

Summary
Several cytisine derivatives have been developed in the search for more selective drugs at nicotinic acetylcholine receptors (nAChR). Binding experiments in transfected cell lines showed that the iodination of cytisine in the position 3 of the pyridone ring increased potency at α7 nAChR and to a lesser extent at the α4β2 subtypes, both of which are widely expressed in the brain. However, no in vivo studies have been published on this compound. Inhibition curves presented here using wild type, β2, and β4 null mutant mice confirm that 3-IC binds to α4β2*, α7* and α3β4* receptors with higher affinity than cytisine (asterisk indicates the receptor may contain additional subunits, Lukas et al., 1999). Intraperitoneal injection of 3-iodocytisine (3-IC) induced considerable dose dependent hypothermia in DBA/2J and C57BL/6J mice. This response was blocked by mecamylamine and partially inhibited by hexamethonium. β4-null mice displayed significantly less 3-IC-induced hypothermia than wild-type mice, β2-null mice were somewhat less affected than wild-types, while responses of α7*-null mice were similar to wild types. Mice treated chronically with 3-IC display a marked increase in α7* and α4β2* binding sites determined by radioligand binding in membrane preparations from cerebral cortex and hippocampus. Quantitative autoradiographic analysis of 28 brain regions of mice treated with 3-IC was consistent with the membrane binding, detecting an increase of cytisine-sensitive [125I]epibatidine binding sites, while cytisine-resistant [125I]epibatidine sites were unchanged. [125I]α-bungarotoxin binding sites also exhibited up-regulation. These results give a first evaluation of in vivo consequences of 3-IC as a potent agonist with marked effects on mice.
Keywords: nicotinic acetylcholine receptors, null mutant mice, 3-IC, hypothermia, epibatidine, α-bungarotoxin
Introduction
Nicotinic acetylcholine receptors (nAChR) constitute a family of ligand-gated ion channels assembled in neurons as pentameric receptors. While mRNA encoding nine nAChR subunits (α2-α7 and β2-β4) is expressed in mammalian brain, the major neuronal subtypes in the central nervous system are homomeric α7*-nAChR and heteromeric α4β2*-nAChR (Dani and Bertrand, 2007). nAChR mediate several simple and complex behavioral and physiological responses (Picciotto, 2003). nAChR also mediates short-term and long-term responses to nicotine and similar drugs indicating a role in nicotine tolerance and dependence (Dani and deBiasi, 2001). nAChRs have also been implicated in several human neurological and psychiatric disorders. Single point mutations in either the α4 or β2 nAChR subunits have been identified as the cause of several different Autosomal Dominant Nocturnal Frontal Lobe Epilepsies (Hogg and Bertrand, 2004). Although the evidence is not as obvious, nAChR also appear to be involved in the etiology of Alzheimer's and Parkinson's diseases as well as in psychopathologies such as anxiety disorders, Tourette's syndrome and schizophrenia (Bourin et al., 2003; Leonard et al., 2001). Because of the relevance of nicotinic physiology to health issues, nAChR ligands are envisioned as potential therapeutic agents for Parkinson's disease (Quik, 2004), obsessive-compulsive disorder (Salin-Pascual and Basanez-Villa, 2003) and schizophrenia (Harris et al., 2004).
Specific and potent nicotinic ligands would facilitate investigation of the multiplicity of in vivo effects modulated by nAChR.
Cytisine is a plant alkaloid with higher affinity for neural α4β2-nAChR than nicotine (Pabreza, 1991). It is a partial agonist at β2*-nAChR but a full agonist at β4*-nAChR (Luetje and Patrick, 1991). Due to its semi-rigid structure cytisine has been used as a template for the preparation of new nicotinic receptor ligands. A group of cytisine derivatives has, indeed, been developed (Imming et al., 2001; Slater et al., 2003). Varenicline, which has recently been approved for smoking cessation, is such an analog (Lam and Patel, 2007). An electrophysiological characterization of the effects of cytisine and its bromo derivatives, in particular 3-bromocytisine, showed a potent effect on inward currents in ACh-activated neurons in cat petrosal ganglion neurons in culture (Varas et al., 2006). The C3-halogenated derivatives of cytisine, and particularly 3-IC (3-IC) display higher binding affinity than cytisine determined either α7* and α4β2* receptors in rat brain with Ki values of 115 nM and 0.17 nM compared to 8 μM and 1.2 nM, respectively (Abin-Carriquiry et al., 2006). Similarly, for human α7 and α4β2 expressed in Xenopus oocytes 3-IC has Ki values of 7.0 and 0.7 nM compared to 30 μM and 0.6 nM for cytisine respectively (Slater et al., 2003). 3-IC is also more potent that cytisine in stimulating [3H]noradrenaline release from rat hippocampal slices and for [3H]dopamine release from rat striatal synaptosomes with EC50 values of 0.22 μM and 0.011 μM for 3-IC compared to 7.4 μM and 0.28 μM for cytisine, respectively (Abin-Carriquiry et al., 2006). Similarly, in Xenopus oocytes expressing human α7 nAChR, the EC50 value for 3-IC was 1.5 μM, compared to 83 μM for cytisine, while EC50 values for the high and low agonist sensitive forms of α4β2-nAChR were 0.8 nM and 86 nM for 3-IC and 5 nM and 2 μM for cytisine (Slater et al., 2003; Abin-Carriquiry et al., 2006). Beyond these in vitro analyses, 3-IC has never been tested in an in vivo model. Here, we describe the acute effect of 3-IC in the modulation of body temperature in mice including evaluation of the effect of some nicotinic antagonists and deletion of either the α7, β2 or β4 nAChR genes on the hypothermic response. Finally we expand the effect of chronic 3-IC treatment on nAChR binding sites in mouse brain.
Materials
[125I]Epibatidine (2200 Ci/mmol) was purchased from Perkin-Elmer Life Science, Boston, MA and [125I]α-bungarotoxin (2000 Ci/mmol) from GE healthcare. A85380, cytisine, mecamylamine, hexamethonium, scopolamine, nicotine, chloral hydrate, pentobarbital, polyethyleneimine (PEI) and bovine serum albumin (BSA) fraction V were purchased from Sigma Chemical Company, St. Louis, MO. 4-(2-Hydroxyethyl)-piperazineethanesulfonic acid (HEPES) half-sodium salt was from Roche Diagnostics Corporation, Indianapolis, IN.
Methods
Synthesis of 3-IC
Cytisine was purified from Sophora secundiflora seeds using standard methodology. Monohalogenated cytisine derivatives were prepared by treating cytisine with iodine monochloride. The iodinated isomers were separated by column chromatography on silica gel, crystallized to homogeneity and characterized by 1H and 13C NMR and HREIMS as reported before (Slater et al., 2003).
Mice
DBA/2J and C57BL/6J mice were bred at the Institute for Behavioral Genetics, University of Colorado, Boulder, CO and housed five per cage prior to testing or surgery and chronic nicotine treatment.
α7-/- (Orr-Urtreger et al., 1997), β2-/- (Picciotto et al., 1995) and β4-/- (Xu et al., 1999) and wild-type littermates were generated by mating heterozygotes for each mutation. Each of these null mutant mice has been backcrossed with C57BL/6JJ mice for at least 10 generations at the time of the experiments. Mice were weaned at 25 days of age and housed with like-sexed littermates. DNA was extracted from tail clippings obtained from 40-day old mice and genotypes were determined as described previously (Salminen et al., 2004). Mice were allowed free access to food (Rodent Chow, Harlan-Teklad, Madison, WI) and water. The animal vivarium was maintained at a temperature of 23 ± 2°C with a 12 h light/12 h dark cycle (lights on 7 AM to 7 PM). All procedures used in this study were reviewed and approved by the Animal Care and Utilization Committee of the University of Colorado.
Temperature measurements
Body temperature measurement was chosen for these experiments because the hypothermic effects of nAChR ligands has been reported before for the two different mice strains used in this study (Marks et al, 1983(a), Tritto et al., 2004). DBA/2J, C57/BL6 and wild type and α7-, β2- and β4-null mutant mice were tested under similar conditions for determination of body temperature. All drugs were administered intraperitoneally (IP) (10 μl/g of body weight in every injection). All antagonists were injected 10 minutes before the 3-IC injections. Temperature was measured with a Thermalert rectal probe lubricated with peanut oil and inserted 1.5 cm into the mouse's rectum.
Chronic 3-IC treatment
DBA/2J mice were anesthetized with pentobarbital (45 mg/kg)-chloral hydrate (63 mg/kg). A cannula was implanted in the right jugular vein (Barr et al., 1979) and after surgery the animals were placed in individual cages (15×15×25 cm). Following 2 days of continuous saline infusion (35 μl/hr), treatment with 3-IC was begun. Fresh drug and saline solution was supplied every day. Mice were randomly assigned to two groups: saline-infused (control) or 3-IC 0.2 mg/kg/h. Stock 3-IC solutions (10 mg/ml) were prepared in sterile saline, neutralized with HCl and stored in the dark at 4°C. Dilutions of this stock solution were prepared to give the appropriate hourly dose. Mice were subsequently infused with 3-IC for 7 days. Following the 7-day treatment, infusion was terminated. Two hours after ending treatment mice were injected IP with 0.2 mg/kg 3-IC and body temperature was measured at 5-minute intervals for the first 20 minutes and at 10 minutes intervals thereafter.
Membrane preparation
Two hours following completion of the tolerance test (five hours after withdrawal from drug infusion), mice that had been treated chronically with saline or 0.2 mg/kg/hr 3-IC were killed by cervical dislocation. Each brain was placed on an ice-cold platform and dissected into the following brain regions: cerebral cortex (Cx), striatum (St), hippocampus (Hp), thalamus (Th), superior colliculus (SC), inferior colliculus (IC) and midbrain (MB). Each brain region was homogenized in ice-cold hypotonic buffer (NaCl, 14.4 mM; KCl, 0.2 mM; CaCl2, 0.2 mM; MgSO4, 0.1 mM; HEPES 2 mM; pH = 7.5) using a Teflon-glass tissue grinder. The particulate fractions were obtained by centrifugation at 20,000 × g (10 min., 4 °C; Sorvall RC-2B centrifuge). The pellets were resuspended in fresh homogenization buffer and washed four times by resuspension/centrifugation, and stored (in pellet form under homogenization buffer) at -70 °C until used.
[125I]Epibatidine binding
[125I]Epibatidine binding was measured as described previously (Whiteaker et al., 2000). Frozen, washed pellets were resuspended in the overlying buffer and centrifuged at 20,000 × g for 20 min. The supernatant was discarded and the pellet was resuspended in ice-cold water. Resuspension volume varied among brain regions, and was adjusted such that less than 10% of the [125I]epibatidine was bound to the protein at the highest ligand concentration. Samples (10-50 μg protein) were incubated in 96-well polystyrene plates for 3 hours at room temperature in a binding buffer of the following composition: NaCl, 144 mM; KCl, 2.2 mM; CaCl2, 2 mM; MgSO4, 1 mM; HEPES hemisodium, 25 mM; pH = 7.5. Final incubation volume was 30 μl. At the completion of the incubation, samples were diluted with 200 μl of ice-cold binding buffer and filtered under vacuum (0.2 atm.) onto glass fiber filters that had been treated with 0.5% PEI (top filter, MFS Type B; bottom filter, Gelman A/E). An Inotech Cell Harvester (Inotech Biosystems International, Rockville, MD) was used to collect the samples, which were subsequently washed five times with ice-cold buffer. Filters containing the washed samples were transferred to glass culture tubes and radioactivity counted at 80% efficiency using a Packard Cobra Auto-Gamma Counter (Packard Instruments, Downers Grove, IL). For all the experiments, nonspecific binding was measured by including 100 μM nicotine in the incubation medium. All brain regions from 3-IC- and saline-treated groups were assayed for [125I]epibatidine binding using 200 pM ligand. Since [125I]epibatidine binds with high affinity to several different nAChR subtypes, differential inhibition by the agonists cytisine (50 and 150 nM) and A85380 (10 and 50 nM) was used to distinguish four binding sites: cytisine-sensitive (primarily α4β2*), cytisine-resistant (mixed population), A85380-sensitive (β2*-nAChR), and A85380-resistant (β4*-nAChR) (Whiteaker et al., 2000).
Inhibition of [125I]epibatidine binding by nicotine, cytisine and 3-IC was measured using a ligand concentration of 200 pM and an incubation volume of 30 μl. Eleven concentrations of drugs (0.01 nM, 0.1 nM, 0.3 nM, 1 nM, 3 nM, 10 nM, 30 nM, 100 nM, 300 nM, 1 μM and 10 μM) were used. Blanks were established by measuring binding in the presence of 100 μM cytisine.
[125I]α-bungarotoxin binding
Binding of [125I]α-bungarotoxin to membrane preparations was performed as described (Whiteaker, 2008). Membrane samples (25-200 mg of membrane preparations) were incubated with 1 nM [125I]α-bungarotoxin in binding buffer supplemented with BSA (0.1 % w/v) for 5 h at 22 °C. Binding reactions were stopped by filtration of samples onto filters treated with 5% (w/v) nonfat dry milk (MFS, Type B) or PEI (0.5 % w/v in binding buffer, Gelman A/E). Samples were subsequently washed six times with ice-cold binding buffer. Total and nonspecific (in the presence of 1 mM nicotine) binding was determined in triplicate. Inhibition of [125I]α-bungarotoxin binding by nicotine, cytisine and 3-IC in Hp total membrane preparation was measured using a ligand concentration of 0.5 nM. An incubation volume of 30 μl was used. Nine concentrations of drugs (1 nM, 10 nM, 30 nM, 100 nM, 300 nM, 1 μM, 3 μM, 10 μM and 100 μM) were used. Blanks were established by measuring binding in the presence of 1 mM nicotine.
Protein determination
Protein was measured according to the method of Lowry et al. (1951) using bovine serum albumin as the standard.
Data calculations
Results of the maximal drop in temperature in the acute dose response experiments were analyzed using the following four parameter logistic equation to obtain estimates of ED50 values; Y=min+(max-min)/(1+(X/ED50)N, where Y is the response after each treatment with each dose of 3-IC, min is the change in temperature after saline treatment, max is the change in temperature observed at the highest dose of 3-IC, X is dose of 3-IC, ED50 is 3-IC dose eliciting 50% of the maximal response and N correspond to the Hill coefficient.
[125I]Epibatidine binding site densities inhibited by cytisine and A85380 were calculated using a two-site inhibition model: BI = B1/(1+I/IC50-1)+B2/(1+I/IC50-2), where, BI = [125I]epibatidine binding at either cytisine or A85380 concentration, I, B1 and B2 are the [125I]epibatidine binding with IC50 values of IC50-1 and IC50-2, respectively. None of the IC50 values differed among the brain regions nor were these values affected by chronic nicotine treatment (average IC50-1 and IC50-2 values were 3.75 and 300 nM for cytisine and 0.26 and 140 nM for A85380, respectively). Densities of cytisine-sensitive and cytisine-resistant sites were calculated from the [125I]epibatidine binding measured with 0, 50 and 150 nM cytisine, while A85380-sensitive and A85380-resistant sites were measured from the [125I]epibatidine binding measured at 0, 5 and 25 nM A85380. Inhibition of [125I]epibatidine binding by nicotine, cytisine and 3-IC in inferior colliculi of wild-type mice was analyzed using a two-site model showed above. Inhibition of [125I]epibatidine binding in the β2-/-, β4-/- mice and inhibition of [125I]α-bungarotoxin binding in wild-type mice was analyzed using a one-site model: BI = B/(1+I/IC50) where B is binding at a drug concentration I. Apparent Ki where obtained using the Cheng and Prusoff equation.
Autoradiography: preparation of sections
Autoradiographic procedures were similar to those described previously (Whiteaker et al., 2000). At the completion of the chronic treatment DBA/2J mice were killed by cervical dislocation, the brains were removed from the skulls and rapidly frozen by immersion in isopentane (-20 °C, 10 sec.). The frozen brains were wrapped in aluminium foil, packed in ice, and stored at -70 °C until sectioning. Tissue sections 14 mm thick prepared using a Leica Model 1850 cryostat refrigerated to -16 °C were thaw-mounted onto Superfrost®/plus microscope slides (Fisher Scientific). Mounted sections were stored, desiccated, at -70 °C until use. Ten series of sections were collected from each mouse brain.
Autoradiography: [125I]α-bungarotoxin binding
A series of sections from each mouse was incubated in binding buffer + 0.1% (w/v) bovine serum albumin at 22 °C for 10 min. The samples were then incubated with 1 nM [125I]α-bungarotoxin in binding buffer + 0.1% (w/v) bovine serum albumin for 4 h at 22 °C. An adjacent series of sections from each mouse was used to determine non-specific [125I]α-bungarotoxin binding in the presence of 1 mM nicotine. The slides were then washed as follows: 10 min. at 22°C in binding buffer (twice), then subsequent washes at 0 °C, 10 sec. in binding buffer (twice), 5 sec. in 0.1× binding buffer (twice), 5 sec. in 5 mM HEPES, pH 7.5 (twice).
Autoradiography: [125I]epibatidine binding
A series of sections from each mouse was incubated in binding buffer at 22 °C for 10 min. The samples were then incubated with 200 pM [125I]epibatidine in binding buffer for 4 h at 22 °C. [125I]Epibatidine was diluted with non-radioactive epibatidine to reduce specific activity to 220 Ci/mmol. An adjacent series of sections from each mouse was used to determine binding in the presence of 10 nM A85380 or 50 nM cytisine. Non-specific [125I]epibatidine binding was determined in the presence of 100 μM (-)-nicotine bitartrate. Following the incubation, the slides were washed as follows (all washes at 0 °C): 10 sec. in binding buffer (twice), 5 sec. in 0.1× binding buffer (twice), 5 sec. in 5 mM HEPES, pH 7.5 (twice).
Autoradiography: image collection
Following the wash, labelled sections were dried with a stream of air and stored overnight at 22 °C under vacuum. Mounted, desiccated sections were apposed to super resolution Cyclone storage phosphor screens (Perkin-Elmer, Shelton, CT) for 2-5 days. Images were captured with a Cyclone phosphorimaging instrument for quantitative analysis of signal intensity by comparison with commercial 125I standards (GE Healthcare, Piscataway, NJ). Signal intensity of selected regions was determined by using Cyclone software. When it was possible, six independent measurements from different tissue sections were made for each brain region, under each incubation condition, for each mouse. The pixel density for each brain area was averaged, and the mean signal was used to calculate the degree of labelling by reference to the relevant standard curve.
Higher resolution images were obtained by exposure of the slides to Kodak MR film for 3-10 days. After the films had been exposed to the sections for an appropriate length of time, they were developed and autoradiographic images of the sections were photographed using a Canon D40 digital camera.
Statistical analyses of data
SPSS software was used for the statistical analysis of data. Effects of 3-IC on body temperature were analyzed using repeated measures ANOVA with time and treatments as within-subjects variables and between-subjects factors for the analysis of the dose-response curves, respectively. Effects of nicotinic receptor null mutations were also analyzed using repeated measures ANOVAs with time and genotype as the within-subjects variable and between subjects factor, respectively. The maximal temperature decrease for each treatment and variations in nAChR binding sites in saline and 3-IC chronically treated mice and values of binding assays for saline or 3-IC treated animals were also analyzed using the unpaired Student test. Statistical significance was assumed for p values < 0.05. All data are presented as mean ± SEM.
Results
Dose dependency of 3-IC-induced hypothermia
To elucidate the effect of 3-IC on thermoregulation, wild-type DBA/2J and C57BL/6J mice were injected intra-peritoneally with 0.05, 0.1, 0.2, 0.5 and 1 mg/kg of the drug. Rectal temperature was measured at 5 minutes intervals through the first 20 minutes and at 10 minutes intervals thereafter. A statistically significant effect of dose and time on hypothermic effect following 3-IC injections was observed for both mice strains (Figure 1A, C, p<0.001 for time variable, p<0.001 for dose factor in DBA/2J and C57BL/6J). The significant time by dose interaction (p<0.001 for both mice strains) confirmed that the time course for hypothermia varied with dose. A dose-dependent increase in magnitude and persistence of hypothermia was observed following administration of 3-IC. Doses of 0.05 and 0.1 mg/kg elicited modest temperature decreases that rapidly recovered. The extent and duration of hypothermia were observed for doses of 0.2 mg/kg and higher and was much longer than that observed for lower doses. The maximal temperature decrease elicited with 1 mg/kg 3-IC (the highest dose tested) was 11.9 ± 0.4 °C in DBA/2J and 13.3 ± 0.9 °C in C57BL/6J (Figure 1). Tremors were seen at doses above 0.5 mg/kg. No statistical difference was observed for an acute injection of 3-IC at 0.2 mg/kg dose in DBA/2J and C57BL/6J mice, producing a decrease of basal temperature of 7.7 ± 0.5 °C and 9.2 ± 1.13 °C respectively (Figure 1). This dose was chosen for further experiments because a clear effect on temperature was observed, without inducing tremors or diarrhea. Despite the remarkable effect of 3-IC on temperature, no seizures were noted at any dose tested. Both mouse strains had similar sensitivity to 3-IC with ED50 values of 0.15 ± 0.04 mg/kg for DBA/2J and 0.16 ± 0.03 mg/kg for C57BL/6J mice. A modest temperature increase was observed at approximately 20 minutes following saline injection (0.67 ± 0.09 °C for DBA/2J and 0.17 ± 0.03 °C for C57BL/6J).
Figure 1. Effect of 3-IC on mouse body temperature.

Time course of hypothermia induced by intraperitoneal injection of several 3-IC doses (0, 0.05, 0.1, 0.2, 0.5, 1.0 mg/kg) in DBA/2J mice (n=3) (A) and C57BL/6J (n=3 to 6) (C). The maximal decrease in temperature in the dose response curve was plotted for DBA/2J (B) and C57BL/6J (D) mice.
Hypothermia produced by 3-IC requires nicotinic receptor activation
To explore the specific role of nicotinic receptors in the modulation of mouse body temperature induced by 3-IC, the nicotinic antagonist mecamylamine was injected 10 minutes before 3-IC injections. A statistically significant difference was obtained for both, the time variable and mecamylamine treatment factor (p<0.001; p<0.001 respectively). DBA/2J and C57BL/6Jmice pretreated with mecamylamine (1 mg/kg) displayed much less hypothermia (1.13 ± 0.42 °C and 0.13 ± 0.09 °C respectively) than did mice pretreated with saline (8.50 ± 0.42 °C and 9.25 ± 0.76 °C respectively) (Figure 2C). For DBA/2J mice following mecamylamine treatment, maximal hypothermia was observed 20 minutes after 3-IC injection while maximal hypothermia following saline pretreated was observed 40 minutes after 3-IC injection (Figure 2A). The blockade of 3-IC-induced hypothermia in C57BL/6J mice was complete, that is, no temperature decrease was noted at any time (Figure 2B). The 1 mg/kg mecamylamine dose did not affect body temperature in both mice strains (Figures 1 A, B and C).
Figure 2. Effect of nAChR inhibitors in 3-IC-induced hypothermia.

Time course of mecamylamine to the hypothermic effect of 3-IC. Mice were injected intraperitoneally with 0.2 mg/kg 3-IC and one group of animals was pre-injected with MEC 1 mg/kg 10 minutes before 3-IC or saline. The time course of hypothermia was registered for DBA/2J (A) (n=3) and C57BL/6J (n=3) (B) mice. Maximal effect on body temperature for each treatment is shown for both mice strains (C) (* p<0.05, Student's t test, comparison to 3-IC alone) Time course of 3-IC-induced hypothermia and the effect of hexamethonium was tested, mice were pre-injected with hexamethonium (HXMT, 10 mg/kg) 10 minutes before 0.2 mg/kg 3-IC or saline and body temperature registered in DBA/2J (n=4) (D) and C57BL/6J mice (n=5) (E). Comparison of maximal hypothermia for each treatment in both mice strains is showed (F) (* p<0.05, Student's t test, comparison to 3-IC alone). The effect of scopolamine on 3-IC-induced hypothermia was tested injecting animals intraperitoneally with 10 mg/kg scopolamine 10 minutes before 0.2 mg/kg 3-IC or saline and body temperature was recording at time intervals for DBA/2J (n=4) (G) and C57BL/6J (n=4) (H). The maximal effect on body temperature for each treatment in both mice strains was plotted (I).
The possible contribution of muscarinic acetylcholine receptors to the hypothermic effect of 3-IC was tested in mice injected with scopolamine (10 mg/kg), a non-specific muscarinic antagonist, 10 minutes before the injection with 3-IC (0.2 mg/kg). No difference in maximal decrease or time course of 3-IC-induced hypothermia was observed between mice of either strain treated with saline or scopolamine (Figures 2 H, I and J).
Central and peripheral nervous system-controlled 3-IC-induced hypothermia
The role of the peripheral nAChR in mediating 3-IC induced hypothermia was also evaluated by using the nicotinic antagonist hexamethonium (HXMT), which has a very low penetration to the brain blod barrier. Mice were injected with 10 mg/kg HXMT 10 minutes before 3-IC (0.2 mg/kg) and rectal temperature was recorded. Injection of 10 mg/kg HXMT did not affect body temperature in C57BL/6J mice compared to saline injected mice, however DBA/2J mice showed a modest, but statistically significant, hypothermia compared to saline injected mice (Figure 1 A and C; p<0.05, comparison of maximal change in temperature). A statistically significant inhibition of maximal 3-IC-induced hypothermia was observed for mice pretreated with hexamethonium (10 mg/kg) compared to those pretreated with saline (for DBA/2J: 5.66 ± 0.45 °C and 8.10 ± 0.63°, and for C57BL/6J: 3.75 ± 0.67 °C and 9.25 ± 0.76 °C, respectively) (Figure 2F, * p<0.05) and also they recovered more rapidly from the hypothermia than did saline-treated mice, which is supported by the statistical analysis in the time by treatment interaction for both mice strains with p values < 0.001 for DBA/2J and < 0.007 for C57BL/6J (Figures 2 D and E). The blockade of 3-IC-induced hypothermia was more profound in C57BL/6J than DBA/2J, where a 60% and 33% of the maximal effect was observed respectively. These results suggest the participation of peripheral nAChR in hypothermia induced by 3-IC administration and that the peripheral effects of 3-IC may be more profound for C57BL/6J mice.
Role of specific nicotinic subunits in 3-IC-induced hypothermia
The results presented above established that the actions of 3-IC are mediated by nAChR and that a significant fraction of this effect appears to be mediated by peripheral nAChR. In order to examine the contribution of different nAChR subtypes, 3-IC was administered to mice in which the α7, β2 or β4 nAChR subunits had been deleted. Previous reports have shown deletion of β4 (Sack et al., 2005) or β2 but not α7 (Tritto et al., 2004) subunits reduced nicotine-induced hypothermia.
Similar to the effects of α7 gene deletion on nicotine-induced hypothermia, no significant differences in maximal temperature decrease (Figure 3B) or in the time course for 3-IC-induced hypothermia were observed following deletion of the α7 subunit (Figure 3A).
Figure 3. Effect of 3-IC on body temperature of α7, β2 and β4 null mutant mice.

Null mutant or wild-type mice were injected intraperitoneally with 3-IC 0.2 mg/kg and rectal temperature was recorded every 5 minutes. Time course of temperature decrease is graphed for C57 α7-/- (n = 4) (A), C57 β2-/- (n = 5) (C) and C57 β4-/- (n = 5) (E). Minimal temperature was plotted for C57 α7-/- (B), C57 β2-/- (D) and C57 β4-/- (F) null mutant and wild type mice (* p<0.05, Student's t test).
A modest effect of β2 gene deletion was observed. Repeated measures ANOVA revealed a significant difference within β2-null and wild-type mice for time variable but not for the genotype factor (p<0.001 and p=0.059, respectively). However deletion of the β2 subunit did reduce maximal 3-IC induced hypothermia with a decrease of temperature of 8.4 ± 1 °C for β2-/-compared to 9.8 ± 0.76 °C for β2+/+, p<0.05 (Figure 3D). β2-/- mice appeared to recover more rapidly from 3-IC induced hypothermia than did β2+/+ (Figure 3C).
In contrast, a substantial contribution of the β4*-nAChR receptors was observed to 3-IC-induced hypothermia. Repeated measures ANOVA revealed statistically significant differences for the time variable and genotype factor between β4-null and wild-type mice (p<0.001 and p<0.001, respectively). A statistically significant time by genotype interaction was also found (p<0.001). This analysis is supported by the observation of a significant decrease in maximal hypothermia for β4 null mutant mice (β4-/-, 2.9 ± 0.4 °C; β4+/+, 7.9 ± 0.3 °C, p<0.05) (Figure 3F) and the faster recovery of basal temperature (Figure 3E).
Inhibition of [125I]epibatidine and [125I]α-bungarotoxin binding
Binding affinities of nicotine, cytisine and 3-IC for [125I]epibatidine binding sites were measured in total membrane preparations from Inferior Colliculus (IC) of C57BL/6J wild-type, as well as β2 and β4 null mutant mice and for [125I]α-bungarotoxin binding sites in hippocampus of wild-type C57BL/6J mice. Cytisine inhibition in IC from wild-type mice was best fit to a two-site model. However, the biphasic nature of the inhibition curves was less obvious for nicotine and even less obvious for 3-IC as the difference in affinity between the two sites for those drugs decreased (Table 1). These differences in the inhibition curves for the three compounds occurred because the differences in affinity of cytisine for the two [125I]epibatidine binding sites was greater (60-fold) than that for either nicotine (20-fold) or 3-IC (1.6 fold). In order to test the contribution of β2*-nAChR and β4*-nAChR to the binding patterns in wild-type mice, inhibition curves were constructed for the three compounds in IC prepared from β4-/- and β2-/- mice, respectively. In tissue prepared from each null mutant mouse, the inhibition of [125I]epibatidine binding was monophasic. In IC prepared from β4-/-mice Ki values were 0.35 nM, 1.67 nM and 0.128 nM and in IC prepared from β2-/- mice Ki values were 9.7 nM, 23.9 nM and 0.25 nM for cytisine, nicotine and 3-IC, respectively. These Ki values and their ratios in the two knockout mice are comparable to those estimated from the biphasic fits obtained with IC prepared from wild-type mice. The experiments using wild-type and null mutant mice establish that, unlike the parent compound, cytisine, 3-IC exhibits very similar affinity for β2*- and β4*-nAChR. Inhibition of [125I]α-bungarotoxin binding was measured in hippocampus prepared from wild-type mice to estimate affinity for α7-nAChR sites. The order of potency for inhibition of [125I]α-bungarotoxin binding was 3-IC (0.0022 μM) >> cytisine (1.58 μM) ≈ nicotine (0.93 μM) (Figure 4). Values of Ki for the different subsets of receptors are presented in table 1.
Table 1.
Comparison of Ki values for nicotine, cytisine and 3-IC inhibition of [125I]epibatidine binding in total membrane preparation from inferior colliculi of wild-type, β2-/- and β4-/- C57BL/6J mice. Same compounds were also tested for inhibition of [125I]α-bungarotoxin binding in hippocampus of wild-type C57BL/6J mice. Data represent mean ± SEM of 3 independent experiments, each with triplicate samples. The concentration of [125I]epibatidine and [125I]α-bungarotoxin was 200 pM and 0.5 nM respectively. The Kd values used to calculate Ki was 30 pM for [125I]epibatidine and 0.5 nM for [125I]α-bungarotoxin.
| [125I]α-bungarotoxin (WT) Ki (μM) | [125I]epibatidine (WT) Ki (nM) | [125I]epibatidine (β2 -/-) Ki (nM) | [125I]epibatidine (β4 -/-) Ki (nM) | ||
|---|---|---|---|---|---|
| low | high | ||||
| nicotine | 0.93 ± 0.10 | 25.0 ± 21.0 | 1.29 ± 0.41 | 23.9 ± 6.2 | 1.67 ± 0.13 |
| cytisine | 1.58 ± 0.18 | 14.4 ± 1.5 | 0.24 ± 0.01 | 9.7 ± 1.5 | 0.35 ± 0.01 |
| 3-IC | 0.0022 ± 0.0012 | 1.36 ± 0.05 | 0.85 ± 0.02 | 0.25 ± 0.04 | 0.128 ± 0.003 |
Figure 4. Inhibition curves for [125I]α-bungarotoxin and [125I]epibatidine binding.

Inhibition of 0.5 nM [125I]α-bungarotoxin binding in total membrane preparation from hippocampus of C57BL/6J mice (A). Data was fitted to a single site inhibition curve. Inhibition of 200 pM [125I]epibatidine binding in total membrane preparation from inferior colliculus of wilt-type (B), β2-/- (C) and β4-/- (D) mice. Data was fitted to a two site inhibition curve for wild-type samples and one site inhibition curve for the two null mutant mice.
4.5. Effect of chronic 3-IC treatment on specific [125I]epibatidine and [125I]α-bungarotoxin binding sites
In order to explore the effect of a chronic administration of 3-IC on nAChR binding sites, we determined [125I]epibatidine and [125I]α-bungarotoxin binding components to several brain regions of DBA/2J mice infused intravenously with a dose of 0.2 mg/kg/h for 7 days. Doses higher than 0.2 mg/kg/h elicited tremors in the animals and had an enormous effect on body temperature (a 13 ± 0.3 °C of decrease after 24 hours of continuous intravenous infusion of 0.5 mg/kg/h 3-IC).
Following chronic treatment mice were tested for response to an acute challenge dose of 3-IC to determine if previous chronic treatment affected the acute sensitivity to the drug; that is, if tolerance had developed. Interestingly, chronic infusion of 0.2 mg/kg/h did not elicit tolerance to 3-IC. No difference in hypothermia elicited by an acute injection of 0.2 mg/kg 3-IC was observed between mice treated chronically with saline or 3-IC (maximal response for saline, 8.25 ± 1.15 °C; and 3-IC, 8.13 ± 0.69 °C).
[125I]Epibatidine binding sites were subsequently measured in membrane preparations from cerebral cortex, hippocampus, striatum, thalamus, inferior colliculi, superior colliculi and midbrain of saline and 3-IC treated mice. Four sets of [125I]epibatidine sites were determined: cytisine-sensitive (primarily α4β2* nAChR), cytisine-resistant (mixed population of nAChR different from α4β2*), A85380-resistant (mainly α3β4* nAChR) and cytisine-resistant minus A85380-resistant (non α4, β2*-nAChR) [125I]epibatidine binding sites (Whiteaker et al., 2000; Marks et al., 2004).
The response of the cytisine-sensitive sites to chronic 3-IC treatment differed among the brain regions. Significant increases in cytisine-sensitive [125I]epibatidine binding sites were only observed in cerebral cortex and hippocampus (70.3 ± 10.7 % and 96.1 ± 12.1 % increase compared to saline infused mice respectively), while there was a tendency for small, but insignificant, increases in binding to occur in several others brain regions (Figure 5).
Figure 5. Determination of subsets of [125I]epibatidine binding sites in whole membranes.

DBA/2J mice were chronically intravenously infused with 3-IC 0.2 mg/kg/h for 7 days. After treatment the brains were removed and total brain membranes were obtained. Cytisine-sensitive, cytisine-resistant, A85380-resistant and cytisine-resistant minus A85380-resistant [125I]epibatidine binding sites were determined in the particulate fractions from brain regions of saline and 3-IC treated mice. Non-specific sites were determined by adding 100 μM nicotine (* p < 0.05, Student's t test, n=4 for saline and n=6 for 3-IC treatments). Abbreviations: cerebral cortex (Cx), striatum (St), hippocampus (Hp), thalamus (Th), superior colliculus (SC), inferior colliculus (IC) and midbrain (MB).
No significant differences in the density of cytisine-resistant [125I]epibatidine binding sites were observed between saline or 3-IC chronically treated mice in any brain region analyzed (Figure 5). The A85380 resistant component of [125I]-epibatidine binding was also unmodified by chronic 3-IC treatment (figure 3). [125I]α-bungarotoxin binding sites appeared to be increased by chronic 3-IC treatment in each of the three brain regions assayed although the effect only attained statistical significance in cerebral cortex detecting a 58% increase compared to saline infused mice (Figure 6).
Figure 6. Determination of [125I]α-bungarotoxin binding sites in whole membranes.
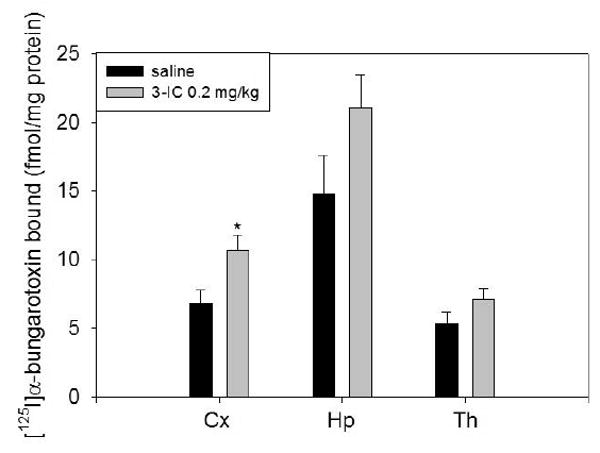
DBA/2J mice were chronically intravenously infused with 3-IC 0.2 mg/kg/h for 7 days. After treatment the brains were removed and total brain membranes were obtained. To determine α7* nAChR binding sites we incubated the particulate fractions from brain regions with 1 nM [125I]α-bungarotoxin. Non-specific sites were determined by adding 1 mM nicotine (* p < 0.05, Student's t test, n=3). Abbreviations: cerebral cortex (Cx), striatum (St), hippocampus (Hp).
4.6. Determination of [125I]epibatidine and [125I]α-bungarotoxin binding sites by autoradiography
Coronal brain slices of DBA/2J mice chronically treated with saline and 0.2 mg/kg/h 3-IC were assayed for binding of [125I]α-bungarotoxin, total [125I]epibatidine, cytisine-resistant and A85380-resistant [125I]-epibatidine sites. This autoradiographic technique allowed determination of nicotinic receptor binding sites at a finer level of resolution than obtained with membrane binding experiments.
Autoradiograms illustrating the effect of chronic 3-IC treatment are shown in Figure 7. There were differences in radioligand binding among brain regions, with high densities of [125I]epibatidine binding expressed in thalamus, superior colliculi, interpeduncular nuclei, fasciculus retroflexus and medial habenula, whereas some have lower density such as parietal cortex, hindbrain or caudate among others. [125I]epibatidine binding sites were differentially regulated by chronic 3-IC treatment; statistically significant increases in binding were observed in parietal cortex, hippocampus, caudate putamen, nucleus accumbens, substantia nigra, olfactory tubercle, external layer of inferior colliculus, superior colliculus and ventral tegmental area, with increases ranging from 45.2% in ventral tegmental area to 126.2% in the optic tract as compared to saline infused mice. Some brain regions (thalamic nuclei, medial habenula, fasciculus retroflexus among others) were unaffected by the chronic 3-IC treatment (Table 1).
Figure 7. Autoradiography of total and cytisine resistant [125I]epibatidine and [125I]bungarotoxin sites.
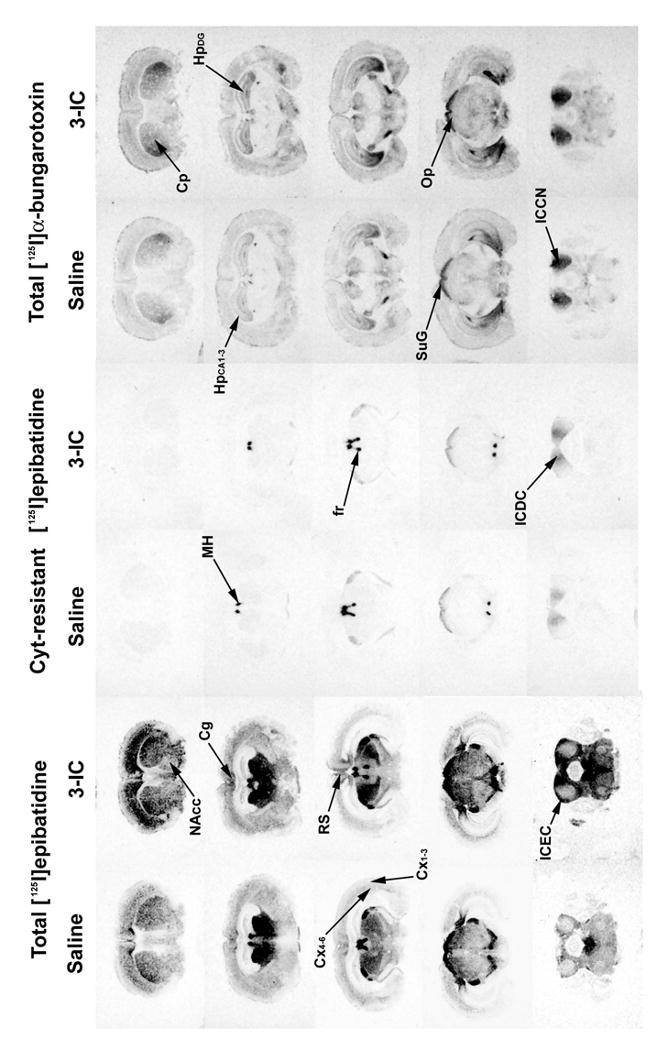
Mice were chronically intravenously infused with 3-IC 0.2 mg/kg/h for 7 days. After treatment, the brains were removed and frozen and coronal sections (14 μm) were obtained and incubated with 1 nM [125I]α-bungarotoxin or 200 pM [125I]epibatidine in presence or absence of cytisine 50 nM. Non-specific binding was determined by adding 1 mM nicotine for [125I]α-bungarotoxin and 100 μM for [125I]epibatidine. Abbreviations: cingulated cortex (Cg), parietal cortex at superficial layers (Cx1-3) and deep layers (Cx4-6), retrosplenial cortex (RS), hippocampus at CA1/CA2/CA3 (HpCA1-3) region and dentate gyrus granule layer (HpDG), caudate putamen (Cp), nucleus accumbens (NAcc), medial habenula (MH), inferior colliculi at dorsal cortex (DCIC), central nucleus (ICCN) and external cortex (ICEC), superior colliculi at superficial gray (SuG) and optic nerve (Op), fasciculus retroflexus (fr).
The cytisine-resistant [125I]epibatidine sites, which are found in areas such as interpeduncular nuclei, medial habenula and colliculi (Figure 7) were not affected by chronic 3-IC treatment (Table 2).
Table 2. Autoradiographic values for subsets of [125I]epibatidine binding.
Mice were chronically intravenously infused with 3-IC 0.2 mg/kg/h for 7 days. After chronic 3-IC treatment, the brains were removed and series of coronal slices were obtained. Subsets of [125I]epibatidine binding were calculated from binding of [125I]epibatidine 200 pM in absence or presence of cytisine 50 nM. Non-specific binding was determined by adding 100 μM nicotine. (* p<0.05, ** p<0.01, *** p<0.001, Student's t test, n=3 for saline and 3-IC treatment)
| Cytisine-sensitive [125I]epibatidine binding (fmol/mg) | Cytisine-resistant [125I]epibatidine binding (fmol/mg) | |||||
|---|---|---|---|---|---|---|
| Saline | 3-iodocytisine | % change | Saline | 3-iodocytisine | % change | |
| cingulate cortex | 16.6 ± 3.3 | 23.6 ± 1.5 | 42.0 | 0.99 ± 0.12 | 1.25 ± 0.13 | 26.9 |
| parietal cortex (layers 1-3) | 5.3 ± 1.1 | 9.1 ± 1.0 | 71.7 * | 0.50 ± 0.01 | 0.62 ± 0.06 | 24.6 |
| parietal cortex (layers 4-6) | 10.6 ± 2.0 | 19.0 ± 1.6 | 79.7 * | 0.89 ± 0.03 | 1.29 ± 0.07 | 45.9 |
| retrosplenial Cortex | 19.6 ± 2.8 | 26.6 ± 2.7 | 35.6 | 1.20 ± 0.05 | 1.63 ± 0.11 | 35.8 |
| anterior olfactory nucleus | 10.5 ± 5.1 | 16.1 ± 1.4 | 53.5 | 17.07 ± 1.30 | 13.27 ± 2.48 | -33.3 |
| hippocampus (CA1-3) | 5.7 ± 0.3 | 9.0 ± 0.2 | 56.3 *** | 0.81 ± 0.06 | 0.98 ± 0.06 | 20.2 |
| dentate gyrus (granular layer) | 8.4 ± 0.9 | 12.3 ± 0.6 | 46.8 * | 0.98 ± 0.05 | 1.15 ± 0.06 | 17.3 |
| subiculum | 32.6 ± 9.1 | 41.7 ± 3.8 | 27.8 | 0.98 ± 0.05 | 1.89 ± 0.10 | 3.0 |
| caudate putamen | 12.1 ± 1.3 | 20.3 ± 1.9 | 68.3 * | 1.84 ± 0.08 | 2.39 ± 0.29 | 18.2 |
| nucleus accumbens | 8.3 ± 1.7 | 15.9 ± 2.1 | 91.8 * | 2.02 ± 0.02 | 1.95 ± 0.21 | 12.9 |
| substantia nigra | 31.6 ± 1.8 | 43.6 ± 7.3 | 38.0 | 1.5 ± 0.1 | 1.6 ± 0.2 | 6.7 |
| ventral tegmental area | 17.3 ± 1.6 | 25.1 ± 1.5 | 45.2 * | 2.82 ± 0.46 | 3.4 ± 0.69 | 20.5 |
| zona incerta | 8.8 ± 0.7 | 13.7 ± 1.2 | 55.3 * | 1.73 ± 0.18 | 2.34 ± 0.22 | 5.2 |
| olfactory tubercle | 7.9 ± 0.3 | 14.5 ± 1.6 | 83.5 * | 3.5 ± 0.3 | 2.9 ± 0.5 | -17.1 |
| optic tract | 6.7 ± 0.6 | 15.1 ± 2.9 | 126.2 * | 1.39 ± 0.05 | 1.75 ± 0.22 | 26.1 |
| anteromedial thalamic nucleus | 50.7 ± 7.5 | 50.3 ± 4.0 | -0.8 | 3.26 ± 0.17 | 3.97 ± 0.25 | 22.0 |
| dorsomedial thalamic nucleus | 55.4 ± 10.6 | 52.9 ± 5.2 | -4.5 | 3.59 ± 0.10 | 3.78 ± 0.50 | 5.3 |
| ventromedial thalamic nucleus | 51.1 ± 3.4 | 50.7 ± 1.5 | -0.8 | 2.90 ± 0.59 | 3.29 ± 0.46 | 13.4 |
| medial habenula | 112.3 ± 31.7 | 171.3 ± 36.4 | 52.6 | 326.93 ± 28.78 | 282.84 ± 31.53 | -13.5 |
| fasciculus retroflexus | 39.8 ± 4.5 | 40.2 ± 4.0 | 1.0 | 61.57 ± 2.35 | 63.47 ± 4.86 | 3.1 |
| dorsolateral geniculate nucleus | 64.5 ± 5.8 | 70.7 ± 3.9 | 9.7 | 10.17 ± 1.26 | 9.89 ± 0.66 | -2.8 |
| medial geniculate nucleus | 38.9 ± 0.7 | 49.4 ± 8.2 | 27.2 | 3.21 ± 0.45 | 4.43 ± 0.80 | 38.1 |
| inferior colliculus (dorsal) | 13.5 ± 4.2 | 22.8 ± 2.1 | 68.7 * | 6.06 ± 1.45 | 10.17 ± 2.62 | 67.7 |
| inferior colliculus (central) | 6.1 ± 1.1 | 12.2 ± 2.5 | 97.2 * | 2.38 ± 0.36 | 4.00 ± 1.42 | 68.2 |
| inferior colliculus (external) | 8.9 ± 1.3 | 16.8 ± 2.1 | 88.9 * | 2.06 ± 0.20 | 2.55 ±0.19 | 24.2 |
| interpeduncular nucleus | 196.0 ±38.4 | 194.7 ± 42.8 | -0.6 | 244.14 ± 31.95 | 211.95 ± 31.83 | 13.2 |
| superior colliculus (sup. gray) | 34.1 ± 1.0 | 51.2 ± 4.4 | 50.2 ** | 10.16 ±1.26 | 11.25 ± 0.12 | 10.8 |
| superior colliculus (optic nerve) | 29.5 ± 0.5 | 47.3 ± 2.2 | 60.6 *** | 3.19 ± 0.25 | 3.21 ± 0.14 | 0.5 |
α7* nAChR binding sites were also analyzed by autoradiography (Figure 7). All regions analyzed tend to show an increase of [125I]α-bungarotoxin binding site density after chronic 3-IC treatment that ranged from 11.1% in cingulate cortex to 81.2% in nucleus accumbens as percentage of saline infused controls (Table 3); statistically significant differences were found in some areas such as parietal cortex, hippocampus, caudate putamen, nucleus accumbens, substantia nigra, ventrolateral geniculate nucleus, interpeduncular nucleus and superior colliculi.
Table 3. Autoradiographic values for [125I]α-bungarotoxin binding.
Mice were chronically intravenously infused with 3-IC 0.2 mg/kg/h for 7 days. After treatment, the brains were removed and frozen, and coronal slices were obtained and incubated with 1 nM [125I]α-bungarotoxin. Non-specific binding was determined by adding 1 mM nicotine. A quantitative analysis of brains regions is presented (* p < 0.05, Student's t test, n=3 for saline and 3-IC treatments).
| [125I]α-bungarotoxin binding (fmol/mg) | |||
|---|---|---|---|
| Saline | 3-iodocytisine | % change | |
| cingulate cortex | 5.7 ± 1.6 | 6.3 ± 1.4 | 11.7 |
| parietal cortex (layers 1-3) | 3.4 ± 0.5 | 5.9 ± 0.3 | 79.3 ** |
| parietal cortex (layers 4-6) | 4.3 ± 0.4 | 7.8 ± 0.4 | 74.4 ** |
| retrosplenial cortex | 3.9 ± 1.0 | 4.8 ± 0.4 | 21.2 |
| hippocampus (CA1-3) | 5.7 ± 0.5 | 10.2 ± 0.8 | 77.5 ** |
| dentate gyrus (granular layer) | 7.2 ± 0.5 | 11.7 ± 1.2 | 63.5 * |
| subiculum | 5.8 ± 1.3 | 7.1 ± 0.7 | 22.0 |
| caudate putamen | 11.8 ± 0.5 | 19.6 ± 2.3 | 65.8 * |
| nucleus accumbens | 3.8 ± 0.1 | 6.8 ± 0.7 | 81.2 ** |
| substantia nigra | 6.1 ± 0.3 | 7.9 ± 0.9 | 29.5 |
| ventral tegmental area | 3.7 ± 0.1 | 5.2 ± 0.4 | 40.5 * |
| zona incerta | 7.2 ± 0.3 | 11.1 ± 1.0 | 52.1 * |
| medial habenula | 9.2 ± 2.0 | 11.6 ± 1.0 | 26.4 |
| supramammillary hypothalamic nucleus | 9.8 ± 1.7 | 12.1 ± 1.2 | 24.4 |
| ventrolateral geniculate nucleus | 13.9 ± 0.7 | 18.1 ± 1.2 | 30.2 * |
| inferior colliculus (dorsal) | 18.9 ± 1.5 | 21.7 ± 3.7 | 14.4 |
| inferior colliculus (central) | 30.9 ± 1.2 | 37.6 ± 4.2 | 21.7 |
| inferior colliculus (externall) | 17.6 ± 1.6 | 23.5 ± 4.1 | 33.1 |
| interpeduncular nucleus | 18.6 ± 0.2 | 23.5 ± 1.4 | 26.2 * |
| superior colliculus (sup. gray) | 17.2 ± 1.3 | 23.1 ± 0.8 | 34.7 ** |
| superior colliculus (optic nerve) | 7.8 ± 0.3 | 11.5 ± 0.5 | 44.2 ** |
Discussion
We report here the effects of 3-IC in mice, including effect of acute doses on body temperature regulation, binding potency at major nAChR populations in brain and the modulation of those receptors after chronic drug treatment. These results are generally consistent with the previous studies of 3-IC in vitro.
Acute effect of 3-IC on body temperature
3-IC elicited a larger hypothermic effect than reported for nicotine in mice (Marks et al, 1983(a), Tritto et al., 2004). The blockade of the hypothermia by mecamylamine and lack of effect of scopolamine established that activation of nicotinic, but not muscarinic, receptors is necessary for 3-IC-induced hypothermia. The effect of 3-IC on body temperature was dependent on the activation of both peripheral and central nAChR as determined by the partial blockade with hexamethonium. Experiments with null mutant mice indicated that the α7* receptor did not significantly mediate 3-IC-induced hypothermia, which is consistent with the observation that deletion of α7 had no effect on nicotine-induced hypothermia (Tritto et al., 2004). Although β2-/- mice displayed significant hypothermia following 3-IC injection, the magnitude and duration of this response was less than that of wild-type mice, suggesting some role for β2*-nAChR. A partial but more pronounced effect of β2 gene deletion was observed for nicotine-induced hypothermia (Tritto et al., 2004). However, β4-/- mice were much less affected by the same dose of 3-IC, indicating that activation of nAChR containing this subunit is responsible for most of the hypothermic response. A significant contribution of β4*-nAChR in the control of body temperature agrees with a previous report performed in mice using nicotine as an agonist (Sack et al., 2005). While the effect of 3-IC may be also associated with desensitization after receptor activation, a mechanism that is common with nAChR, α3β4*-nAChR desensitize more slowly and require higher ligand concentrations than α4β2*-nAChR (for review see Quick and Lester, 2002). Prolonged activation of α3β4*nAChR may contribute to the large effect of 3-IC. The assumption that the effect of 3-IC might be due the activation of α3β4*-nAChR is supported also by the observation that 3-IC is approximately 100-fold more potent as an inhibitor of [125I]epibatidine binding in β2-/- mice than cytisine or nicotine. The α3β4* receptor has been shown to be the most abundant among β4 subunit-containing nAChR expressed in autonomic ganglia (Conroy and Berg, 1995), suggesting a role for these receptors in the hypothermic response, a result consistent with the partial sensitivity to hexamethonium. However the contribution of central β4*-nAChR cannot be completely discounted.
3-bromocytisine is a full agonist at human α3β4-nAChR expressed in Xenopus oocytes, being more potent than either acetylcholine or cytisine (Abin-Carriquiry et al., 2006), suggesting that 3-IC would have a similar effect on α3β4*-nAChR because both C3-halogenated compounds exhibit a similar pharmacology (Slater et al. 2003, Imming et al., 2001). The higher potency and efficacy of 3-IC relative to cytisine can be explained by the influence of the halogen atom on the affinity of 3-IC for α4β2* and α7* nAChR as discussed before (Slater et al., 2003), although pharmacokinetic factors may also be involved.
The pharmacokinetic of 3-IC is not known. However, cytisine has a longer half-life than nicotine in rats (Sloan et al., 1988). Nonetheless, further work is necessary in order to reveal pharmacokinetic parameters such as distribution, blood-brain barrier penetration, metabolism and excretion of 3-IC.
Effect of chronic 3-IC on nAChR binding sites
The up-regulation of nAChR binding sites by chronic nicotine treatment is well established (Marks et al., 1983(b); Flores et al., 1992; Sparks and Pauly, 1999; Nguyen et al., 2003; Marks et al., 2004; McCallum et al., 2006; Rasmussen and Perry, 2006). We observed here a differential increase in α4β2* nAChR, principally in dopaminergic brain areas, cerebral cortex and hippocampus. In contrast, A85380-resistant [125I]epibatidine biding sites (likely β4*-nAChR) were unchanged after chronic 3-IC treatment. A similar insensitivity to upregulation of β4*-nAChR following nicotine treatment has been noted for adrenal gland, superior cervical ganglion and pineal (Davila-Garcia et al., 2003) and also following nicotine treatment in mice (Marks et al., 2004). Perhaps, the absence of tolerance to the hypothermic effect of 3-IC observed after chronic treatment reflects the major contribution of β4*-nAChR to 3-IC-induced hypothermia and the insensitivity of β4*-nAChR to up-regulation (Nguyen et al., 2003; McCallum et al., 2006).
The pattern of binding sites observed in the autoradiography for the α7* nAChR population is consistent with the previous reports (Pauly et al., 1989; Sparks and Pauly, 1999; Rasmussen and Perry, 2006), showing a high density in hippocampus, caudate putamen, superior and inferior colliculus and some structures in the mesencephalon and hindbrain as well. [125I]α-Bungarotoxin binding sites (α7-nAChR) are less responsive to chronic nicotine treatment than are α4β2*-nAChR binding sites (Marks et al., 1983(b)). The up-regulation of α7* receptors depends on the route of administration and the doses required are higher than those that elicit increase in α4β2*-nAChR (Sparks and Pauly, 1999; Rasmussen and Perry, 2006).
The increases in [125I]α-bungarotoxin binding sites produced by chronic 3-IC treatment are greater than those reported in mice treated chronically with nicotine, i.e. in dentate gyrus there was an increase of 63.5% compared with the 12.6% increase detected in the same DBA/2J mice with a ten days 2 mg/kg/h nicotine treatment (Pauly et al., 1991); zona incerta 52.1% compared to 17.6%; hippocampus 77.5% compared to 33%; caudate putamen 65.8% compared to 14.2%. However, there were regions in these two studies that showed similar α7* upregulation such as medial habenula 26.4% compared to 22.6%; ventral tegmental area 40.5% compared to 32.4%; substantia nigra 29.5% compared to 25.6%. The greater effect on binding sites elicited by chronic 3-IC treatment may be related to relatively high affinity of the C3 halo-derivatives of cytisine on α7* receptors (Abin-Carriquiry et al., 2006). This postulate is supported by the observation that 3-IC is a much more potent inhibitor of [125I]α-bungarotoxin binding than cytisine or nicotine.
Concluding remarks
In the present we report an in vivo effect of 3-IC on the population of central nervous system and peripheral nAChR has been demonstrated. In the case of new drugs for smoking cessation the central effects are desired and 3-IC has profound autonomic effects. Our results indicate that 3-IC may not be the best choice for this application. However, owing to the profound hypothermia elicited, 3-IC may be useful in prevention of degenerative processes observed in nervous tissue after ischemic damage, since a decrease in core body temperature is used to mitigate damage (Olsen et al., 2003; Gordon C.J., 2001), however, an extensive exploration of systemic effects including pharmacological interactions with anaesthetic drugs is needed. Furthermore, 3-IC may be a useful compound for the study of the effects of activation of β4*-nAChR.
Acknowledgments
This study was supported by research grant DA003194 and animal resource grant DA015663 from the National Institute on Drug Abuse. C.A. Zambrano was supported by MECESUP 0208 fellowship program from the Ministry of Education of Chile. Authors also thank Sharon R. Grady and Allan C. Collins for their assistance in preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abin-Carriquiry JA, Voutilainen MH, Barik J, Cassels BK, Iturriaga-Vasquez P, Bermudez I, Durand C, Dajas F, Wonnacott S. C3-halogenation of cytisine generates potent and efficacious nicotinic receptor agonists. European Journal of Pharmacology. 2006;536:1–11. doi: 10.1016/j.ejphar.2006.02.012. [DOI] [PubMed] [Google Scholar]
- Barr JE, Holmes DB, Ryan LJ, Sharpless SK. Techniques for the chronic cannulation of the jugular vein in mice. Pharmacology, Biochemistry and Behavior. 1979;11:115–118. doi: 10.1016/0091-3057(79)90307-1. [DOI] [PubMed] [Google Scholar]
- Bourin M, Ripoll N, Dailly E. Nicotinic receptors and Alzheimer's disease. Current Medical Research and Opinion. 2003;19:169–177. doi: 10.1185/030079903125001631. [DOI] [PubMed] [Google Scholar]
- Conroy WG, Berg DK. Neurons can maintain multiple classes of nicotinic acetylcholine receptors distinguished by different subunit compositions. Journal of Biological Chemistry. 1995;270:4424–4431. doi: 10.1074/jbc.270.9.4424. [DOI] [PubMed] [Google Scholar]
- Dani JA, Bertrand D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annual Review of Pharmacology and Toxicology. 2007;47:699–729. doi: 10.1146/annurev.pharmtox.47.120505.105214. [DOI] [PubMed] [Google Scholar]
- Dani JA, DeBiasi M. Cellular mechanism of nicotine addiction. Pharmacology Biochemistry and Behavior. 2001;70:439–446. doi: 10.1016/s0091-3057(01)00652-9. [DOI] [PubMed] [Google Scholar]
- Davila-Garcia MI, Musachio JL, Kellar KJ. Chronic nicotine administration does not increase nicotinic receptors labeled by [125I]epibatidine in adrenal gland, superior cervical ganglia, pineal or retina. Journal of Neurochemistry. 2003;85:1237–1246. doi: 10.1046/j.1471-4159.2003.01774.x. [DOI] [PubMed] [Google Scholar]
- Flores CM, Rogers SW, Pabreza LA, Wolfe BB, Kellar KJ. A subtype of nicotinic cholinergic receptor in rat brain is composed of alpha 4 and beta 2 subunits and is up-regulated by chronic nicotine treatment. Molecular Pharmacology. 1992;41:31–37. [PubMed] [Google Scholar]
- Gotti C, Moretti M, Gaimarri A, Zanardi A, Clementi F, Zoli M. Heterogeneity and complexity of native brain nicotinic receptors. Biochemical Pharmacology. 2007;74:1102–1111. doi: 10.1016/j.bcp.2007.05.023. [DOI] [PubMed] [Google Scholar]
- Gordon CJ. The therapeutic potential of regulated hypothermia. Emergency Medicine Journal. 2001;18:81–89. doi: 10.1136/emj.18.2.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JG, Kongs S, Allensworth D, Martin L, Tregellas J, Sullivan B, Zerbe G, Freedman R. Effects of nicotine on cognitive deficits in schizophrenia. Neuropsychopharmacology. 2004;29:1378–1385. doi: 10.1038/sj.npp.1300450. [DOI] [PubMed] [Google Scholar]
- Hogg RC, Bertrand D. Neuronal nicotinic receptors and epilepsy, from genes to possible therapeutic compounds. Bioorganic and Medicinal Chemistry Letters. 2004;14:1859–1861. doi: 10.1016/j.bmcl.2003.10.075. [DOI] [PubMed] [Google Scholar]
- Imming P, Klaperski P, Stubbs MT, Seitz G, Gündisch D. Syntheses and evaluation of halogenated cytisine derivatives and of bioisosteric thiocytisine as potent and selective nAChR ligands. European Journal of Medicinal Chemistry. 2001;36:375–388. doi: 10.1016/s0223-5234(01)01222-3. [DOI] [PubMed] [Google Scholar]
- Lam S, Patel PN. Varenicline: a selective alpha4beta2 nicotinic acetylcholine receptor partial agonist approved for smoking cessation. Cardiology in Review. 2007;15:154–161. doi: 10.1097/01.crd.0000260270.12829.45. [DOI] [PubMed] [Google Scholar]
- Leonard S, Adler LE, Benhammou K, Berger R, Breese CR, Drebing C, Gault J, Lee MJ, Logel J, Olincy A, Ross RG, Stevens K, Sullivan B, Vianzon R, Virnich DE, Waldo M, Walton K, Freedman R. Smoking and mental illness. Pharmacology Biochemistry and Behavior. 2001;70:561–570. doi: 10.1016/s0091-3057(01)00677-3. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. Journal of Biological Chemistry. 1951;193:265–275. [PubMed] [Google Scholar]
- Luetje CW, Patrick J. Both alpha- and beta-subunits contribute to the agonist sensitivity of neuronal nicotinic acetylcholine receptors. The Journal of Neuroscience. 1991;11:837–845. doi: 10.1523/JNEUROSCI.11-03-00837.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas RJ, Changeux JP, Le Novère N, Albuquerque EX, Balfour DJ, Berg DK, Bertrand D, Chiappinelli VA, Clarke PB, Collins AC, Dani JA, Grady SR, Kellar KJ, Lindstrom JM, Marks MJ, Quik M, Taylor PW, Wonnacott S. International Union of Pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacological Reviews. 1999;51:397–401. [PubMed] [Google Scholar]
- Marks MJ, Burch JB, Collins AC. Genetics of nicotine response in four inbred strains of mice. The Journal of Pharmacology and Experimental Therapeutics. 1983a;226:291–302. [PubMed] [Google Scholar]
- Marks MJ, Burch JB, Collins AC. Effects of chronic nicotine infusion on tolerance development and nicotinic receptors. The Journal of Pharmacology and Experimental Therapeutics. 1983b;226:817–825. [PubMed] [Google Scholar]
- Marks MJ, Rowell PP, Cao JZ, Grady SR, McCallum SE, Collins AC. Subsets of acetylcholine-stimulated 86Rb+ efflux and [125I]-epibatidine binding sites in C57BL/6J mouse brain are differentially affected by chronic nicotine treatment. Neuropharmacology. 2004;46:1141–1157. doi: 10.1016/j.neuropharm.2004.02.009. [DOI] [PubMed] [Google Scholar]
- McCallum SE, Collins AC, Paylor R, Marks MJ. Deletion of the beta 2 nicotinic acetylcholine receptor subunit alters development of tolerance to nicotine and eliminates receptor upregulation. Psychopharmacology (Berl) 2006;184:314–327. doi: 10.1007/s00213-005-0076-6. [DOI] [PubMed] [Google Scholar]
- Millar NS. Assembly and subunit diversity of nicotinic acetylcholine receptors. Biochemical Society Transactions. 2003;31:869–874. doi: 10.1042/bst0310869. [DOI] [PubMed] [Google Scholar]
- Nguyen HN, Rasmussen BA, Perry DC. Subtype-selective up-regulation by chronic nicotine of high-affinity nicotinic receptors in rat brain demonstrated by receptor autoradiography. The Journal of Pharmacology and Experimental Therapeutics. 2003;307:1090–1097. doi: 10.1124/jpet.103.056408. [DOI] [PubMed] [Google Scholar]
- Olsen TS, Weber UJ, Kammersgaard LP. Therapeutic hypothermia for acute stroke. Lancet Neurology. 2003;2:410–416. doi: 10.1016/s1474-4422(03)00436-8. [DOI] [PubMed] [Google Scholar]
- Orr-Urtreger A, Goldner FM, Saeki M, Lorenzo I, Goldberg L, De Biasi M, Dani JA, Patrick JW, Beaudet AL. Mice deficient in the alpha7 neuronal nicotinic acetylcholine receptor lack alpha-bungarotoxin binding sites and hippocampal fast nicotinic currents. The Journal of Neuroscience. 1997;17:9165–9171. doi: 10.1523/JNEUROSCI.17-23-09165.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauly JR, Stitzel JA, Marks MJ, Collins AC. An autoradiographic analysis of cholinergic receptors in mouse brain. Brain Research Bulletin. 1989;22:453–459. doi: 10.1016/0361-9230(89)90072-5. [DOI] [PubMed] [Google Scholar]
- Pauly JR, Marks MJ, Gross SD, Collins AC. An autoradiographic analysis of cholinergic receptors in mouse brain after chronic nicotine treatment. Pharmacology and Experimental Therapeutics. 1991;258:1127–1136. [PubMed] [Google Scholar]
- Pabreza LA, Dhawan S, Kellar KJ. [3H]cytisine binding to nicotinic cholinergic receptors in brain. Molecular Pharmacology. 1991;39:9–12. [PubMed] [Google Scholar]
- Picciotto MR. Nicotine as a modulator of Behavior: beyond the inverted U. Trends in Pharmacological Sciences. 2003;24:493–499. doi: 10.1016/S0165-6147(03)00230-X. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Zoli M, Léna C, Bessis A, Lallemand Y, Le Novère N, Vincent P, Pich EM, Brulet P, Changeux JP. Abnormal avoidance learning in mice lacking functional high-affinity nicotine receptor in the brain. Nature. 1995;374:65–67. doi: 10.1038/374065a0. [DOI] [PubMed] [Google Scholar]
- Quick MW, Lester RAJ. Desensitization of neuronal nicotinic receptors. Journal of Neurobiology. 2002;53:457–478. doi: 10.1002/neu.10109. [DOI] [PubMed] [Google Scholar]
- Quik M. Smoking, nicotine and Parkinson's disease. Trends Neuroscience. 2004;27:561–568. doi: 10.1016/j.tins.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Rasmussen BA, Perry DC. An autoradiographic analysis of [125I]alpha-bungarotoxin binding in rat brain after chronic nicotine exposure. Neuroscience Letters. 2006;404:9–14. doi: 10.1016/j.neulet.2006.05.010. [DOI] [PubMed] [Google Scholar]
- Sack R, Gochberg-Sarver A, Rozovsky U, Kedmi M, Rosner S, Orr-Urtreger A. Lower core body temperature and attenuated nicotine-induced hypothermic response in mice lacking the beta4 neuronal nicotinic acetylcholine receptor subunit. Brain Research Bulletin. 2005;66:30–36. doi: 10.1016/j.brainresbull.2005.02.032. [DOI] [PubMed] [Google Scholar]
- Salminen O, Murphy KL, McIntosh JM, Drago J, Marks MJ, Collins AC, Grady SR. Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Molecular Pharmacology. 2004;65:1526–1535. doi: 10.1124/mol.65.6.1526. [DOI] [PubMed] [Google Scholar]
- Salin-Pascual RJ, Basanez-Villa E. Changes in compulsion and anxiety symptoms with nicotine transdermal patches in non-smoking obsessive-compulsive disorder patients. Revista de Investigacion Clinica. 2003;55:650–654. [PubMed] [Google Scholar]
- Slater YE, Houlihan LM, Maskell PD, Exley R, Bermudez I, Lukas RJ, Valdivia AC, Cassels BK. Halogenated cytisine derivatives as agonists at human neuronal nicotinic acetylcholine receptor subtypes. Neuropharmacology. 2003;44:503–515. doi: 10.1016/s0028-3908(03)00025-x. [DOI] [PubMed] [Google Scholar]
- Sloan JW, Martin WR, Bostwick M, Hook R, Wala E. The comparative binding characteristics of nicotinic ligands and their pharmacology. Pharmacology Biochemistry and Behavior. 1988;30:255–267. doi: 10.1016/0091-3057(88)90454-6. [DOI] [PubMed] [Google Scholar]
- Sparks JA, Pauly JR. Effects of continuous oral nicotine administration on brain nicotinic receptors and responsiveness to nicotine in C57BL/6J mice. Psychopharmacology (Berl) 1999;141:145–153. doi: 10.1007/s002130050818. [DOI] [PubMed] [Google Scholar]
- Tritto T, McCallum SE, Waddle SA, Hutton SR, Paylor R, Collins AC, Marks MJ. Null mutant analysis of responses to nicotine: Deletion of β2 nicotinic acetylcholine receptor subunit but not α7 subunit reduces sensitivity to nicotine-induced locomotor depression and hypothermia. Nicotine and Tobacco Research. 2004;6:145–157. doi: 10.1080/14622200310001656966. [DOI] [PubMed] [Google Scholar]
- Varas R, Valdés V, Iturriaga-Vásquez P, Cassels BK, Iturriaga R, Alcayaga J. Electrophysiological characterization of nicotinic acetylcholine receptors in cat petrosal ganglion neurons in culture: effects of cytisine and its bromo derivatives. Brain Research. 2006;1072:72–78. doi: 10.1016/j.brainres.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Whiteaker P, Jimenez M, McIntosh JM, Collins AC, Marks MJ. Identification of a novel nicotinic binding site in mouse brain using [125I]-epibatidine. British Journal of Pharmacology. 2000;131:729–739. doi: 10.1038/sj.bjp.0703616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteaker P, Marks MJ, Christensen S, Dowell C, Collins AC, McIntosh JM. Synthesis and characterization of 125I-alpha-conotoxin ArIB[V11L;V16A], a selective alpha7 nicotinic acetylcholine receptor antagonist. The journal of Pharmacology and Experimental Therapeutic. 2008;325:910–919. doi: 10.1124/jpet.108.136895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Orr-Urtreger A, Nigro F, Gelber S, Sutcliffe CB, Armstrong D, Patrick JW, Role LW, Beaudet AL, De Biasi M. Multiorgan autonomic dysfunction in mice lacking the beta2 and the beta4 subunits of neuronal nicotinic acetylcholine receptors. The Journal of Neuroscience. 1999;19:9298–9305. doi: 10.1523/JNEUROSCI.19-21-09298.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]