

Abstract
We have explored a series of trisubstituted acridine-peptide conjugates for their ability to recognize and discriminate between DNA quadruplexes derived from the human telomere, and the c-kit and N-ras proto-oncogenes. Quadruplex affinity was measured as the peptide sequences were varied, together with their substitution position on the acridine, and the identity of the C-terminus (acid or amide). Surface plasmon resonance measurements revealed that all compounds bound to the human telomeric quadruplex with sub-micromolar affinity. Docking calculations from molecular modelling studies were used to model the effects of substituent orientation and peptide sequence. Modelling and experiment were in agreement that placement of the peptide over the face of the acridine is detrimental to binding affinity. The highest degrees of selectivity were observed towards the N-ras quadruplex by compounds capable of forming simultaneous contacts with their acridine and peptide moieties. The ligands that bound best displayed quadruplex affinities in the 1-5 nM range and at least 10-fold discrimination between the quadruplexes studied.
Introduction
Quadruplexes are four-stranded nucleic acid secondary structures in which guanine bases form stacked planar tetrads stabilized by hydrogen bonding and cation coordination.1 Folding rules predict many potential quadruplex sequences within the human genome, in addition to those at telomeric repeats.2 Sequences with a high propensity to form quadruplexes are not distributed evenly throughout the genome but are more likely to be found in gene promoters, proto-oncogenes and the first intron.3,4 Quadruplex-forming sequences have been found in genes including c-myc,5 c-kit,6,7 N-ras,8,9 Ki-ras,10,11 Rb,12 RET,13 VEGF,14 HIF-1α,15 BCL2,16 PDGF-A,17 and c-myb.18 We and others have shown that quadruplex-binding ligands can regulate the expression of such genes.11,19-22 This evidence, together with a genome wide analysis of expression levels23 of genes containing putative quadruplex forming sequences in the vicinity of the transcription start site suggests that quadruplexes may naturally act as regulatory elements. Quadruplex-binding ligands will enable us to further address this hypothesis and artificially regulate the expression of genes of medical interest. The human telomeric quadruplex is also a target for antitumour compounds that disrupt the telomere nucleoprotein complex and/or inhibit the action of the enzyme telomerase.24 However a major challenge is to design ligands with specificity for individual quadruplexes amongst the multitude of potential quadruplex sequences.
We describe here the development of a general quadruplex-binding scaffold with substituents that confer affinities of 1-5 nM together with up to an order of magnitude discrimination between the quadruplexes studied. Molecular modelling has been used to rationalize the binding to the telomeric quadruplex and to suggest how discrimination between quadruplexes might occur.
Monomolecular quadruplexes display a wide range of topologies differing in the nature and positions of the loops connecting the strands, and thus in relative strand orientation. The loops and peripheral grooves provide a pattern of hydrogen bonding, hydrophobic π surfaces, and negative charges which are unique to the sequence of a particular quadruplex. The guanine tetrads themselves are conserved amongst quadruplexes hence ligand motifs that interact predominantly with the tetrad should display broad quadruplex affinity. It has been found that ligands can show a degree of discrimination between the c-myc25 or c-kit21,26,27 quadruplexes and human telomeric quadruplex or even perturb quadruplex topology.27,28 Our strategy for the discovery of high affinity ligands with selectivity between quadruplexes employs a common core that targets the planar surface of a G-tetrad, appended with variable substituents to contact the loops and grooves which distinguish quadruplexes from each other. As core units from which to build quadruplex binding ligands according to this design template, we and others have investigated various fused tricyclic planar heterocycles including ethidium derivatives,29 quindolines,22 isoalloxazines,21 carbazoles30 and acridines.31-35 The dimension of the long axis of these planar cores approximately matches the width of a G-tetrad, such that substituents are expected to be positioned correctly to interact with up to three of the quadruplex loops and grooves. Here we describe peptide-acridine conjugates designed according to these principles, with structural variation to test the binding mode hypothesis. The affinity of the compounds for three proto-oncogene quadruplexes and the human telomeric quadruplex is compared, and interpreted in terms of the compound structures and models of complexes with the telomeric quadruplex.
Results and discussion
Ligand design
Previous studies have demonstrated that 3,6,9-substituted acridine derivatives recognize quadruplexes,32,34 and it has been confirmed by a co-crystal structure of a bimolecular telomeric sequence with the compound BRACO-19 that the acridine stacks on a terminal guanine tetrad.35 The ability of heterocyclic ligands to interact also with quadruplex grooves has been suggested by circular dichroism spectroscopy.36 We have previously shown37 that acridines bearing peptides at the 3,6-positions, but lacking a 9-substituent, bind a human telomeric monomolecular quadruplex with dissociation constants in the range 0.1-10 μM. 9-Anilino substituents bearing simple tertiary alkylamines have enhanced interactions with the telomeric quadruplex relative to the parent compound with an N-dimethylphenyldiamine substituent.38 Affinity for the telomeric quadruplex, as judged by a FRET melting assay, was also comparable or enhanced when a 9-anilino substituent was replaced with extended and more flexible benzylamines.31 This substituent provides a point for variation to adjust selectivity whilst maintaining the constant tetrad binding motif and we anticipated that a peptide would present a more complex recognition surface and hence augment discrimination between quadruplexes. The effect of a short peptide substituent at this position had not previously been investigated. In the current work, we have therefore investigated trisubstituted peptide acridine conjugates, varying the location and orientation of the peptide substituent with respect to the acridine core which is proposed to act as an anchor group by stacking on to the terminal guanine tetrad.
In the first series of compounds, 7-9, the peptides are appended to the 3- and 6-positions of the acridine core and the 9- substituent is N-dimethylphenyldiamine from the parent compound BRACO-19. In contrast to the approach of Carlson et al. in which an acridine amino acid residue is incorporated into the backbone of peptides,39 we have opted for a bis-carboxylic acid-substituted acridine for symmetrical conjugation to the N-termini of two peptides.40 Peptide sequences were chosen from our previous screening experiments against the human telomeric quadruplex.41
In the second series, 10-21, pyrrolidine propionamide substituents at the 3,6-positions were retained from earlier optimal designs34 and the peptide was conjugated through its N-terminus to the 9-position via an aminobenzoyl linker. The linker permits variation in the orientation of the peptide with respect to the acridine, and ortho, meta and para variants were synthesized. The peptide C-terminus is free, and amide and acid variants were prepared in anticipation of charge effects on quadruplex affinity.
Ligand synthesis
The acridine core of the ligands was derived ultimately from 3,6-diaminoacridone by acylation with chloropropionyl chloride, followed by alkylation with pyrrolidine and conversion to an acridine by treatment with phosphoryl chloride according to a literature route. Displacement of the 9-chloro group using aminobenzoic acids (Scheme 1) afforded the acridine building blocks required for conjugation at the N-terminus of a peptide to yield compounds 10-21. Compounds 7-9 were prepared from bisacid 6 which was synthesised according to Scheme 2. Alkylation of the chloropropionyl side chains of a substituted acridone with sarcosine ethylester, supplied as a hydrochloride salt, was found to proceed most effectively using an in situ deprotonation by dropwise addition of sodium ethoxide. Conversion of 4 to acridine 5 using POCl3 and reaction with N-dimethylphenyldiamine proceeded smoothly. Careful hydrolysis of the ethyl esters of 5, to avoid reformation of the acridone by hydrolytic loss of the 9-substituent, afforded the bis-carboxylic acid. All peptides were prepared by Fmoc solid phase synthesis and after terminal Fmoc deprotection, the acridines were conjugated to their N-termini using the same reagents (PyBOP, HOBt, DIPEA) as for standard amino acid couplings. This strategy was successfully employed to prepare the 3,6-bis peptide conjugates by one step intersite solid phase cross linking.40
Scheme 1.

Reagents and conditions: (a) aminobenzoic acid, CHCl3, Δ.
Scheme 2.

Reagents and conditions: (a) sarcosine ethylester hydrochloride, NaI, DMF, NaOEt, Δ; (b) (i) POCl3, Δ (ii) N-dimethylphenyldiamine, CHCl3, Δ; (c) LiOH, THF, H2O, rt.
Binding studies
Surface plasmon resonance (SPR) was used to determine dissociation constants against various folded DNA targets that included the human telomeric quadruplex (Htelo), quadruplexes from the promoter of the human c-kit gene, c-kit1,6 c-kit2,7 and from the 5′-untranslated region of the human N-ras gene.9 As a second measure of quadruplex interaction, the ability of ligands to stabilize a telomeric quadruplex to thermal unfolding was determined,42 by measuring the change in its melting temperature, Tm.
The magnitudes of the dissociation constants of 3,6-bis peptide acridines (7-9) and the previously reported 3,6-bis peptide acridones for Htelo correspond well,37 indicating that inclusion of the additional 9-dimethylphenyldiamine substituent has little effect on the Htelo affinity (Table 3). The apparent stoichiometries differ between compounds, with 8 and 9 adopting a 2:1 binding mode whereas compound 7 adopts a 1:1 mode, suggesting that the former compounds bind to both terminal tetrads raising the possibility that the latter either distinguishes between them or binds in a strongly negatively cooperative manner. Compounds 7-9 display no significant discrimination between the three quadruplexes tested, binding to all with similar affinity indicating that interactions occur primarily through features shared by the quadruplexes such as the presence of the guanine tetrads. We hypothesise that the relatively flexible linkers at the 3- and 6-position are not conducive to the formation of structure specific interactions between the peptides and quadruplex.
Table 3.
Dissociation constants (Kd ± std. error) and stoichiometries (n) of compounds 7-21 with the Htelo, c-kit1, c-kit2 and N-ras quadruplexes, determined by surface plasmon resonance
|
Htelo
|
c-kit1
|
c-kit2
|
N-ras
|
|||||
|---|---|---|---|---|---|---|---|---|
| Compound | Kd/nM | n | Kd/nM | n | Kd/nM | n | Kd/nM | n |
| 7 | 770 ± 80 | 1.2 | a | a | 800 ± 100 | 1.3 | 440 ± 70 | 0.7 |
| 8 | 250 ± 40 | 2.2 | a | a | 290 ± 40 | 2.0 | 240 ± 40 | 1.5 |
| 9 | 380 ± 50 | 2.3 | a | a | 400 ± 50 | 1.9 | 460 ± 70 | 1.6 |
| 10 | 140 ± 60 | 0.9 | 25 ± 8 | 0.4 | 120 ± 40 | 0.6 | 129 ± 3 | 0.7 |
| 11 | 50 ± 10 | 1.1 | 50 ± 20 | 0.9 | 130 ± 50 | 1.3 | 60 ± 40 | 0.6 |
| 12 | 28 ± 5 | 0.8 | 138 ± 5 | 1.3 | 50 ± 10 | 0.7 | 21 ± 2 | 0.8 |
| 13 | 38 ± 9 | 0.7 | 37 ± 7 | 0.8 | 37 ± 7 | 0.7 | 16 ± 3 | 0.8 |
| 14 | 22 ± 4 | 1.2 | 50 ± 20 | 1.2 | 40 ± 10 | 1.2 | 4 ± 1 | 0.5 |
| 15 | 23 ± 4 | 1.0 | 90 ± 10 | 1.3 | 40 ± 10 | 1.1 | 7 ± 1 | 0.8 |
| 16 | 20 ± 8 | 0.4 | 49 ± 9 | 0.4 | 60 ± 30 | 0.4 | 69.3 ± 0.8 | 0.9 |
| 17 | 25 ± 3 | 0.8 | 40 ± 10 | 0.7 | 50 ± 10 | 0.9 | 17 ± 5 | 0.6 |
| 18 | 19 ± 3 | 0.7 | 35 ± 8 | 0.6 | 40 ± 10 | 0.7 | 5 ± 1 | 0.5 |
| 19 | 30 ± 3 | 0.6 | 37 ± 3 | 0.6 | 40 ± 20 | 0.6 | 29 ± 2 | 1.1 |
| 20 | 13 ± 2 | 0.8 | 33 ± 6 | 0.8 | 29 ± 9 | 1.0 | 3.9 ± 0.8 | 0.6 |
| 21 | 17 ± 2 | 0.8 | 50 ± 10 | 0.8 | 30 ± 10 | 0.9 | 4.4 ± 0.9 | 0.6 |
Not determined.
The affinities of the 9-peptide conjugates (10-21) for Htelo are higher than the 3,6-bis-peptide acridine conjugates (7-9) by approximately an order of magnitude confirming the importance of the acridine substitution pattern for affinity. We have therefore focussed our attention on these compounds. The orientation of the 9-aminobenzoyl substitution was varied between ortho, meta and para in the two series of compounds 10-12 and 13-15. Dissociation constants for the Htelo, c-kit1, c-kit2 and N-ras quadruplexes, as determined by SPR, are given Table 3. The concentrations of ligand required to induce a change in the melting temperature of the telomeric quadruplex of 25 °C as determined in the FRET melting assay42 are given in Table 4. Of the two peptide categories, the KRSR compounds typically displayed higher affinity than their FRHR counterparts. Compound 10 showed an almost 6-fold greater affinity for the c-kit1 quadruplex compared to the Htelo one, and similar discrimination against the c-kit2 and N-ras quadruplexes. This selectivity is reversed for compounds 12 and 15, which have a 1:1 stoichiometry with both Htelo and c-kit1 quadruplexes. Compounds 10-15 display little discrimination between Htelo and c-kit2, although overall there is a slight (<3-fold) bias towards Htelo.
Table 4.
Ligand concentration required to stabilize F21T quadruplex by 25 °C in a FRET melting assay
| Ligand | [Ligand] for ΔTm of 25 °C/μM |
|---|---|
| 10 | 1.16 |
| 11 | 0.36 |
| 12 | 0.32 |
| 13 | 0.89 |
| 14 | 0.10 |
| 15 | 0.17 |
| 16 | 3.27 |
| 17 | 0.34 |
| 18 | 0.29 |
| 19 | 1.06 |
| 20 | 0.09 |
| 21 | 0.12 |
The affinities for the N-ras quadruplex are some of the highest reported to date for quadruplex complexes. There is a general bias, with the exception of compound 16, for preferential binding to the N-ras quadruplex over Htelo. The selectivity is greatest for compound 14. Significant selectivities were observed with the two c-kit quadruplexes; compound 14 shows an order of magnitude discrimination for the N-ras quadruplex over both the c-kit1 and c-kit2 ones. Compounds 15 and 21 also show selectivity for N-ras over c-kit1 and to a lesser extent c-kit2, whereas compound 10 also shows a five-fold preference for c-kit1 over N-ras. The highest (>5-fold) selectivities towards N-ras over the two c-kit quadruplexes are all achieved by ligands with a meta or para substitution pattern. The ortho compounds show little or no selectivity for N-ras, and in the case of 10 actually discriminates against this quadruplex in favour of the c-kit1 one. Overall the KRSR peptide substituent leads to greater N-ras selectivity than FRHR. It is interesting to note that the apparent stoichiometry of binding in some instances is less than 1:1. A possible interpretation of this phenomenon could be heterogeneity in the folding of the quadruplex, with ligands exhibiting preferential binding to a particular folded species out of several simultaneously present.
Molecular modelling
There is evidence from a considerable number of studies for dynamic inter-converting parallel, anti-parallel and hybrid folds of the human telomeric quadruplex.43,44 Cations, flanking sequences and the presence of ligands have all been found to influence the folding topology. Recent X-ray structural studies of co-crystals of human telomeric quadruplexes with porphyrin,45 naphthalene diimide46 and BRACO-1935 ligands have shown that these ligands bind to a parallel topology with its feature of exposed terminal G-tetrads. Additionally it has been reported that the parallel topology is stabilized in K+ solution relative to the hybrid forms under the influence of molecular crowding induced by polyethylene glycol which is intended to mimic biologically relevant intracellular conditions.47 Hence here we have used the parallel native crystal structure44 as a basis for the modelling studies, especially since parallel folds are also relevant to the c-kit and N-ras quadruplexes. Based on circular dichroism spectra, the N-ras DNA quadruplex appears to adopt a parallel fold with a single nucleotide loop,9 contrasting with Htelo in which all loops are three nucleotides in length. The 1H-NMR of the c-kit1 quadruplex was reported to be sharp and consistent with a well-defined single fold, which subsequent studies have shown to correspond to a parallel topology and structure, albeit with novel loop arrangements.6 The native c-kit2 sequence forms a mixture of predominantly parallel-stranded conformations with both single and five-nucleotide loops.7 These major structural differences between quadruplexes can be expected to provide a basis for ligand discrimination between them. The Htelo quadruplex is the sole member of this group for which extensive structural data are currently available, and so modelling studies have been restricted to this one.
We have assumed a binding mode for the molecular modelling in which the acridine stacks against the terminal G-tetrad of the parallel form of the human monomolecular quadruplex. It was reasoned that with the 9-aminobenzoyl substituent oriented to avoid steric clash, meta and para substitution would direct the peptide away from the tetrad to favour interactions with loops and grooves. This would explain both the lower affinity and selectivity of ortho-substituted compounds. In addition to experimental measurements of association constants, we probed this hypothesis using molecular modelling with the telomeric quadruplex. Models of compounds 10-15 were docked to the quadruplex, and the interaction energy (Table 5) was calculated as a Boltzmann weighting of the 25 lowest-energy structures.
Table 5.
Interaction energies of compounds 10-15 with the human parallel telomeric quadruplex, determined by docking calculations and Boltzmann weighting (at 300 K) of the 25 lowest-energy complexes
| Compound | Energy/kcal mol-1 |
|---|---|
| 10 | −110.34 |
| 11 | −127.32 |
| 12 | −133.85 |
| 13 | −117.34 |
| 14 | −141.19 |
| 15 | −142.38 |
The models were classified according to the binding mode as “stacked” where the acridine core makes π-π contacts with the G-tetrad, “groove” where there are interactions between the acridine and the DNA groove, or “displaced” when both types of interaction are absent. The resulting models predominantly exhibited the “stacked” binding mode with the acridine group stacked on the exposed tetrad, offset from the centre to locate the pyrrolidine substituents in the vicinity of adjacent grooves (Fig. 1). In these models the peptide typically either lies along a single groove, or basic amino acid side chains contact adjacent loops or grooves as shown in Fig. 1a and b respectively.
Fig. 1.
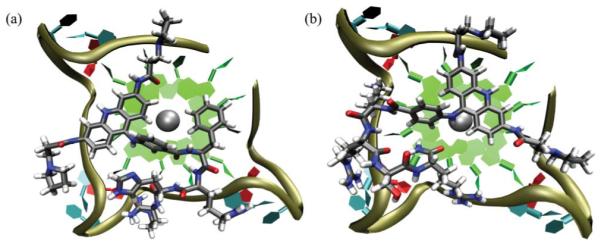
Representative models of peptide-acridine conjugates docked to the parallel human telomeric quadruplex (a) Compound 11, showing the acridine and Phe groups stacking against a tetrad, with pyrrole and peptide contacts in three adjacent grooves. (b) Compound 15, showing the peptide side chains making contacts with two adjacent grooves.
There is competition between the acridine, the 9-aminobenzoyl substituent and a Phe side chain (of FRHR) for stacking on the tetrad. In several models the 9-aminobenzoyl substituent, instead of the acridine, stacks on the tetrad, as these two groups are sterically inhibited from coplanarity. In some cases this leads to complete displacement of the acridine from the tetrad and into the quadruplex groove. Compound 11 showed the highest propensity to adopt the “displaced” mode, with a weighted population of 32%. The models of ortho-substituted 13 indicate an especial tendency towards acridine groove binding, exhibiting “stacked” and “groove” binding modes of similar energy that could coexist at room temperature with only 25% of the population in the “stacked” binding mode, and the remainder in the groove.
A plot (Fig. 2) of the calculated interaction energy against experimental log(Ka) for the Htelo quadruplex demonstrates the trend that the ortho substitution pattern gives rise to the lowest affinity compounds, which can be ascribed to the difficulty for the quadruplex to simultaneously contact both the acridine and peptide moieties. The greater affinity of the KRSR peptides for Htelo relative to their FRHR counterparts is also reproduced by the modelling. An explanation is that favourable hydrogen bonding and electrostatic interactions from the lysine residue contribute to binding to a greater extent than any hydrophobic and π contacts formed by the phenylalanine, which must compete for these interactions with the acridine and aminobenzoyl moieties. The trends observed by SPR are reflected in data from the thermal melting assay (Table 4), namely a higher concentration of ligand is required to achieve ΔTm 25 °C for ortho-substituted ligands than meta or para, and for FRHR-substituted ligands than their KRSR analogues.
Fig. 2.

Plot of Ka (± std. error) for binding to the Htelo quadruplex, shown on a logarithmic scale against calculated interaction energies, for compounds 10-15.
Surprisingly, the carboxyl-terminated compounds 16-21 bound to Htelo as tightly as the corresponding amides, and induced similar shifts in Tm. It had been anticipated that at pH 7.4 the carboxyl terminus would be deprotonated, leading to electrostatic repulsions with the oligonucleotide. This view was supported by docking calculations on 17 and 20 with Htelo which revealed that the weighted interaction energies of the deprotonated forms were 15 and 23 kcal mol-1 less favourable than the protonated forms respectively. The experimentally-observed binding affinities of acids 16-21 relative to the amides could arise from protonation of the carboxylates due to an environmental perturbation in the pKa value owing to the close proximity of the oligonucleotide, or intramolecular interactions involving the peptide terminus which preorganize the conformation of the peptide for quadruplex binding.
Conclusion
This study has demonstrated the principle that it is possible to devise molecules with a common G-tetrad interacting acridine core and variable peptide substituents that have the ability to discriminate between different quadruplex types. The substitution pattern and orientation of the peptide substituent with respect to the acridine appears to be important for affinity and selectivity of the ligands. Just a single peptide at the 9-position when combined with 3,6-pyrrolidine propionamide substituents is sufficient to result in ligands with high affinity and selectivity for particular quadruplexes. Molecular modelling suggested that the most successful compounds permit the acridine core to simultaneously stack on a tetrad and the peptide to make loop and groove contacts. Compounds with meta and para substitution typically exhibited selectivity towards a parallel quadruplex derived from the human N-ras gene in preference to the human telomeric quadruplex. In terms of both selectivity and affinity, the peptide substituent KRSR proved marginally superior to FRHR. These results demonstrate the feasibility of constructing high-affinity quadruplex selective ligands by appending a variable substituent to a generic quadruplex binding scaffold. It is hoped that the future availability of structural data on non-telomeric quadruplexes will enable rational design to reinforce these conclusions and permit further enhancements to selectivity to be achieved.
Experimental
DNA Sequences
Desalted biotinylated oligonucleotides were supplied by Invitrogen.
Htelo 5′ Biotin GTT AGG GTT AGG GTT AGG GTT AGG GTT AGG 3′
c-kit1 5′ Biotin AGA GGG AGG GCG CTG GGA GGA GGG GCT G 3′
c-kit2 5′ Biotin CCC GGG CGG GCG CGA GGG AGG GGA GG 3′
N-ras 5′ Biotin TGT GGG AGG GGC GGG TCT GGG TGC 3′
F21T 5′ FAM-GGG TTA GGG TTA GGG TTA GGG-TAMRA 3′
Synthetic Protocols
N-[9-Chloro-6-(3-pyrrolidin-1-yl-propionylamino)-acridin-3-yl]-3-pyrrolidin-1-yl-propionamide and 3-chloro-N-[6-(3-chloro-propionylamino)-9-oxo-4a,9,9a,10-tetrahydro-acridin-3-yl]-propionamide were prepared according to published procedures.33,34 Resins for peptide synthesis were supplied by Novabiochem. HPLC was performed using Phenomenex C18 Luna columns (250 × 4.6 mm for analytical, 250 × 10 mm for preparative) unless stated otherwise. HPLC solvent A = water, 0.1% TFA, solvent B = MeCN, 0.1% TFA. NMR spectra were recorded on Bruker Avance 500 (1H 500 MHz, 13C 126 MHz) or DRX-400 (1H 400 MHz, 13C 101 MHz) spectrometers. J values are given in Hz.
2-[3,6-Bis-(3-pyrrolidin-1-yl-propionylamino)-acridin-9-ylamino]-benzoic acid 1
A suspension of anthranilic acid (26 mg) and N-[9-chloro-6-(3-pyrrolidin-1-yl-propionylamino)-acridin-3-yl]-3-pyrrolidin-1-yl-propionamide in chloroform (5 mL) were refluxed for 24 h. Methanol (0.5 mL) was added and the red solution stirred for a further 2 d at r.t. The solvent was evaporated and the product purified by HPLC on a Phenomenex C18 Luna (250 × 10 mm) column, eluting with a gradient 0.5-2.5-13-13.5 min, 10-10-30-95% solvent B at 4.6 mL min-1. The product eluted at r.t. 13.7 min and was obtained as an orange solid after lyophilization (49 mg, 49%). Found: C, 49.3; H, 4.5; N, 8.6. C34H38N6O4·3TFA·2H2O requires C, 49.4; H, 4.7; N, 8.6%; δH(500 MHz, DMSO) 13.87 (1 H, br, OH), 11.14 (1 H, br, NH), 10.99 (2 H, s, NH), 9.87 (2 H, br, NH), 8.50 (2 H, d, J 2, Acr 4,5-H), 8.06 (1 H, dd, J 8 and 2, Ar 6-H); 7.98 (2 H, d, J 9, Acr 1,8-H), 7.63 (1 H, td, J 8 and 2 Hz, Ar 4-H), 7.50 (1 H, t, J 8, Ar 5-H), 7.37 (1 H, d, J 8, Ar 3-H), 7.31 (2 H, dd, J 9 and 2, Acr 2,7-H), 7.11 (1 H, t, J 51, NH), 3.07 (2 H, m, pyrrolidine NCH), 2.94 (4 H, t, J 7, CH2), 2.02 (4 H, m, pyrrolidine CH), 1.87 (4 H, m, pyrrolidine CH); δC(126 MHz, DMSO) 169.8, 167.5, 153.4, 144.6, 142.3, 141.4, 134.1, 132.2, 127.7, 127.1, 126.2, 125.9, 117.4, 110.4, 105.3, 54.0, 49.9, 33.0, 23.1; m/z (ES+) [M + H]+ requires 595.3033, found 595.2957.
3-[3,6-Bis-(3-pyrrolidin-1-yl-propionylamino)-acridin-9-ylamino]-benzoic acid 2
Prepared analogously to 1, substituting with 3-aminobenzoic acid instead of anthranilic acid. HPLC purification yielded 2 as an orange solid (24 mg, 24%). Found: C, 48.6; H, 4.3; N, 8.4. C34H38N6O4·3TFA·3H2O requires C, 48.5; H, 4.8; N, 8.5%; δH(400 MHz, D2O) 8.02 (2 H, d, J 2, Acr 4,5-H), 7.88 (1 H, d, J 8, Ar 6-H), 7.67 (3 H, m, Ar 2-H, Acr 1,8-H), 7.44 (1 H, t, J 8, Ar 5-H), 7.29 (1 H, d, J 8, Ar 4-H), 7.07 (2 H, dd, J 9 and 2, Acr 2,7-H), 3.67 (4 H, m, pyrrolidine NCH), 3.55 (4 H, t, J 7, CH2), 3.11 (4 H, m, pyrrolidine NCH), 2.97 (4 H, t, J 7, CH2), 2.13 (4 H, m, pyrrolidine CH), 1.99 (4 H, m, pyrrolidine CH); δC(126 MHz, D2O) 170.6, 170.2, 162.9 (q, J 35), 152.9, 143.2, 141.1, 140.4, 133.0, 130.2, 128.1, 128.0, 126.3, 124.4, 117.4, 116.2 (q, J 292), 110.1, 105.6, 54.4, 50.1, 32.3, 22.6; m/z (ES+) [M + H]+ requires 595.3033, found 595.3051.
4-[3,6-Bis-(3-pyrrolidin-1-yl-propionylamino)-acridin-9-ylamino]-benzoic acid 3
Prepared analogously to 1, substituting with 4-aminobenzoic acid instead of anthranilic acid. The product was obtained as an orange solid after HPLC purification (18.5 mg, 18%). Found: C, 48.25; H, 4.4; N, 8.4. C34H38N6O4·3TFA·3H2O requires C, 48.5; H, 4.8; N, 8.5%; δH(400 MHz, D2O, 45 °C) 8.30 (2 H, d, J 2, Acr 4,5-H), 8.09 (2 H, d, J 9, Ar H), 7.95 (2 H, d, J 9, Acr 1,8-H), 7.33 (4 H, m, Ar H and Acr 2,7-H), 3.89 (4 H, br m, pyrrolidine NCH), 3.76 (4 H, t, J 7, CH2), 3.33 (4 H, br m, pyrrolidine NCH), 3.19 (4 H, t, J 7, CH2), 2.34 (4 H, br, pyrrolidine CH), 2.20 (4 H, br, pyrrolidine CH); δC(126 MHz, D2O) 170.5, 169.6, 162.9 (q, J 35), 152.1, 144.8, 143.4, 141.1, 131.2, 127.7, 126.3, 117.7, 116.2 (q, J 292), 110.8, 105.4, 54.4, 50.1, 32.3, 22.6; m/z (ES+) [M + H]+ requires 595.3033, found 595.2984.
[(2 - {6 - [3 - (Ethoxycarbonylmethyl - methyl - amino) - propionyl - amino]-9-oxo-9,10-dihydro-acridin-3-ylcarbamoyl}-ethyl)-methyl-amino]-acetic acid ethyl ester 4
3-Chloro-N-[6-(3-chloro-propionylamino)-9-oxo-4a,9,9a,10-tetrahydro-acridin-3-yl]-propionamide (200 mg), sodium iodide (145 mg), sarcosine ethylester hydrochloride (1.14 g) were mixed in DMF (14 mL). The suspension was heated with stirring to 80 °C while a solution of sodium ethoxide (566 mg) in ethanol (20 mL) was added over 20 h using a syringe pump. Heating was continued for a further 10 h, after which the suspension was cooled to r.t., filtered and the solvent evaporated. The residues were triturated with Et2O, and the washings discarded. The remaining solids were purified by HPLC to afford 4 as a pale yellow solid (146 mg, 39%). HPLC conditions: Phenomenex C18 Luna (250 × 10 mm) column, gradient 0-2.5-11 min, 13-13-32% solvent B at 4.6 mL min-1, retention time 11.3 min. Found: C, 48.65; H, 4.95; N, 8.6. C29H37N5O7·2TFA·H2O requires C, 48.7; H, 5.1; N, 8.6%; δH(400 MHz, D2O) 7.74 (2 H, d, J 9, Acr 1,8-H), 7.10 (2 H, d, J 2, Acr 4,5-H), 6.80 (2 H, dd, J 9 and 2, Acr 2,7-H), 4.27 (4 H, q, J 7, OCH2), 4.17 (4 H, s, CH2CO2), 3.56 (4 H, t, J 7, NCH2), 3.00 (6 H, s, NCH3), 2.83 (4 H, t, J 7, CH2), 1.23 (6 H, t, J 7, CH2CH3); δC(101 MHz, D2O) 175.7, 168.9, 166.3, 162.6 (q, J 36), 141.5, 140.6, 126.0, 116.3 (q, J 293), 115.4, 113.6, 104.7, 63.7, 56.1, 52.4, 41.5, 30.3, 13.1; m/z (ES+) [M + H]+ requires 568.2771, found 568.2785.
[(2-{9-(4-Dimethylamino-phenylamino)-6-[3-(ethoxycarbonyl-methyl-methyl-amino)-propionylamino]-acridin-3-ylcarbamoyl}-ethyl)-methyl-amino]-acetic acid ethyl ester 5
Phosphoryl chloride (15 mL) was added to 4 (140 mg) and the suspension heated to 100 °C for 1 h under N2. After cooling to room temperature the orange suspension was poured into Et2O (200 mL), cooled to −20 °C and the yellow solids collected by filtration and washed with Et2O. To the solids were added N-dimethylphenyldiamine (1.20 g) followed by chloroform (20 mL) and the dark solution stirred at r.t. for 24 h. The solvent was evaporated to afford a brown oil to which Et2O (100 mL) was added. After standing at −20 °C for 1 h, the precipitate was collected by filtration, washed with Et2O and dried in vacuo. The residues were dissolved in water (5 mL) acidified with TFA (330 μL) and passed in 500 μL aliquots through an Isolute C18EC cartridge (1 g) eluting with water (0.1% TFA, 10 mL), 10% MeCN (0.1% TFA, 20 mL), 20% MeCN (0.1% TFA,10 mL). The 10% MeCN and 20% MeCN eluents were combined and evaporated. The residues were purified by HPLC on a Phenomenex C18 Jupiter column (250 × 21.2 mm) with a gradient 0-2.5-32.5 min, 5-5-100% solvent B at 10 mL min-1, retention time 16.5 min. Lyophilization afforded the product as a dark red solid (153 mg, 84%). Found: C, 48.2; H, 4.65; N, 8.9. C37H47N7O6·3TFA·2H2O requires C, 48.5; H, 5.1; N, 9.2%; δH(400 MHz, D2O) 7.91 (2 H, d, J 2, Acr 4,5-H), 7.64 (2 H, d, J 9, Acr 1,8-H), 7.54 (2 H, d, J 9, Ar H), 7.26 (2 H, d, J 9, Ar H), 7.05 (2 H, dd, J 9 and 2, Acr 2,7-H), 4.21 (4 H, q, J 7, OCH2), 4.15 (4 H, s, CH2CO2), 3.62 (4 H, br m, CH2), 3.21 (6 H, s, NCH3), 3.00 (10 H, m, CH2 and NCH3), 1.17 (6 H, t, J 7, CH2CH3); δC(101 MHz, D2O) 170.3, 166.4, 162.5 (q, J 36), 152.7, 143.5, 142.4, 141.1, 139.9, 126.3, 125.1, 122.2, 117.7, 116.2 (q, J 293), 110.6, 105.5, 63.7, 56.2, 52.4, 46.4, 41.7, 30.8, 13.1; m/z (ES+) [M + H]+ requires 686.3661, found 686.3677.
({2-[6-[3-(Carboxymethyl-methyl-amino)-propionylamino]-9-(4 - dimethylamino-phenylamino)-acridin-3-ylcarbamoyl]-ethyl}-methyl-amino)-acetic acid 6
To a solution of 5 (153 mg) in a mixture of water (3.8 mL) and THF (3.8 mL) was added LiOH (34 mg). After stirring 1 h at r.t., TFA (350 μL) was added and the solution concentrated to 1.5 mL. The product was purified by HPLC on a Phenomenex C18 Luna column (250 × 10 mm) with gradient 0-2.5-11 min, 5-5-30% solvent B, 4.6 mL min-1, retention time 10.6 min. Lyophilization afforded the product as an orange solid (62 mg, 42%). Found: C, 45.4; H, 4.2; N, 9.25. C33H39N7O6·3TFA·3H2O requires C, 45.7; H, 4.7; N, 9.6%; δH(400 MHz, D2O) 7.98 (2 H, d, J 2, Acr 4,5-H), 7.74 (2 H, d, J 9, Acr 1,8-H), 7.59 (2 H, d, J 9, Ar H), 7.33 (2 H, d, J 9, Ar H), 7.10 (2 H, dd, J 9 and 2, Acr 2,7-H), 4.01 (4 H, s, CH2CO2), 3.62 (4 H, br, CH2), 3.26 (6 H, s, NCH3), 3.04 (4 H, t, J 7, CH2), 2.90 (6 H, s, NCH3); δC(101 MHz, D2O) 170.2, 168.0, 162.5 (m), 143.4, 142.2, 140.9, 139.9, 126.1, 125.1, 122.1, 117.6, 116.1 (q, J 293), 110.3, 105.3, 56.3, 52.3, 46.3, 41.5, 30.7; m/z (ES+) [M + H]+ requires 630.3040, found 630.3028.
Peptide synthesis
Peptides were prepared on Rink amide (amide terminated) or preloaded Wang (acid terminated) resins using Fmoc protected amino acids and PyBOP, HOBt and DIPEA in N-methylpyrrolidone for couplings and 50% piperidine in DMF for deprotections. The final acridine residue was coupled using identical reagents. Deprotection and cleavage from the resin was performed using 95% TFA, 2.5% H2O, 2.5% triisopropylsilane. After concentration of the TFA by nitrogen blow-down, the products were recovered by Et2O precipitation. Purification by HPLC afforded compounds 7-21 (Tables 1, 2) which were dissolved in water to form stock solutions. Peptides were characterized by LCMS and stock solutions standardized by amino acid analysis (Department of Biochemistry, University of Cambridge). Retention times and amino acid analyses are given in the supporting information.
Table 1.
Sequences of 3,6-bis peptide acridines
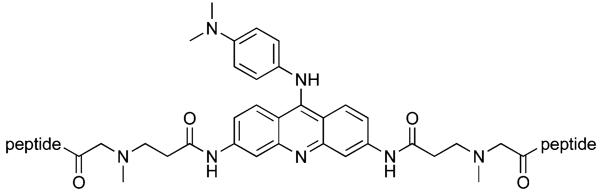
| |
|---|---|
|
| |
| Compound | Peptide |
| 7 | FRHR |
| 8 | KRSR |
| 9 | RKKV |
Table 2.
Substitution patterns and sequences of 9-peptide acridines
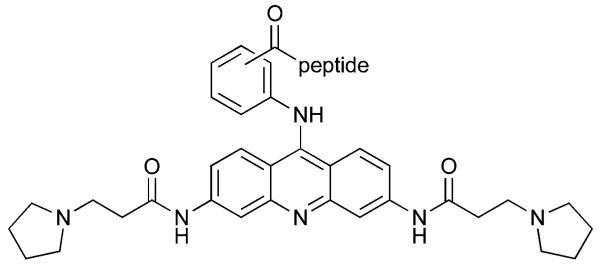
| |||
|---|---|---|---|
|
| |||
| Compound | Substitution | Peptide | C-terminus |
| 10 | o | FRHR | NH2 |
| 11 | m | FRHR | NH2 |
| 12 | p | FRHR | NH2 |
| 13 | o | KRSR | NH2 |
| 14 | m | KRSR | NH2 |
| 15 | p | KRSR | NH2 |
| 16 | o | FRHR | OH |
| 17 | m | FRHR | OH |
| 18 | p | FRHR | OH |
| 19 | o | KRSR | OH |
| 20 | m | KRSR | OH |
| 21 | p | KRSR | OH |
Surface plasmon resonance
Surface plasmon resonance was performed on a Biacore 2000 instrument, using degassed phosphate running buffer (50 mM potassium phosphate, 100 mM KCl, pH 7.4). Sensor chips (Type SA, Biacore) were loaded with approximately 500 RU of biotinylated oligonucleotides. Serial dilutions of compound were injected at a flow rate of 20 μL min-1 and the equilibrium response determined relative to the baseline.
3,6 Bis-peptide-substituted acridines (7-9) were varied over a nominal range of concentration of 10-0.08 μM, whereas acridines with a peptide substitution on the 9-position (10-21) were varied over the concentration range 2.5-640 nM. Compounds 10-21 were handled in glass vials as they were found to adsorb to plastic tubes. Injections were each performed in duplicate on two separate occasions. Between injections the sensor surface was refreshed with injections of 1 M KCl and buffer. After subtraction of the background response from the blank flow cell, the responses were fitted to a BET isotherm (Eq. 1) using the BIAevaluation software. The BET equation permits binding of a monolayer with association constant Ka followed by multiple bindings with association constant Km. The monolayer dissociation constant Kd = Ka-1 is analogous to the dissociation constant of the simpler Langmuir model which is often used for fitting SPR data. The multilayer constant Km permits modelling of non-specific adsorption which was observed at higher ligand concentrations. The stoichiometry of binding was estimated from the response corresponding to a monolayer coverage relative to the DNA loading of the chip.
| (1) |
Req is the equilibrium response, Rmon is the response for a single monolayer, Ka is the association constant for the first monolayer, Km is the association constant for multilayers, and c is the concentration of ligand. Data have been expressed in terms of the dissociation constant Kd = Ka-1.
FRET melts
The FRET melting assay was conducted using the dual labelled (fluorescein, tetramethylrhodamine) oligonucleotide F21T according to our modification48 of the procedure of Mergny et al.42
Molecular modelling
The crystal structure44 of the G-quadruplex formed from the 22-mer human telomeric DNA sequence d(AGGG[TTAGGG]3) (PDB code 1KF1) was used as the binding host to the various acridine-peptide conjugates. Ligands were built using the Insight II package (Accelrys Inc.) on an SGI workstation. Partial charges for the central scaffold were derived by fitting the HF/6-31G* electrostatic potential obtained with the GAMESS ab initio software49 to the atomic centres with the RESP program.50 Atomic parameters for the peptide side chains were taken directly from the Amber force field,51 as implemented within Insight II. The protonation state of the amino acid side chains was determined based on an assumed physiological pH of 7.0. The N10 position of the acridine was also protonated with a +1 charge based on its role as a hydrogen bond donor in a quadruplex-ligand crystal structure (1L1H).52
Docking was performed with the Affinity Docking module of Insight II using a previously-described protocol.36 In this way, a multi-phase docking protocol was used. First the ligand was manually displaced along the binding host while interactively calculating their interaction energy. Once a favourable initial relative conformation was found a binding pocket which included hydrogen atoms 5 Å from the ligand was defined, and then the ligand was randomly orientated with respect to the binding host 200 times. Van der Waals radii were set to 10% of the full value, charges were not considered and non-bonded cut-offs were set to 8 Å. The system was minimized for 300 steps using the conjugate gradient method. The maximum allowable change for succeeding structures was set to 10000 kcal mol-1 and the energy range was set to 40 kcal mol-1. The 75 lowest-energy structures were used for the second phase of the modelling. The second phase of the docking protocol consisted of a simulated annealing procedure in which the van der Waals radii were adjusted to their full values, charges were included with a distance-dependent dielectric of 4*rij and the non-bonded cut-off was set to 18 Å. Each of the 75 lowest-energy structures was again minimized for 300 steps of conjugate-gradient minimization, and then molecular dynamics calculations were performed, starting at a temperature of 500 K and cooling the system to 300 K over 10 ps. The resulting structures were minimized for 1000 steps of conjugate gradients and the 25 structures with lowest total energy were used for further evaluation. These 25 filtered structures were further refined in order to perform an optimal conformational sampling of the peptide side-chains while maintaining the ligand orientation. Hence, the structures were subjected to another run of simulated annealing in which the binding pocket was extended to contain all atoms 5 Å from the ligand while the bases of the G-quartet were tethered to their original position. The system was cooled down over 10 ps from 500 K to 300 K, and the resulting structures were subjected to a 1000 steps of conjugate gradient minimization.
Acknowledgements
We thank Cancer Research UK for programme funding (to SB and to SN).
Footnotes
Electronic supplementary information (ESI) available: HPLC conditions, amino acid analysis and mass spectral data for compounds 7-21. UV thermal melt and CD spectrum of N-ras DNA quadruplex.
References
- 1.Keniry MA. Biopolymers. 2001;56:123–146. doi: 10.1002/1097-0282(2000/2001)56:3<123::AID-BIP10010>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 2.Huppert JL, Balasubramanian S. Nucleic Acids Res. 2005;33:2908–2916. doi: 10.1093/nar/gki609. [DOI] [PMC free article] [PubMed] [Google Scholar]; Todd AK, Johnston M, Neidle S. Nucleic Acids Res. 2005;33:2901–2907. doi: 10.1093/nar/gki553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eddy J, Maizels N. Nucleic Acids Res. 2006;34:3887–3896. doi: 10.1093/nar/gkl529. [DOI] [PMC free article] [PubMed] [Google Scholar]; Huppert JL, Balasubramanian S. Nucleic Acids Res. 2007;35:406–413. doi: 10.1093/nar/gkl1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eddy J, Maizels N. Nucleic Acids Res. 2008;36:1321–1333. doi: 10.1093/nar/gkm1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simonsson T, Pecinka P, Kubista M. Nucleic Acids Res. 1998;26:1167–1172. doi: 10.1093/nar/26.5.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rankin S, Reszka AP, Huppert J, Zloh M, Parkinson GN, Todd AK, Ladame S, Balasubramanian S, Neidle S. J. Am. Chem. Soc. 2005;127:10584–10589. doi: 10.1021/ja050823u. [DOI] [PMC free article] [PubMed] [Google Scholar]; Phan AT, Kuryavyi V, Burge S, Neidle S, Patel DJ. J. Am. Chem. Soc. 2007;129:4386–4392. doi: 10.1021/ja068739h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fernando H, Reszka AP, Huppert J, Ladame S, Rankin S, Venkitaraman AR, Neidle S, Balasubramanian S. Biochemistry. 2006;45:7854–7860. doi: 10.1021/bi0601510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumari S, Bugaut A, Huppert JL, Balasubramanian S. Nature Chem. Biol. 2007;3:218–221. doi: 10.1038/nchembio864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu S, Huppert JL, Balasubramanian S. unpublished results. See Supplementary Information for details.
- 10.Cogoi S, Quadrifoglio F, Xodo LE. Biochemistry. 2004;43:2512–2523. doi: 10.1021/bi035754f. [DOI] [PubMed] [Google Scholar]
- 11.Cogoi S, Xodo LE. Nucleic Acids Res. 2006;34:2536–2549. doi: 10.1093/nar/gkl286. [DOI] [PMC free article] [PubMed] [Google Scholar]; Cogoi S, Paramasivam M, Spolaore B, Xodo LE. Nucleic Acids Res. 2008;36:3765–3780. doi: 10.1093/nar/gkn120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Y, Sugiyama H. Nucleic Acids Res. 2006;34:949–954. doi: 10.1093/nar/gkj485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo K, Pourpak A, Beetz-Rogers K, Gokhale V, Sun D, Hurley LH. J. Am. Chem. Soc. 2007;129:10220–10228. doi: 10.1021/ja072185g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun D, Guo K, Rusche JJ, Hurley LH. Nucleic Acids Res. 2005;33:6070–6080. doi: 10.1093/nar/gki917. [DOI] [PMC free article] [PubMed] [Google Scholar]; Sun D, Liu W-J, Guo K, Rusche JJ, Ebbinghaus S, Gokhale V, Hurley LH. Mol. Cancer Ther. 2008;7:880–889. doi: 10.1158/1535-7163.MCT-07-2119. [DOI] [PMC free article] [PubMed] [Google Scholar]; Guo K, Gokhale V, Hurley LH, Sun D. Nucleic Acids Res. 2008;36:4598–4608. doi: 10.1093/nar/gkn380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Armond R, Wood S, Sun D, Hurley LH, Ebbinghaus SW. Biochemistry. 2005;44:16341–16350. doi: 10.1021/bi051618u. [DOI] [PubMed] [Google Scholar]
- 16.Dai J, Chen D, Jones RA, Hurley LH, Yang D. Nucleic Acids Res. 2006;34:5133–5144. doi: 10.1093/nar/gkl610. [DOI] [PMC free article] [PubMed] [Google Scholar]; Dexheimer TS, Sun D, Hurley LH. J. Am. Chem. Soc. 2006;128:5404–5415. doi: 10.1021/ja0563861. [DOI] [PMC free article] [PubMed] [Google Scholar]; Dai J, Dexheimer TS, Chen D, Carver M, Ambrus A, Jones RA, Yang D. J. Am. Chem. Soc. 2006;128:1096–1098. doi: 10.1021/ja055636a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qin Y, Rezler EM, Gokhale V, Sun D, Hurley LH. Nucleic Acids Res. 2007;35:7698–7713. doi: 10.1093/nar/gkm538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palumbo SL, Memmott RM, Uribe DJ, Krotova-Khan Y, Hurley LH, Ebbinghaus SW. Nucleic Acids Res. 2008;36:1755–1769. doi: 10.1093/nar/gkm1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siddiqui-Jain A, Grand CL, Bearss DJ, Hurley LH. Proc. Natl. Acad. Sci. USA. 2002;99:11593–11598. doi: 10.1073/pnas.182256799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mikami-Terao Y, Akiyama M, Yuza Y, Yanagisawa T, Yamada O, Yamada H. Cancer Lett. 2008;261:226–234. doi: 10.1016/j.canlet.2007.11.017. [DOI] [PubMed] [Google Scholar]
- 21.Bejugam M, Sewitz S, Shirude PS, Rodriguez R, Shahid R, Balasubramanian S. J. Am. Chem. Soc. 2007;129:12926–12927. doi: 10.1021/ja075881p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ou T-M, Lu Y-J, Zhang C, Huang Z-S, Wang X-D, Tan J-H, Chen Y, Ma D-L, Wong K-Y, Tang JC-O, Chan AS-C, Gu L-Q. J. Med. Chem. 2007;50:1465–1474. doi: 10.1021/jm0610088. [DOI] [PubMed] [Google Scholar]
- 23.Du Z, Zhao Y, Li N. Genome Res. 2008;18:233–241. doi: 10.1101/gr.6905408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kelland L. Clin. Cancer Res. 2007;13:4960–4963. doi: 10.1158/1078-0432.CCR-07-0422. [DOI] [PubMed] [Google Scholar]; De Cian A, Lacroix L, Douarre C, Temime-Smaali N, Trentesaux C, Riou J-F, Mergny J-L. Biochimie. 2008;90:131–155. doi: 10.1016/j.biochi.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 25.Lemarteleur T, Gomez D, Paterski R, Mandine E, Maillet P, Riou J-F. Biochem. Biophys. Res. Commun. 2004;323:802–808. doi: 10.1016/j.bbrc.2004.08.150. [DOI] [PubMed] [Google Scholar]
- 26.Waller ZAE, Shirude PS, Rodriguez R, Balasubramanian S. Chem. Commun. 2008:1467–1469. doi: 10.1039/b718854d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tera M, Ishizuka H, Takagi M, Suganuma M, Shin-ya K, Nagasawa K. Angew. Chem. Int. Ed. 2008;47:5557–5560. doi: 10.1002/anie.200801235. [DOI] [PubMed] [Google Scholar]
- 28.Han H, Langley DR, Rangan A, Hurley LH. J. Am. Chem. Soc. 2001;123:8902–8913. doi: 10.1021/ja002179j. [DOI] [PubMed] [Google Scholar]; Rezler EM, Seenisamy J, Bashyam S, Kim M-Y, White E, Wilson WD, Hurley LH. J. Am. Chem. Soc. 2005;127:9439–9447. doi: 10.1021/ja0505088. [DOI] [PubMed] [Google Scholar]; Seenisimasy J, Bashyam S, Gokhale V, Vankayalapati H, Sun D, Siddiqui-Jain A, Streiner N, Shin-ya K, White E, Wilson WD, Hurley LH. J. Am. Chem. Soc. 2005;127:2944–2959. doi: 10.1021/ja0444482. [DOI] [PubMed] [Google Scholar]; Gonçalves DPN, Rodriguez R, Balasubramanian S, Sanders JKM. Chem. Commun. 2006:4685–4687. doi: 10.1039/b611731g. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zhang W-J, Ou T-M, Lu Y-J, Huang Y-Y, Wu W-B, Huang Z-S, Zhou J-L, Wong K-Y, Gu L-Q. Bioorg. Med. Chem. 2007;15:5493–5501. doi: 10.1016/j.bmc.2007.05.050. [DOI] [PubMed] [Google Scholar]; Fu B, Huang J, Ren L, Weng X, Zhou Y, Du Y, Wu X, Zhou X, Yang G. Chem. Commun. 2007:3264–3266. doi: 10.1039/b704599a. [DOI] [PubMed] [Google Scholar]; Rodriguez R, Pantoş GD, Gonçalves DPN, Sanders JKM, Balasubramanian S. Angew. Chem. Int. Ed. 2007;46:5405–5407. doi: 10.1002/anie.200605075. [DOI] [PMC free article] [PubMed] [Google Scholar]; Galezowska E, Masternak A, Rubis B, Czyrski A, Rybczyńska M, Hermann TW, Juskowiak B. Int. J. Biol. Macromol. 2007;41:558–563. doi: 10.1016/j.ijbiomac.2007.07.008. [DOI] [PubMed] [Google Scholar]; Zhou J, Yuan G. Chem. Eur. J. 2007;13:5018–5023. doi: 10.1002/chem.200601605. [DOI] [PubMed] [Google Scholar]; Ren L, Zhang A, Huang J, Wang P, Weng X, Zhang L, Liang F, Tan Z, Zhou X. ChemBioChem. 2007;8:775–780. doi: 10.1002/cbic.200600554. [DOI] [PubMed] [Google Scholar]; Monchaud D, Yang P, Lacrois L, Teulade-Fichou M-P, Mergny J-L. Angew. Chem. Int. Ed. 2008;47:4858–4861. doi: 10.1002/anie.200800468. [DOI] [PubMed] [Google Scholar]
- 29.Koeppel F, Riou J-F, Laoui A, Maillet P, Arimondo PB, Labit D, Petitgenet O, Hélène C, Mergny J-L. Nucleic Acids Res. 2001;29:1087–1096. doi: 10.1093/nar/29.5.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]; Rosu F, De Pauw E, Guittat L, Alberti P, Lacroix L, Mailliet P, Riou J-F, Mergny J-L. Biochemistry. 2003;42:10361–10371. doi: 10.1021/bi034531m. [DOI] [PubMed] [Google Scholar]
- 30.Chang C-C, Wu J-Y, Chien C-W, Wu W-S, Liu H, Kang C-C, Yu L-J, Chang T-C. Anal. Chem. 2003;75:6177–6183. doi: 10.1021/ac034789i. [DOI] [PubMed] [Google Scholar]; Dias N, Jacquemard U, Baldeyrou B, Tardy C, Lansiaux A, Colson P, Tanious F, Wilson WD, Routier S, Mérour J-Y, Bailly C. Biochemistry. 2004;43:15169–15178. doi: 10.1021/bi048474o. [DOI] [PubMed] [Google Scholar]
- 31.Martins C, Gunaratnam M, Stuart J, Makwana V, Greciano O, Reszka AP, Kelland LR, Neidle S. Bioorg. Med. Chem. Lett. 2007;17:2293–2298. doi: 10.1016/j.bmcl.2007.01.056. [DOI] [PubMed] [Google Scholar]
- 32.Harrison RJ, Cuesta J, Chessari G, Read MA, Basra SK, Reszka AP, Morrell J, Gowan SM, Incles CM, Tanious FA, Wilson WD, Kelland LR, Neidle S. J. Med. Chem. 2003;46:4463–4476. doi: 10.1021/jm0308693. [DOI] [PubMed] [Google Scholar]; Moore MJB, Schultes CM, Cuesta J, Cuenca F, Gunaratnam M, Tanious FA, Wilson WD, Neidle S. J. Med. Chem. 2006;49:582–599. doi: 10.1021/jm050555a. [DOI] [PubMed] [Google Scholar]
- 33.Read MA, Wood AA, Harrison JR, Gowan SM, Kelland LR, Dosanjh HS, Neidle S. J. Med. Chem. 1999;42:4538–4546. doi: 10.1021/jm990287e. [DOI] [PubMed] [Google Scholar]; Harrison RJ, Gowan SM, Kelland LR, Neidle S. Bioorg. Med. Chem. Lett. 1999;9:2463–2468. doi: 10.1016/s0960-894x(99)00394-7. [DOI] [PubMed] [Google Scholar]
- 34.Read M, Harrison RJ, Romagnoli B, Tanious FA, Gowan SH, Reszka AP, Wilson WD, Kelland LR, Neidle S. Proc. Natl. Acad. Sci. USA. 2001;98:4844–4849. doi: 10.1073/pnas.081560598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Campbell NH, Parkinson GN, Reszka AP, Neidle S. J. Am. Chem. Soc. 2008;130:6722–6724. doi: 10.1021/ja8016973. [DOI] [PubMed] [Google Scholar]
- 36.White EW, Tanious F, Ismail MH, Reszka AP, Neidle S, Boykin DW, Wilson WD. Biophysical Chem. 2007;126:140–153. doi: 10.1016/j.bpc.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 37.Ladame S, Schouten JA, Stuart J, Roldan J, Neidle S, Balasubramanian S. Org. Biol. Chem. 2004;2:2925–2931. doi: 10.1039/B409698C. [DOI] [PubMed] [Google Scholar]
- 38.Schultes CM, Guyen B, Cuesta J, Neidle S. Bioorg. Med. Chem. Lett. 2004;14:4347–4351. doi: 10.1016/j.bmcl.2004.05.090. [DOI] [PubMed] [Google Scholar]
- 39.Carlson CB, Beal PA. Bioorg. Med. Chem. Lett. 2000;10:1979–1982. doi: 10.1016/s0960-894x(00)00388-7. [DOI] [PubMed] [Google Scholar]
- 40.Ladame S, Harrison RJ, Neidle S, Balasubramanian S. Org. Lett. 2002;4:2509–2512. doi: 10.1021/ol026130p. [DOI] [PubMed] [Google Scholar]
- 41.Schouten JA, Ladame S, Mason SJ, Cooper MA, Balasubramanian S. J. Am. Chem. Soc. 2003;125:5594–5595. doi: 10.1021/ja029356w. [DOI] [PubMed] [Google Scholar]
- 42.Mergny J-L, Lacroix L, Teulade-Fichou M-P, Hounsou C, Guittat L, Hoarau M, Arimondo PB, Vigneron J-P, Lehn J-M, Riou J-F, Garestier T, Hélène C. Proc. Natl. Acad. Sci. USA. 2001;98:3062–3067. doi: 10.1073/pnas.051620698. [DOI] [PMC free article] [PubMed] [Google Scholar]; Mergny J-L, Maurizot J-C. ChemBioChem. 2001;2:124–132. doi: 10.1002/1439-7633(20010202)2:2<124::AID-CBIC124>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]; De Cian A, Guittat L, Kaiser M, Saccà B, Amrane S, Bourdoncle A, Alberti P, Teulade-Fichou M-P, Lacroix L, Mergny J-L. Methods. 2007;42:183–195. doi: 10.1016/j.ymeth.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 43.Wang Y, Patel DJ. Structure. 1993;1:263–282. doi: 10.1016/0969-2126(93)90015-9. [DOI] [PubMed] [Google Scholar]; Ying L, Green JJ, Li H, Klenerman D, Balasubramanian S. Proc. Natl. Acad. Sci. USA. 2003;100:14629–14634. doi: 10.1073/pnas.2433350100. [DOI] [PMC free article] [PubMed] [Google Scholar]; Li J, Correia JJ, Wang L, Trent JO, Chaires JB. Nucleic Acids. Res. 2005;33:4649–4659. doi: 10.1093/nar/gki782. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ambrus A, Chen D, Dai J, Bialis T, Jones RA, Yang D. Nucleic Acids Res. 2006;34:2723–2735. doi: 10.1093/nar/gkl348. [DOI] [PMC free article] [PubMed] [Google Scholar]; Xu Y, Noguchi Y, Sugiyama H. Bioorg. Med. Chem. 2006;14:5584–5591. doi: 10.1016/j.bmc.2006.04.033. [DOI] [PubMed] [Google Scholar]; Luu KN, Phan AT, Kuryavyi V, Lacroix L, Patel DJ. J. Am. Chem. Soc. 2006;128:9963–9970. doi: 10.1021/ja062791w. [DOI] [PMC free article] [PubMed] [Google Scholar]; Phan AT, Luu KN, Patel DJ. Nucleic Acids Res. 2006;34:5715–5719. doi: 10.1093/nar/gkl726. [DOI] [PMC free article] [PubMed] [Google Scholar]; Phan AT, Kuryavyi V, Luu KN, Patel DJ. Nucleic Acids Res. 2007;35:6517–6525. doi: 10.1093/nar/gkm706. [DOI] [PMC free article] [PubMed] [Google Scholar]; Dai J, Punchihewa C, Ambrus A, Chen D, Jones RA, Yang D. Nucleic Acids Res. 2007;35:2440–2450. doi: 10.1093/nar/gkm009. [DOI] [PMC free article] [PubMed] [Google Scholar]; Chang C-C, Chien C-W, Lin Y-H, Kang C-C, Chang T-C. Nucleic Acids Res. 2007;35:2846–2860. doi: 10.1093/nar/gkm155. [DOI] [PMC free article] [PubMed] [Google Scholar]; Dai J, Carver M, Punchihewa C, Jones RA, Yang D. Nucleic Acids Res. 2007;35:4927–4940. doi: 10.1093/nar/gkm522. [DOI] [PMC free article] [PubMed] [Google Scholar]; Antonacci C, Chaires JB, Sheardy RD. Biochemistry. 2007;46:4654–4660. doi: 10.1021/bi602511p. [DOI] [PubMed] [Google Scholar]; Gray RD, Chaires JB. Nucleic Acids Res. 2008;36:4191–4203. doi: 10.1093/nar/gkn379. [DOI] [PMC free article] [PubMed] [Google Scholar]; Okamoto K, Sannohe Y, Mashimo T, Sugiyama H, Terazima M. Bioorg. Med. Chem. 2008;16:6873–6879. doi: 10.1016/j.bmc.2008.05.053. [DOI] [PubMed] [Google Scholar]; Gaynutdinov TI, Neumann RD, Panyutin IG. Nucleic Acids Res. 2008;36:4079–4087. doi: 10.1093/nar/gkn351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parkinson GN, Lee MPH, Neidle S. Nature. 2002;417:876–880. doi: 10.1038/nature755. [DOI] [PubMed] [Google Scholar]
- 45.Parkinson GN, Ghosh R, Neidle S. Biochemistry. 2007;46:2390–2397. doi: 10.1021/bi062244n. [DOI] [PubMed] [Google Scholar]
- 46.Parkinson GN, Cuenca F, Neidle S. J. Mol. Biol. 2008;381:1145–1156. doi: 10.1016/j.jmb.2008.06.022. [DOI] [PubMed] [Google Scholar]
- 47.Xue Y, Kan Z, Wang Q, Yao Y, Liu J, Hao Y, Tan Z. J. Am. Chem. Soc. 2007;129:11185–11191. doi: 10.1021/ja0730462. [DOI] [PubMed] [Google Scholar]
- 48.Guyen B, Schultes CM, Hazel P, Mann J, Neidle S. Org. Biomol. Chem. 2004;2:981–988. doi: 10.1039/b316055f. [DOI] [PubMed] [Google Scholar]
- 49.Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su S, Windus TL, Dupuis M, Montgomery JA. J. Comput. Chem. 1993;14:1347–1363. [Google Scholar]
- 50.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Jr., Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- 51.Weiner SJ, Kollman PA, Nguyen DT, Case DA. J. Comput. Chem. 1986;7:230–252. doi: 10.1002/jcc.540070216. [DOI] [PubMed] [Google Scholar]
- 52.Haider SM, Parkinson GN, Neidle S. J. Mol. Biol. 2003;326:117–125. doi: 10.1016/s0022-2836(02)01354-2. [DOI] [PubMed] [Google Scholar]