

Abstract
Mammalian cardiomyocytes withdraw from the cell cycle soon after birth. This process is called terminal differentiation. The c-kit, a receptor tyrosine kinase, is expressed on cardiomyocytes immediately after birth but for only a few days. In mice with genetic c-kit dysfunction, adult cardiomyocytes are phenotypically indistinguishable from those of wild type mice, except that they are capable of proliferation in vivo after acute pressure overload. This review explores the idea that postnatal cardiomyocyte differentiation and cell cycle withdrawal are distinct processes and that terminal differentiation may not simply be due to altered expression of genes that regulate the cell cycle but could involve c-kit induced epigenetic change.
Keywords: Cardiomyocytes, c-Kit, Terminal differentiation
Introduction
Mammalian cardiomyocytes undergo developmental changes required for the differentiated adult phenotype. These include downregulation of several fetal genes and upregulation of genes responsible for the adult phenotype. Some of these changes regulate structural differentiation of cardiomyocytes for better adaptation of the heart to the increased systolic load experienced after birth. These involve the altered expression of genes, causing a switch in the isoforms or types of several proteins involved in cardiac structure and contractility such as the replacement of skeletal actin and β-myosin heavy chain in the fetal heart with cardiac actin and α-myosin heavy chain (note, in humans compared with rodents, the opposite switch occurs after birth, namely, β-myosin heavy chain replaces α-myosin heavy chain), respectively, in the adult heart [7, 14, 27]. As a result of these changes, sarcomeric organization becomes more developed and compact in the early neonatal stage.
These important aspects of cardiomyocyte differentiation have been the subject of several reviews and thus will not be discussed in this report [2, 5, 23]. Concomitant with this differentiation, cardiomyocytes withdraw from the cell cycle (also known as terminal differentiation). Several studies suggest that cell cycle withdrawal is produced by downregulation of cell cycle entry genes, such as cyclins, and upregulation of cell cycle inhibitors, such as p27Kip [19]. In cardiomyocytes, progressive differentiation increases complexity in the structural organization of myofibrils [4, 9]. If this complexity physically hinders cytokinesis, as suggested, for example, by Ahuja et al. [1], then differentiation could help to reinforce cell cycle withdrawal during early postnatal life.
Mouse fetal cardiomyocytes readily divide in vivo and in vitro. In the initial hours to days after birth, mouse cardiomyocytes maintain their ability to re-enter the cell cycle, displaying DNA synthesis, as evidenced, for example, by [3H]thymidine- or BrdU-labeling of nuclei and karyokinesis (nuclear division). However, they can no longer undergo cell division (cytokinesis). As a result of this continued karyokinesis in the absence of cytokinesis, cardiomyocytes rapidly shift from being mononucleated to most of them (>80%) being binucleated.
After the first week of neonatal life, even cell cycle reentry ceases, although in response to certain physiologic stresses, such as left ventricular hypertrophy, low levels of DNA synthesis have been observed even in adult cardiomyocytes [19]. Nevertheless, mitotic figures and definitive evidence of cytokinesis (i.e., the presence of a well-defined cleavage furrow) are very rarely, if ever, observed. The absence of mitosis and cytokinesis is permanent as well as the discerning phenotypic properties of the adult differentiated mammalian cardiomyocyte that identify them as terminally differentiated. Moreover, they explain why the response of the adult mammalian heart to increased systolic load is limited to hypertrophic growth of cardiomyocytes. Although this increases muscle mass and helps to restore cardiac performance, it is nonetheless maladaptive [6].
Why mammalian cardiomyocytes undergo terminal differentiation soon after birth whereas those of other species such as newts and teleost fish maintain regenerative competence is unclear [17, 22]. Recently, we found that, surprisingly, mice with inactivation of c-kit signaling show a hyperplastic response to pressure overload. This allowed us to then identify c-kit as a developmental cue that causes postnatal terminal differentiation in mouse cardiomyocytes in vivo. In this review we explore the implications of cardiomyocyte terminal differentiation, using the signaling inactive c-kit mouse as a model, in which the adult state is characterized by nonterminally differentiated cardiomyocytes.
Characteristics of Terminal Differentiation in Adult Cardiomyocytes
Terminal differentiation of mammalian cardiomyocytes appears to activate various cell cycle checkpoints so that even if there is a stimulus to divide, cell division is prevented. Checkpoints G1/S and G2/M stop cell cycle reentry and progression to the M phase. Karyokinesis is required for successful completion of the M phase. In the absence of karyokinesis, nuclear accumulation of duplicated chromosomes results in increased ploidy, and karyokinesis without cytokinesis results in multinucleation. Ploidy and multinucleation, as observed in adult mammalian cardiomyocytes [8, 24–26], are collectively termed endoreduplication. This indicates that mammals have evolved a multilayered mechanism to prevent cytokinesis in adult cardiomyocytes. The evolutionary pressures that lead to the acquisition of these checkpoints are unknown.
In contrast, cardiomyocytes in certain lower vertebrates such as urodele amphibians and teleost are differentiated but not terminally differentiated. In these species, cardiomyocytes retain the potential to proliferate in response to injury. Could terminal differentiation have evolved to check excessive heart growth leading to tumors? This seems unlikely because mammalian fetal cardiomyocytes divide readily with little propensity to tumor formation.
Furthermore, no evidence of excess tumor formation exists in those amphibian hearts composed of nonterminally differentiated cardiomyocytes. Of course, from a clinical viewpoint the acquisition of terminal differentiation by mammalian cardiomyocytes is an extremely undesirable trait because it contributes in a major way to the development of heart failure in patients who have lost a substantive number of cardiomyocytes due to myocardial infarction.
To define the developmental cues that may lead to terminal differentiation, we focused our attention on the c-kit. We found that the c-kit, the receptor for stem cell factor, is not expressed in fetal cardiomyocytes until half-a-day after birth (Fig. 1). Thereafter, its expression is transient, waning after only a few days, coincident with the onset of their terminal differentiation [11]. Indeed, this close temporal relationship between these two responses led us to hypothesize that the c-kit may be the developmental cue for cardiomyocyte terminal differentiation.
Fig. 1.
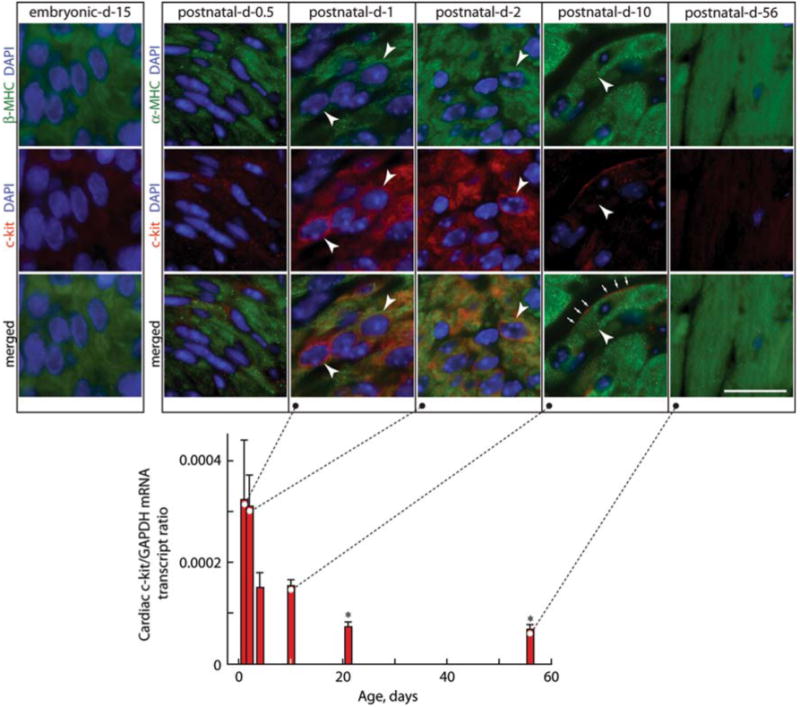
Expression of the c-kit in the mouse heart. Cardiac c-kit mRNA levels by quantitative reverse transcriptase-polymerase chain reaction (RT-PCR) are normalized to glyceraldehyde phosphate dehydrogenase (GAPDH) mRNA levels in postnatal (1- to 10-day-old) and adult (21- and 56-day-old) mice. The levels of cardiac c-kit expression are high 1 day after birth, fall by postnatal day 4, and are barely evident by day 10. Values are mean ± standard error of the mean, and comparisons were made by analysis of variance (ANOVA) followed by Tukey's test. *p < 0.05 relative to day 1. Insets show photomicrographs of left ventricle sections from embryonic day 15 and postnatal days 0.5, 1, 2, 10, and 56 mice stained for α- or β-MHC to identify cardiomyocytes and 4′,6′-diamidino-2-phenylindole (DAPI) to detect nuclei and c-kit, with dual staining or merged images as indicated. Arrowheads show some c-kit+ LV cardiomyocytes in postnatal hearts. Arrows show cell-surface c-kit+. Scale bar is 20 μm. (From Li et al. [11])
Role of the c-Kit in Cardiomyocyte Terminal Differentiation
The c-kit is a type 3 transmembrane receptor tyrosine kinase activated by stem cell factor-induced binding and homodimerization. This results in the transphosphorylation of two tyrosines (Y568 and Y570) in the juxtamembranous region [10, 12, 13]. As a consequence, a large conformational change in the activation loop from a compact structure to an extended structure abolishes the autoinhibitory role of the juxtamembrane domain on the c-kit kinase activity. Transphosphorylation of Y823 in the activation loop further stabilizes the receptor in its active form [13].
The tyrosine kinase activity of the c-kit also leads to autophosphorylation of other tyrosine residues in the cytoplasmic domain of the receptor. This alters the conformation of this domain to expose phosphorylated tyrosine residues that are targets for binding of the Src homology 2 domain containing proteins (Fig. 2). The phosphorylation and interaction of these proteins including phosphoinositide 3′-kinase, phospholipase Cγ, the Src family of tyrosine kinases, Janus kinase/signal transducers and activators of transcription, and p21ras GTP-activating protein/mitogen-activated protein kinase are important features of c-kit signaling (Fig. 3). These signaling pathways regulate multiple organismal and cellular processes such as fertility, skin pigmentation, stem cell mobilization, and cellular differentiation, which are evident by characteristic phenotypic changes that result from germ line loss-of-function mutations in the c-kit [15, 16].
Fig. 2.
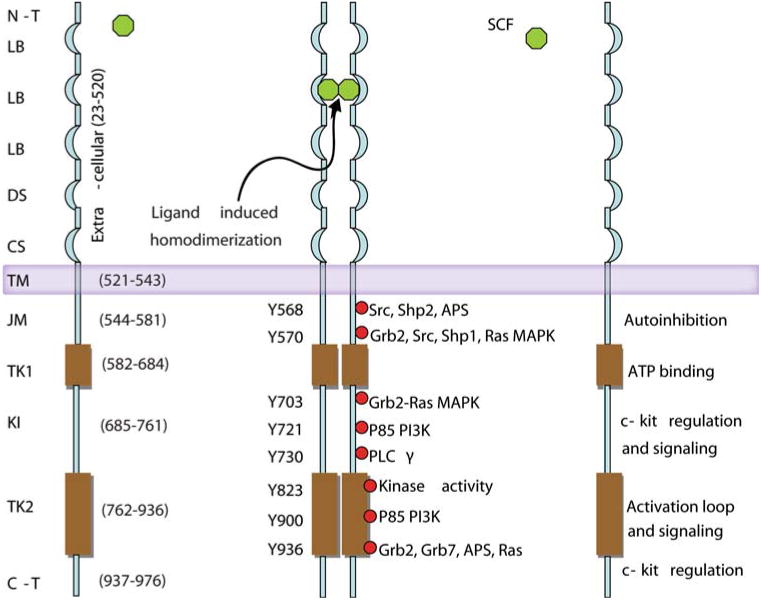
Schematic representation of the c-kit protein showing the known function of each of its domains. N-T N-terminal; LB ligand binding; DS dimer stabilization; CS cleavage site; TM transmembrane; JM juxtamembrane; TK1 tyrosine kinase domain 1, also known as the proximal kinase domain; KI kinase insert; TK2 tyrosine kinase domain 2 or distal kinase domain; C-T c-terminal domain. Furthermore, after stem cell factor-induced homodimerization, all the tyrosine residues that are phosphorylated and the adaptor proteins that bind to these phosphorylated tyrosine residues also are represented
Fig. 3.
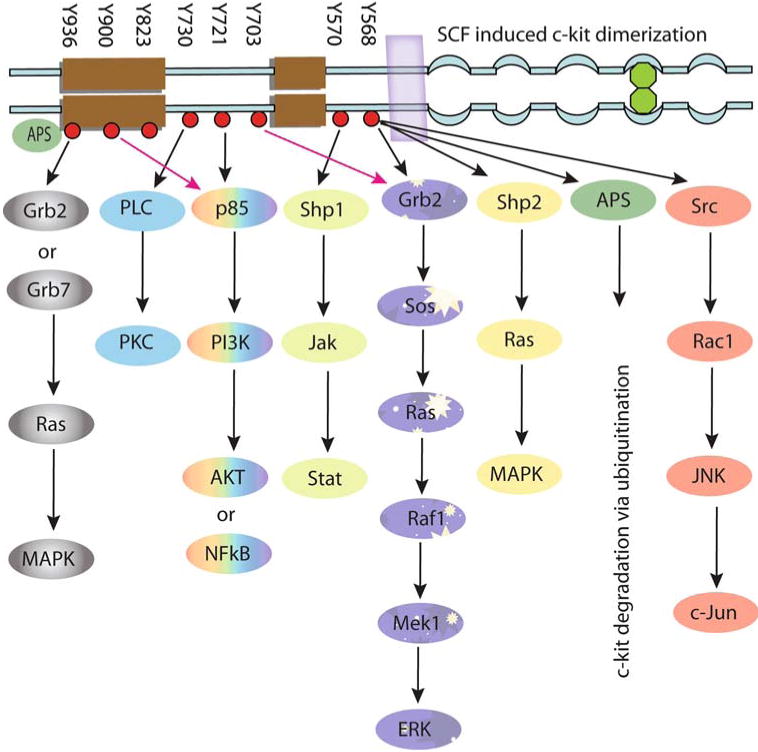
Schematic representation of the signal transduction pathways originating from intracellular phosphorylated tyrosine residues of the c-kit. Activation of these pathways is shown to be involved in cell survival, proliferation, differentiation, and ubiquitination. All these signaling molecules are individually explained in the text
We chose the compound heterozygous c-kit W/Wv loss-of-function mouse model for study the c-kit role in cardiomyocytes terminal differentiation [11]. Biochemical and phenotypic changes associated with the W and Wv mutations in the c-kit have been reported by Nocka et al. [15, 16]. Despite defective c-kit signaling, cardiomyocytes from adult W/Wv mice are phenotypically indistinguishable from those of wild type (WT) hearts. Both WT and W/Wv mice have similar mean proximal aortic blood pressures, left ventricles in terms of weight (left ventricle-to-body weight ratio) and morphology (LV wall thickness-to-diameter ratio), and isovolemic (±dP/dtmax) and ejection-phase function (rate-corrected velocity of circumferential shortening). In addition, the left ventricle (LV) cardiomyocytes of adult animals of both genotypes are similar in cross-sectional area, and both are predominantly binucleated.
Thus, under basal conditions, there appears to be no overt phenotypic difference between W/Wv and WT cardiomyocytes. Pressure overload (PO), produced by suprarenal aortic constriction, resulted in similar LV growth in WT and W/Wv mice. In W/Wv mice, this LV growth was due to cardiomyocyte hyperplasia, which caused an approximate 34% increase in the number of cardiomyocytes after just 7 days of PO, whereas in the WT mice, LV growth was limited exclusively to cardiomyocyte hypertrophy [11]. Cytochemical evaluation indicated an absence of endoreduplication in W/Wv LVs subjected to PO. Furthermore there was no evidence of cardiomyocyte apoptosis in W/Wv LVs subjected to PO (Li, Naqvi, Yahiro, Husain, unpublished observation).
These findings suggest that all cell cycle checkpoints that normally prevent hypertrophy-induced cardiomyocyte proliferation are disabled during PO-induced hyperplastic growth of the W/Wv heart. Importantly, the cardiomyocyte hyperplastic response to PO in W/Wv suprarenal aortic constriction LVs appeared to be linked to improved LV contractility and survival [11].
Morphometric, immunohistochemical, and immunocytochemical analysis indicated no difference in W/Wv cardiomyocyte size or sarcomeric organization relative to that of WT cardiomyocytes. Although fully capable of cytokinesis, W/Wv LV cardiomyocytes do not show increased expression of fetal genes such as β-myosin heavy chain, skeletal actin, and atrial natriuretic factor. Indeed, gene expression profiling of WT and W/Wv LV cardiomyocytes from unstressed hearts showed that only 8 unrelated genes out of more than 40,000 transcripts analyzed were different between WT and W/Wv LV cardiomyocytes, suggesting that W/Wv LV cardiomyocytes are virtually identical to those of WT animals.
It is thought that proliferation of differentiated cardiomyocytes requires dedifferentiation to the fetal phenotype [5, 23]. Our findings, however, do not support this view, given that gene expression profiling of cardiomyocytes from WT and W/Wv LVs subjected to 7 days of PO showed that more than 150 genes were differentially expressed [11]. The upregulated genes were required mainly for transit through the G1/S and G2/M phases of the cell cycle and for karyokinesis and cytokinesis (Table 1). However, the expression of fetal genes such as β-myosin heavy chain, skeletal actin, and atrial natriuretic factor was unchanged. Furthermore, proliferating W/Wv LV cardiomyocytes subjected to PO had a well-defined sarcomeric organization such as that found in WT adult cardiomyocytes. Indeed, lack of a disorganized sarcomeric structure in these proliferating cardiomyocytes is congruent with the finding that PO does not diminish systolic function in W/Wv LVs subjected to PO. Rather, LV systolic function of hyperplastic W/Wv hearts was found to be robust. Importantly, the hyperplastic LV response in W/Wv mice subjected to PO appears to be a direct effect of c-kit dysfunction because cardiomyocyte proliferation also was observed with PO in animals who had cardiomyocyte-restricted inhibition of c-kit induced by transgenic overexpression of a dominant-negative c-kit mutant.
Table 1.
c-Kit dysfunction regulates cell-cycle gene expression in cardiomyocytes from hypertensive mice [11]
| Gene | x-fold increasea (W/Wv-SAC/WT-SAC) (n = 5–7/group) | Comment |
|---|---|---|
| G1/S phase transition and DNA replication | ||
| Thymidine kinase 1 (TK1) | 2.6b | TK1 regulation (upregulated during S and M phases) provides the pool of dTTP for DNA synthesis in the cell cycle |
| Ribonucleotide reductase M2 (Rrm2) | 2.4b | Rrm2 converts ribonucleoside diphosphates to deoxyribonucleoside diphosphates, which are essential for DNA synthesis |
| E2F transcription factor 1 (E2f1) | 1.7b | E2f1, a key target of the retinoblastoma protein, activates cell cycle progression, but also apoptosis |
| E2F transcription factor 2 (E2f2) | 2.6b | E2f2, a key target of the retinoblastoma protein, induces DNA synthesis in cardiomyocytes but without activation of apoptosis |
| Cytoskeleton-associated protein 2 (ckap2) | 3.3c | Ckap2 is a microtubule component. It is involved in apoptosis, ploidy, and cell cycle progression |
| DBF4 homolog (DBF4) | (2.5b) | DDK, composed of the catalytic subunit Cdc7 and the regulatory subunit Dbf4, promotes timely S phase progression |
| Chromatin assembly factor 1, subunit B (Chaf1b) | (3.8b) | Chromatin assembly factor 1 is required for the assembly of histone octamers onto newly replicated DNA |
| Chromosome transmission fidelity factor 18 (Chtf18) | (4.3b) | Chtf18 is required for sister chromatid cohesion in S phase that is a critical step in high-fidelity cell division |
| Histone 1h2af (Hist1h2af) | 2.9b | Hist1h2af is involved in chromosome organization, biogenesis, and nucleosome assembly |
| Histone 1h2an (Hist1h2an) | 2.8b | Hist1h2an is involved in chromosome organization, biogenesis, and nucleosome assembly |
| G2/M phase regulation | ||
| Cyclin B1 (Ccnb1) | 2.6b | The G2 to M phase transition is triggered by the dimerization of cyclin B/cdc2, which are subunits of mitosis-promoting factor |
| Cell division cycle 25 homolog C (cdc25c) | 2.8d | Cyclin B/cdc2 is activated by the phosphatase Cdc25 |
| Cell division cycle 2 homolog A (cdc2) | 3.1c | Cdc2, a Ser/Thr kinase, is essential for G1/S and G2/M phase cell cycle transitions |
| Cell division cycle 20 homolog (cdc20) | (3.0b) | Cdc20 activates anaphase-promoting complex/cyclosome from mitosis through the metaphase-anaphase transition |
| Forkhead box M1 (Foxm1) | (3.9b) | Foxm1 regulates transcription of proteins involved in G1/S and G2/M phase transitions |
| Cell division cycle–associated 5 (cdca5) | (2.2b) | Cdca5, a nuclear protein associated with cohesin complex, is involved in chromatin binding and G1/S phase transition |
| Topoisomerase (DNA) 2 alpha (Top2a) | 3.2b | Top2a catalyzes topologic genomic changes needed for chromosome segregation and DNA replication |
| Thymidylate synthase (Tyms) | 1.5b | Tyms is essential for DNA synthesis and repair. Tyms is regulated by E2F-1 and thus is linked to the cell cycle pathway |
| Cell division cycle–associated 7-like (cdca7 l) | 2.1b | Cdca7 l interacts with c-myc and leads to cell proliferation |
| Sister chromatid separation, kinetocore, and centrosome associated protein | ||
| Aurora kinase A (Aurka) | 2.6b | Aurora-A and Plk1, which localize aurora-A to centrosomes, are involved in centrosome maturation and spindle assembly |
| Kinetochore associated 1 (Kntc1) | 3.2b | Kntc1 plays important roles in ensuring proper chromosome segregation during cell division |
| Budding uninhibited by benzimidazoles 1β (Bub1b) | (4.7b) | The Bub1b gene encodes BubR1. BubR1 regulates spindle checkpoint |
| Centromere protein E (CenpE) | 3.3c | CenpE is needed for organization of stable microtubule–kinetochore assembly for proper chromosome segregation in mitosis |
| Centromere protein M (CenpM) | 3.4b | CenpM is required for proper assembly of the CenpA nucleosome–associated complex |
| Actin and microtubules assembly | ||
| Anillin (Anln) | 3.1b | Anillin, a cross-linker, binds to filaments in the contractile ring that constricts to form a cleavage furrow during cytokinesis |
| Diaphanous homolog 3 (Drosophila) (Diap3) | 3.5c | Diap3 recruits profilin to the membrane, where it promotes actin polymerization. It is required for cytokinesis |
| Central spindle and aurora kinase complex/microtubule–associated protein | ||
| Protein-regulating cytokinesis 1 (Prc1) | 3.5b | Microtubule-associated Prc1 is an anaphase-specific binding partner for Plk1. This interaction is required for cytokinesis |
| Kinesin family member 22 (Kif22) | 2.7c | Kif22, a microtubule-dependent molecular motor, is essential for metaphase chromosome alignment and maintenance |
| Kinesin family member 4 (Kif4) | 2.9b | Kif4 is a microtubule-based motor protein that plays a crucial role in cell division |
| Kinesin family member C5A (Kifc1) | 2.5c | Kifc1 is a minus-end-directed microtubule-dependent motor involved in the control of centrosome duplication |
| Kinesin family member 2C (Kif2c) | (3.6b) | Kif2c, a microtubule-dependent molecular motor, is important for anaphase chromosome segregation |
| Aurora kinase B (Aurkb) | 2.6b | Aurora B phosphorylates the microtubule-binding proteins, which leads to detachment of microtubules from kinetochores |
| Inner centromere protein (Incenp) | 3.0c | Incenp, which activates aurora-B kinase, complexes with survivin to attach chromosomes correctly to spindle microtubules |
| Survivin (Birc5) | 2.9c | Survivin, a subunit of the chromosomal passenger complex, is essential for proper chromosome segregation and cytokinesis |
| Polo-like kinase 1 (Drosophila) (Plk1) | 2.4b | Plk1 plays a critical role in centrosome maturation, mitotic spindle assembly, mitotic entry and exit, and cytokinesis |
| Nucleolar and spindle–associated protein 1 (Nusap1) | 3.2b | NuSAP is an essential microtubule-stabilizing and -bundling protein enriched at the central part of the spindle. |
| Meiosis-specific nuclear structural protein 1 (Mns1) | 2.3b | MNS1 maintains appropriate nuclear morphology during meiotic prophase |
| Claspin (Clspn) | 2.9c | Plk1 phosphorylates claspin, releasing it from DNA, thereby contributing to Chk1 (a cell cycle checkpoint) inactivation |
SAC suprarenal aortic constriction, WT wild type, dTTP deoxythymidine triphosphate thymidine 5′-triphosphate, DDK DBF4 dependent kinase
mRNA transcript levels normalized to glyceraldehyde phosphate dehydrogenase (GAPDH) mRNA, were determined using q-reverse transcriptase-polymerase chain reaction (RT-PCR). Values are x-fold increases in the normalized mRNA expression level in W/Wv 7-day SAC cardiomyocytes relative to that in WT 7-day SAC cardiomyocytes. Values in parentheses are the x-fold changes determined by microarray analyses
p<0.05
p<0.01
p<0.001, Student's t test
These findings demonstrate a vital role for the c-kit in cardiomyocyte terminal differentiation. The absence of dysregulated cell cycle gene expression in the unstressed W/Wv LV relative to its WT control could suggest that the decrease in cell cycle protein levels or an increase of their inhibitors in cardiomyocytes after birth is not the cause of terminal differentiation. Indeed, the phenotype of mice in which cell cycle proteins are overexpressed or cell cycle inhibitors are suppressed in postnatal cardiomyocytes is markedly different from that seen in adult W/Wv hearts. For example, p27 null mice display an extra round of cardiomyocyte division in the early postnatal period leading to an enlarged heart even in the absence of PO [21]. Overexpression of cyclins also leads to an enlarged heart phenotype with increased endoreduplication [3, 18, 26]. Endoreduplication and an increase in cyclins also are seen in WT rat cardiomyocytes subjected to PO [20].
How the c-kit produces terminal differentiation remains unknown, although the paucity of differentially expressed genes in unstressed W/Wv compared with WT LV cardiomyocytes suggests that an epigenetic mechanism may be operative. This could result in placement of a permanent methylation mark on regulatory sequences of genes required for cell cycle reentry, mitosis, and cytokinesis. As a consequence, basal cell cycle gene expression would not necessarily be altered between WT and W/Wv LV cardiomyocytes, but still could be induced to increase in W/Wv LV cardiomyocytes in response to PO, an effect resulting in hyperplasia. In trying to achieve efficient cardiac regeneration, therefore, the key to successful cardiomyocyte cytokinesis without G2/M phase arrest, mitosis without cytokinesis, or cytokinesis with induction of apoptosis may be the simultaneous removal of multiple cell cycle checkpoints rather than an increase in the levels of a single cell cycle regulator without disablement of the many checkpoints involved in maintaining cardiomyocyte terminal differentiation.
Contributor Information
Nawazish Naqvi, Division of Cardiology, Department of Medicine, Emory University School of Medicine, 101 Woodruff Circle, 319 Woodruff Memorial Research Building, Atlanta, GA 30322, USA.
Ming Li, Division of Cardiology, Department of Medicine, Emory University School of Medicine, 101 Woodruff Circle, 319 Woodruff Memorial Research Building, Atlanta, GA 30322, USA.
Eiji Yahiro, Division of Cardiology, Department of Medicine, Emory University School of Medicine, 101 Woodruff Circle, 319 Woodruff Memorial Research Building, Atlanta, GA 30322, USA.
Robert M. Graham, Victor Chang Cardiac Research Institute, Darlinghurst, NSW, Australia
Ahsan Husain, Email: ahusai2@emory.edu, Division of Cardiology, Department of Medicine, Emory University School of Medicine, 101 Woodruff Circle, 319 Woodruff Memorial Research Building, Atlanta, GA 30322, USA.
References
- 1.Ahuja P, Sdek P, MacLellan WR. Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol Rev. 2007;87:521–544. doi: 10.1152/physrev.00032.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ausma J, Schaart G, Thoné F, et al. Chronic ischemic viable myocardium in man: aspects of dedifferentiation. Cardiovasc Pathol. 1995;4:29–37. doi: 10.1016/1054-8807(94)00028-p. [DOI] [PubMed] [Google Scholar]
- 3.Chaudhry HW, Dashoush NH, Tang H, et al. Cyclin A2 mediates cardiomyocyte mitosis in the postmitotic myocardium. J Biol Chem. 2004;279:35858–35866. doi: 10.1074/jbc.M404975200. [DOI] [PubMed] [Google Scholar]
- 4.Ehler E, Rothen BM, Hämmerle SP, et al. Myofibrillogenesis in the developing chicken heart: assembly of Z-disk, M-line, and the thick filaments. J Cell Sci. 1999;112:1529–1539. doi: 10.1242/jcs.112.10.1529. [DOI] [PubMed] [Google Scholar]
- 5.Engel FB, Schebesta M, Duong MT, et al. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. 2005;19:1175–1187. doi: 10.1101/gad.1306705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Esposito G, Rapacciuolo A, Naga Prasad SV, et al. Genetic alterations that inhibit in vivo pressure-overload hypertrophy prevent cardiac dysfunction despite increased wall stress. Circulation. 2002;105:85–92. doi: 10.1161/hc0102.101365. [DOI] [PubMed] [Google Scholar]
- 7.Gulick J, Subramaniam A, Neumann J, Robbins J. Isolation and characterization of the mouse cardiac myosin heavy chain genes. J Biol Chem. 1991;266:9180–9185. [PubMed] [Google Scholar]
- 8.Herget GW, Neuburger M, Plagwitz R, Adler CP. DNA content, ploidy level, and number of nuclei in the human heart after myocardial infarction. Cardiovasc Res. 1997;36:45–51. doi: 10.1016/s0008-6363(97)00140-5. [DOI] [PubMed] [Google Scholar]
- 9.Hirschy A, Schatzmann F, Ehler E, Perriard JC. Establishment of cardiac cytoarchitecture in the developing mouse heart. Dev Biol. 2006;289:430–441. doi: 10.1016/j.ydbio.2005.10.046. [DOI] [PubMed] [Google Scholar]
- 10.Hubbard SR. Juxtamembrane autoinhibition in receptor tyrosine kinases. Nat Rev Mol Cell Biol. 2004;5:464–471. doi: 10.1038/nrm1399. [DOI] [PubMed] [Google Scholar]
- 11.Li M, Naqvi N, Yahiro E, Liu K, et al. C-kit is required for cardiomyocyte terminal differentiation. Circ Res. 2008;102:677–685. doi: 10.1161/CIRCRESAHA.107.161737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mol CD, Lim KB, Sridhar V, et al. Structure of a c-kit product complex reveals the basis for kinase transactivation. J Biol Chem. 2003;278:31461–31464. doi: 10.1074/jbc.C300186200. [DOI] [PubMed] [Google Scholar]
- 13.Mol CD, Dougan DR, Schneider TR, et al. Structural basis for the autoinhibition and STI-571 inhibition of c-kit tyrosine kinase. J Biol Chem. 2004;279:31655–31663. doi: 10.1074/jbc.M403319200. [DOI] [PubMed] [Google Scholar]
- 14.Ng WA, Grupp IL, Subramaniam A, Robbins J. Cardiac myosin heavy chain mRNA expression and myocardial function in the mouse heart. Circ Res. 1991;68:1742–1750. doi: 10.1161/01.res.68.6.1742. [DOI] [PubMed] [Google Scholar]
- 15.Nocka K, Majumder S, Chabot B, Ray P, et al. Expression of c-kit gene products in known cellular targets of W mutations in normal and W mutant mice: evidence for an impaired c-kit kinase in mutant mice. Genes Dev. 1989;3:816–826. doi: 10.1101/gad.3.6.816. [DOI] [PubMed] [Google Scholar]
- 16.Nocka K, Tan JC, Chiu E, et al. Molecular bases of dominant negative and loss of function mutations at the murine c-kit/white spotting lOCUS: W37, W′, W41, and W. EMBO J. 1990;9:1805–1813. doi: 10.1002/j.1460-2075.1990.tb08305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oberpriller JO, Oberpriller JC. Response of the adult newt ventricle to injury. J Exp Zool. 1974;187:249–253. doi: 10.1002/jez.1401870208. [DOI] [PubMed] [Google Scholar]
- 18.Pasumarthi KB, Nakajima H, Nakajima HO, et al. Targeted expression of cyclin D2 results in cardiomyocyte DNA synthesis and infarct regression in transgenic mice. Circ Res. 2005;96:110–118. doi: 10.1161/01.RES.0000152326.91223.4F. [DOI] [PubMed] [Google Scholar]
- 19.Poolman RA, Brooks G. Expressions and activities of cell cycle regulatory molecules during the transition from myocyte hyperplasia to hypertrophy. J Mol Cell Cardiol. 1998;10:2121–2135. doi: 10.1006/jmcc.1998.0808. [DOI] [PubMed] [Google Scholar]
- 20.Poolman RA, Gilchrist R, Brooks G. Cell cycle profiles and expressions of p21CIP1 AND P27KIP1 during myocyte development. Int J Cardiol. 1998;67:133–142. doi: 10.1016/s0167-5273(98)00320-9. [DOI] [PubMed] [Google Scholar]
- 21.Poolman RA, Li JM, Durand B, Brooks G. Altered expression of cell cycle proteins and prolonged duration of cardiac myocyte hyperplasia in p27KIP1 knockout mice. Circ Res. 1999;85:117–127. doi: 10.1161/01.res.85.2.117. [DOI] [PubMed] [Google Scholar]
- 22.Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science. 2002;298:2188–2190. doi: 10.1126/science.1077857. [DOI] [PubMed] [Google Scholar]
- 23.Rumyantsev PP. Interrelations of the proliferation and differentiation processes during cardiact myogenesis and regeneration. Int Rev Cytol. 1977;51:186–273. [PubMed] [Google Scholar]
- 24.Soonpaa MH, Field LJ. Assessment of cardiomyocyte DNA synthesis in normal and injured adult mouse hearts. Am J Physiol. 1997;272:H220–H226. doi: 10.1152/ajpheart.1997.272.1.H220. [DOI] [PubMed] [Google Scholar]
- 25.Soonpaa MH, Kim KK, Pjak L, et al. Cardiomyocyte DNA synthesis and binucleation during murine development. Am J Physiol. 1996;271:2183–2189. doi: 10.1152/ajpheart.1996.271.5.H2183. [DOI] [PubMed] [Google Scholar]
- 26.Soonpaa MH, Koh GY, Pajak L, et al. Cyclin D1 overexpression promotes cardiomyocyte DNA synthesis and multinucleation in transgenic mice. J Clin Invest. 1997;99:2644–2654. doi: 10.1172/JCI119453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeller R, Bloch KD, Williams BS, et al. Localized expression of the atrial natriuretic factor gene during cardiac embryogenesis. Genes Dev. 1987;1:693–698. doi: 10.1101/gad.1.7.693. [DOI] [PubMed] [Google Scholar]