [Google Scholar]
- 22.Nikolova PV, Henckel J, Lane DP, Fersht AR. Semirational design of active tumor suppressor p53 DNA binding domain with enhanced stability. Proc Natl Acad Sci USA. 1998;95:14675–14680. doi: 10.1073/pnas.95.25.14675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Joerger AC, Allen MD, Fersht AR. Crystal structure of a superstable mutant of human p53 core domain. Insights into the mechanism of rescuing oncogenic mutations. J Biol Chem. 2004;279:1291–1296. doi: 10.1074/jbc.M309732200. [DOI] [PubMed] [Google Scholar]
- 24.Tidow H, et al. Quaternary structures of tumor suppressor p53 and a specific p53 DNA complex. Proc Natl Acad Sci USA. 2007;104:12324–12329. doi: 10.1073/pnas.0705069104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joerger AC, Ang HC, Veprintsev DB, Blair CM, Fersht AR. Structures of p53 cancer mutants and mechanism of rescue by second-site suppressor mutations. J Biol Chem. 2005;280:16030–16037. doi: 10.1074/jbc.M500179200. [DOI] [PubMed] [Google Scholar]
- 26.Bullock AN, Henckel J, Fersht AR. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: Definition of mutant states for rescue in cancer therapy. Oncogene. 2000;19:1245–1256. doi: 10.1038/sj.onc.1203434. [DOI] [PubMed] [Google Scholar]
- 27.Aquilina JA, Benesch JL, Bateman OA, Slingsby C, Robinson CV. Polydispersity of a mammalian chaperone: Mass spectrometry reveals the population of oligomers in alphaB-crystallin. Proc Natl Acad Sci USA. 2003;100:10611–10616. doi: 10.1073/pnas.1932958100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Friedler A, Veprintsev DB, Hansson LO, Fersht AR. Kinetic instability of p53 core domain mutants: Implications for rescue by small molecules. J Biol Chem. 2003;278:24108–24112. doi: 10.1074/jbc.M302458200. [DOI] [PubMed] [Google Scholar]
- 29.Veprintsev DB, Fersht AR. Algorithm for prediction of tumour suppressor p53 affinity for binding sites in DNA. Nucleic Acids Res. 2008;36:1589–1598. doi: 10.1093/nar/gkm1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Veprintsev DB, et al. Core domain interactions in full-length p53 in solution. Proc Natl Acad Sci USA. 2006;103:2115–2119. doi: 10.1073/pnas.0511130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Joerger AC, Ang HC, Fersht AR. Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc Natl Acad Sci USA. 2006;103:15056–15061. doi: 10.1073/pnas.0607286103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tidow H, Veprintsev DB, Freund SM, Fersht AR. Effects of oncogenic mutations and DNA response elements on the binding of p53 to p53-binding protein 2 (53BP2) J Biol Chem. 2006;281:32526–32533. doi: 10.1074/jbc.M604725200. [DOI] [PubMed] [Google Scholar]
- 33.Weinberg RL, Veprintsev DB, Fersht AR. Cooperative binding of tetrameric p53 to DNA. J Mol Biol. 2004;341:1145–1159. doi: 10.1016/j.jmb.2004.06.071. [DOI] [PubMed] [Google Scholar]
- 34.Rajagopalan S, Jaulent AM, Wells M, Veprintsev DB, Fersht AR. 14–3-3 activation of DNA binding of p53 by enhancing its association into tetramers. Nucleic Acids Res. 2008;36:5983–5991. doi: 10.1093/nar/gkn598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sobott F, Hernandez H, McCammon MG, Tito MA, Robinson CV. A tandem mass spectrometer for improved transmission and analysis of large macromolecular assemblies. Anal Chem. 2002;74:1402–1407. doi: 10.1021/ac0110552. [DOI] [PubMed] [Google Scholar]
- 36.Nettleton EJ, et al. Protein subunit interactions and structural integrity of amyloidogenic transthyretins: Evidence from electrospray mass spectrometry. J Mol Biol. 1998;281:553–564. doi: 10.1006/jmbi.1998.1937. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.