

Abstract
The measurement of yeast's intracellular pH (ICP) is a proven method for determining yeast vitality. Vitality describes the condition or health of viable cells as opposed to viability, which defines living versus dead cells. In contrast to fluorescence photometric measurements, which show only average ICP values of a population, flow cytometry allows the presentation of an ICP distribution. By examining six repeated propagations with three separate growth phases (lag, exponential, and stationary), the ICP method previously established for photometry was transferred successfully to flow cytometry by using the pH-dependent fluorescent probe 5,6-carboxyfluorescein. The correlation between the two methods was good (r2 = 0.898, n = 18). With both methods it is possible to track the course of growth phases. Although photometry did not yield significant differences between exponentially and stationary phases (P = 0.433), ICP via flow cytometry did (P = 0.012). Yeast in an exponential phase has a unimodal ICP distribution, reflective of a homogeneous population; however, yeast in a stationary phase displays a broader ICP distribution, and subpopulations could be defined by using the flow cytometry method. In conclusion, flow cytometry yielded specific evidence of the heterogeneity in vitality of a yeast population as measured via ICP. In contrast to photometry, flow cytometry increases information about the yeast population's vitality via a short measurement, which is suitable for routine analysis.
Yeast plays an important role in the food industry. It is primarily used for making bread, beer, and wine, and the flavor and aromatic compounds it produces are characteristic of these fermented products. The physiological state of the yeast biomass influences the fermentation performance and thus the quality of the resulting product, e.g., of beer (1, 32). Food processors who depend on yeast health for consistent fermentations demand a method to measure yeast's physiological condition during yeast growth, that is, its vitality. The term “vitality” refers to the health of living biomass where high vitality results in a fast fermentation with minimal undesired by-products, while low vitality results in sluggish or poorly attenuating fermentation. In contrast, the term “viability” only distinguishes between dead and alive cells.
Several methods exist to measure yeast vitality such as carbon dioxide production (10), vicinal diketone reduction (3), glycogen and trehalose staining (16-18), vital titration (30), measurement of the specific oxygen uptake rate (27, 38), and the acidification power test (12, 22). One important method for vitality measurement is the detection of intracellular pH (ICP) (9, 19, 20, 31).
Intracellular metabolic reactions in yeast are catalyzed by enzymes that have their optimum working pH in a neutral range (23). During fermentation or propagation, yeast produces carbon dioxide and organic acids and releases protons to the medium. To maintain a desired ICP, yeast continuously pumps protons against a gradient from the cytosol to the extracellular medium. The regulating proton pump is plasma-membrane-ATPase (4). The transmembrane proton gradient has to be sustained since the transport of important nutrients such as maltose or amino acids depends on it. The efficiency of glycolysis and gluconeogenesis is affected by ICP. The higher the proton extrusion rate, the more active is the metabolism. Thus, a high ICP indicates high yeast vitality. Yeast with the highest ICP ferment the best (1).
Techniques using fluorescence photometry and the pH-dependent dye, 5,6-carboxyfluorescein (CF), for measuring yeast ICP have been published (1, 20, 21, 33). These proved reliable but only yield an average value of ICP of a yeast population. Based on the method of Imai (21), a shorter method for measuring ICP was developed by Thiele and Back (33) using photometry whereby the sample preparation time was reduced from approximately 3.5 to 1.25 h. This reduction made it suitable for routine analysis.
In the present study and according to Thiele and Back (33), the nonfluorescent, esterified form of CF, 5,6-carboxyfluorescein-diacetate (CF-DA), was used for cell loading because it is more capable of passing through cell membranes than its nonesterified counterpart (6). After the cells were infused with CF-DA, yeast esterase enzymes split the molecule into acetate residues and the actual fluorescent probe CF. Excitation with a certain wavelength results in different fluorescent intensities that depend on both the pH of the cell cytoplasm and the dye concentration. In order to receive results independent on probe concentration, a ratio between pH-dependent and pH-independent fluorescence intensities of the sample can be used to produce data depending on pH only (20, 35).
In addition to measuring ICP via fluorescence photometry, flow cytometry can also serve as detection system of CF fluorescent intensities corresponding to ICP. Flow cytometry is becoming a standard tool in biotechnology research and has been successfully applied to analyze and optimize yeast processes (13, 14, 15, 24, 36). Cytometric measurements provide the opportunity to detect single cells. With this method it is possible to describe a whole yeast population in detail by illustrating the distribution of ICP (35, 39). In contrast, photometry only reflects an average value of the population.
We compare here the application of the short ICP method executed via fluorescence photometry to flow cytometry. During the propagation of brewer's yeast, three different growth phases (the lag, exponential, and stationary phases) were compared to determine whether the vitality varied during growth and whether this variation was detectable by both methods. Since flow cytometry offers the opportunity to identify subpopulations (35, 36), we hypothesized that it was possible to detect yeast subpopulations with differing ICP during propagation.
MATERIALS AND METHODS
Chemicals and buffers.
CF-DA (Sigma Chemicals Co., St. Louis, MO) was dissolved in dimethyl sulfoxide to 10 mM and used for yeast cell dyeing. The de-esterified probe CF (Sigma) was dissolved in dimethyl sulfoxide to 100 mM and used for calibration. A 50 mM citric disodium hydrogen phosphate buffer (pH 3.0, containing 110 mM NaCl, 5 mM KCl, and 1 mM MgCl2 at 1°C) was used as loading buffer. For calibration purposes, appropriate mixtures of citric acid buffer (50 mM) and disodium hydrogen phosphate buffer (50 mM Na2HPO4) plus 110 mM NaCl, 5 mM KCl, and 1 mM MgCl2 at 1°C were prepared to achieve a range of buffers from pH 4.6 to 7.6 in steps of 0.2 pH units.
ICP determination. (i) Sample preparation and staining.
A 200-ml aliquot of propagator yeast was centrifuged (2,300 × g for 5 min at room temperature); the resulting pellet was resuspended in 10 ml of saline solution (NaCl 0.9%) and stored at 1°C for a maximum of 24 h. During the cell loading procedure, samples and loading buffer were kept on ice. The yeast slurry was sieved with a wire netting (32-μm-pore-size mesh) to remove precipitates and afterwards centrifuged (3,600 × g for 4 min at 4°C). The same conditions were set for further centrifugation steps if not otherwise mentioned. Precipitates were removed from top of the pellet and 0.5 cm3 of the pellet bottom was resuspended in 3 ml of loading buffer. After centrifugation, the yeast sample was washed three times with 3 ml of cold loading buffer and resuspended in 0.75 ml of the same.
After the addition of 0.075 ml of 10 mM CF-DA stock solution, the suspension was immediately shaken vigorously for 1 min. A portion (0.25 ml) of this suspension was added to 8 ml of loading buffer (0.0275 mM CF-DA) and placed immediately in a water bath of 30°C and incubated for 14 min. After incubation the sample was centrifuged and washed two times with 6 ml of loading buffer and resuspended in 3 ml of loading buffer at room temperature.
(ii) Photometer analysis.
Photometer analyses were carried out with a fluorescence photometer F-2000 (Hitachi, Japan). Samples were excited with a xenon lamp two times at 441 and 488 nm, while fluorescent emissions were measured at 518 nm. The ICP was determined as the ratio of the emission signal from 488-nm excitation divided by the emission signal from 441-nm excitation, the former being pH and concentration dependent and the latter being only concentration dependent. For any sample, the ICP was calculated by using the calibration described below.
(iii) Flow cytometric analysis.
Flow cytometric analyses were performed with a Coulter Epics XL-MLC (Beckmann Coulter, Krefeld, Germany). Before measurement, each sample was diluted 1:11 in loading buffer. Samples were excited with a 488-nm argon ion laser, while the fluorescence emission was measured through a 525-nm (±20-nm) band-pass filter and a 575-nm (±15-nm) band-pass filter. The fluorescence intensity of CF was pH dependent at 525 nm, while at 575 nm it was not. All data were acquired in a logarithmic mode with total of 20,000 cells measured for every sample. Subsequent data analysis was performed by using WinMDI software (Windows Multiple Document Interface for Flow Cytometry, version 2.9; Scripps Research Institute, La Jolla, CA) in which the fluorescence intensities at 525 and 575 nm were recorded for every single yeast cell as mean data. These fluorescence intensities were divided to build the ratio of the fluorescence emission (ratio 525 nm/575 nm). For any sample, the fluorescence ratio of every cell was rounded to two decimal places and applied to the calibration curve described below. Further data sorting and histogram preparation was performed by using Microsoft Excel.
(iv) Device settings of flow cytometer.
With a flow cytometer, the detected fluorescence signals are converted to voltages and can be amplified and further analyzed. Each fluorescence signal (in our case, 525 and 575 nm) was treated individually. The amplification of these signals had a significant influence on the final result because different combinations of these signal voltages produced variation in the fluorescence ratios (525 nm/575 nm), thereby affecting the resolution of the ICP distribution. It was necessary to find a combination of these signal voltages, where fluorescence intensities of both the samples and the calibration substances were within the measuring range. The combination of 459 V for 525 nm and 707 V for 575 nm was determined to be optimal for flow cytometric analyses. Using these settings, comparative measurements between the cytometer and photometer were carried out.
Instrument calibration.
Calibration measurements were executed with the same instrument settings used for ICP measurement with the photometer and cytometer. For photometer calibration, the fluorescent dye CF was added to the calibration buffers of different pH increments as described earlier in order to span a range of potential ICP values. The measured fluorescence ratios of the yeast sample were converted to ICP values using these calibration data. The same solutions were used for cytometer calibration; however, the dye was first adsorbed onto Superdex 200 particles (GE Healthcare Bio-Sciences, Uppsala, Sweden). The stained particles were subsequently added to the calibration buffers of different pH, followed by incubation for 4 days on a reciprocal shaker before measurements were made. The data analysis was performed as described for flow cytometric analysis.
Yeast strain and growth conditions.
For the ICP correlation procedure the bottom-fermenting Saccharomyces cerevisiae strain W34/70 (Fermentis, Marque-en-Baroeul, France) was used. The propagation experiments were performed in a laboratory fermentor type L1523 (Bioengineering, Wald, Switzerland) with a constant temperature of 25°C, a dissolved oxygen concentration of 0.5 ppm, a stirring rate of 500 rpm, and without pH control. A Bavarian Pilsener Malt Extract (Weyermann Malzfabrik, Bamberg, Germany), diluted with water to an extract content of 11.5%, was used as the growth medium. Every propagation began with a working volume of 8 liters. Polypropylene glycol P2000 (Fluka, Switzerland) was added as an antifoam in a concentration of 1 ml/liter. Before pitching, the growth medium was pasteurized for 45 min at 85°C. Yeast samples were taken from different growth phases after 3 h (lag phase), 14 h (exponential phase), and 26 h (stationary phase) for ICP determination. The propagations were independently replicated six times.
Stress and storage experiments.
To demonstrate the influence of the device settings on the measuring range, propagated yeast from the stationary phase was stored and stressed under three different conditions. Yeast was cropped after 26 h of propagation with standard settings of growth conditions. Storage was performed with saline solution (0.9% NaCl) for 45 h. First stress condition was osmotic shock with 18% (wt/wt) D(−)-Sorbitol (VWR International, Leuven, Belgium). Yeast was kept for 45 h in this high osmotic solution at room temperature. The second stress was via heat in which yeast that was stored for 42 h in saline solution at 1°C was heated in a water bath to 35°C for 3 h and then chilled to 1°C. The third stress was the application of a high ethanol concentration. Ethanol (96%; VWR International) was diluted to a final concentration of 10% (vol/vol). After storage in saline solution for 41 h at 1°C, centrifuged yeast was transferred to the 10% ethanol solution and kept at room temperature for 4 h.
Experimental design and statistical analyses.
Six independent yeast propagations were performed to produce ICP data for comparing fluorescence photometry and flow cytometry. Only single replicate of the stress conditions was performed. Summary statistics were performed using Microsoft Excel while means comparisons and correlation analyses were performed by using XLSTAT (version 2007.6; Addinsoft).
RESULTS
Instrument calibration.
An essential step to measuring yeast ICP is to properly calibrate the instrument in order to correlate fluorescent emission intensity with environmental pH. For photometry, the buffer pH was plotted against the natural logarithm of the CF fluorescence ratios. The calibration was highly linear, with r2 = 0.991. The flow cytometer displayed a different response (Fig. 1), which was adequately described by a third-degree polynomial: r2 = 0.989.
FIG. 1.

Flow cytometer calibration curve for measuring yeast ICP using CF-stained Superdex 200 particles.
ICP measured by photometry.
For the purpose of examining different growth phases, yeast was sampled at three different time points during six independent propagations. Yeast from lag phase (ICP range, 5.68 to 5.93) displayed the lowest ICP mean value, while exponential-phase cells (ICP range, 6.04 to 6.19) displayed the highest value, and stationary-phase cells were in between (ICP range, 5.97 to 6.17). Means comparisons using a Tukey test indicated the ICP values of lag-phase yeast were significantly smaller than those of yeast in the exponential growth phase (P < 0.0001) and stationary phase (P = 0.001). Differences between the lag phase and the stationary phase yeast were insignificant (P = 0.433).
ICP distributions from flow cytometry.
Cells from different growth phases resulted in three different ICP distributions (Fig. 2). Lag-phase yeast displayed the maximum number of cells at an ICP of 6.7, whereas yeast from the exponential and stationary phases had maximum peaks at ∼6.9 and thus a higher vitality. Stationary-phase yeast presented a broader distribution, with a shoulder at a lower ICP, and therefore differed from yeast at the exponential phase. This shoulder in the ICP distribution was centered at ∼6.7, which corresponds with the peak ICP of yeast from the lag phase. With cytometry it was possible to distinguish clearly between the exponential and stationary phases.
FIG. 2.

ICP cytometer distributions for different growth phases during yeast propagation: lag phase at 3 h (•), exponential phase at 14 h (▾), and stationary phase at 26 h (▪). Error bars represent ± 1 standard deviation (n = 6).
In order to produce a single ICP value from a distribution of values, the geometric mean of each population's ICP distribution was calculated (see the equation below), where x̄ is the calculated mean value of the distribution, wi is the number of events (cells), and xi is the corresponding pH ratio. In addition, the geometric mean was used for comparison with photometric mean values (Table 1) .
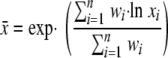 |
Unlike the comparisons among the three growth phases using photometry, all three distributions displayed significant differences (P < 0.05) based on a Tukey test comparison of the geometric means of the ICP distributions. Measurement by cytometry led to higher mean ICP values than those obtained by photometry (Table 1). Flow cytometry also resulted in reduced variability among replicates than photometry, as seen in smaller coefficients of variation across all three growth phases.
TABLE 1.
Comparison of ICP values measured via photometry and flow cytometry for different growth phases during yeast propagation
| Growth phase | ICP method | Mean ICP value ± SDa | Coefficient of variation (%) |
|---|---|---|---|
| Lag | Photometer | 5.83 ± 0.09 | 1.48 |
| Cytometer | 6.61 ± 0.03 | 0.49 | |
| Exponential | Photometer | 6.13 ± 0.07 | 1.08 |
| Cytometer | 6.83 ± 0.03 | 0.40 | |
| Stationary | Photometer | 6.07 ± 0.08 | 1.28 |
| Cytometer | 6.77 ± 0.03 | 0.44 |
Arithmetic means were calculated from six independent replications. Mean ICP values from photometry are means of duplicate readings. Mean ICP values from flow cytometry distributions are calculated as the geometric mean.
Method transfer.
To determine the suitability of implementing flow cytometry ICP measurements in place of those based on photometry, data from these two techniques were correlated with each other (Fig. 3). The correlation of cytometric and photometric measurements utilized mean values from all six propagations. Furthermore, data from all growth phases were utilized for correlations resulting in 18 pairs of variates.
FIG. 3.

Correlation of ICP values measured with cytometry versus photometry (r2 = 0.894; P < 0.0001).
The significant linear correlation between the two different methods shows the applicability of flow cytometry to the short ICP method. The chemical substance CF-DA in combination with flow cytometry appears to be appropriate for identifying ICP. Finally, the correlation was independent of growth phase.
Stress and storage experiments.
To demonstrate the wide range of ICP detection, further experiments were carried out with stressed yeast (Fig. 4). Exposure to heat, ethanol, and osmotic pressure was intended to result in low vitality, while storage in saline solution for 45 h was intended to imitate the lack of nutrients. Before treatment, all yeast samples were taken from the stationary-growth phase and compared to the freshly propagated stationary yeast.
FIG. 4.

ICP distributions of cytometric measurements for stationary yeast treated by different stress events, including osmotic pressure (▿), ethanol (▪), heat (⋄), and untreated stored yeast (▴). For comparison, results obtained with fresh propagated yeast (•) are also shown.
All stored and stressed yeast samples showed lower ICP values than the corresponding fresh propagated stationary-phase yeast. Osmotic pressure had the most negative effect on yeast ICP, represented by a nearly flat curve. Ethanol treatment did not have as such strong effect as osmotic pressure but left the yeast in a less vital state than heat (35°C) or storage for 2 days in saline solution. Stress by heat or lack of nutrients resulted in similar ICP values and distributions. Furthermore, these two stressors resulted in the same ICP maximum (i.e., 6.7) as freshly propagated yeast in the lag phase. The total number of counted, viable cells, however, is lower after heat stress or storage. Examining the geometric mean of the ICP distribution for the nonstressed lag-phase, heat-stressed, and stored yeast resulted in similar values. Consequently, measuring these three samples of yeast by using photometry would not lead to a differentiation even though the distribution of cell vitality measured with cytometry was evident.
DISCUSSION
Propagations.
At the beginning of the propagations in this study, yeast was in a lag phase. Adaptation to the media had not yet been completed after 3 h. The yeast had therefore low vitality, as represented by low ICP values. After 14 h of propagation, yeast was in exponential growth phase, and the adaptation to the environment was complete. During this phase, the yeasts' proliferation activity and growth is at maximum, and the ATPase of the cell extrudes more protons to maintain a higher ICP than in other growth states. Enzymes catalyzing proliferation and growth reactions have their optimum at higher ICP values (23); hence, they work better and reactions in the cells run faster in the exponential phase than in other growth states. Thus, yeast is in the most vital state during the exponential phase, which is demonstrated by high ICP values (1). Our findings here confirm this observation.
There was no further increase of cell density after 26 h of propagation marking the end of yeast growth and attainment of the stationary phase. The broad distribution in the population's ICP was indicative of the yeast dividing into two subpopulations in terms of differences in vitality. These results support those of Valli et al. (35), who reported that yeast from the stationary phase divides into two subpopulations. The subpopulation with the low ICP was a victim of the incipient lack of nutrition during the end of propagation. These cells had low vitality similar to lag-phase cells and were potentially dying. With incipient cell lyses, nutrition returned to the media and was assimilated by other yeast cells. The second subpopulation of yeast cells were profiting from the starving cells. This subpopulation maintained high ICP values close to those from exponential yeasts. Valli et al. (35) also reported that exposure to low-pH buffer caused a quick ICP drop in stationary-phase yeast cells, while exposure of exponential yeast to the same buffer caused much slower ICP drops. This result affirms the hypothesis that proton pumps of exponential yeast cells must work better, thereby maintaining higher ICP, which in turn yields higher reaction rates in exponential-phase yeast metabolism.
Method correlation.
In the present study, we successfully demonstrated that a yeast population's ICP can be measured with flow cytometry in combination with the probe CF-DA. The main benefit to this new method was the ability to visualize the distribution in ICP for a population of yeast. With the flow cytometric measurements, it was possible to distinguish between all three growth phases during propagation. In contrast to the lag and exponential phases, stationary-phase yeast displayed a broader ICP distribution. Lag-phase yeast possessed a considerably lower vitality than the other two growth phases and was distinguishable from the other two phases, regardless of the measurement method. With photometry it was not possible to distinguish between exponential and stationary phases. The low ICP subpopulation of stationary-phase yeast could only be discovered when each single cell was captured and analyzed during flow cytometric measurements. Valli et al. (35) could also detect subpopulations with flow cytometer, but in combination with the stain SNARF-4F. This stain is a commonly used for flow cytometry, whereas CF or fluorescein has been established primarily for photometric analysis.
The correlation between the two methods exhibited a significantly high coefficient of determination (P < 0.0001), implying the transferability of fluorescence photometry ICP measurements to flow cytometry irrespective of the growth phase. Furthermore, flow cytometry produced more reproducible results, as indicated by substantially lower coefficients of variation among replicate propagations in comparison to photometry.
ICP values gained with cytometry.
The calibration with Superdex 200 particles promises to be appropriate as a universal calibration procedure for flow cytometry. Differences between yeast cells and Superdex 200 particles led no difference in fluorescence detection. The stain displayed the same fluorescence characteristics (intensity, excitation/emission wavelength) from dye-impregnated Superdex 200 particles as in the yeast cytoplasm. Visser et al. (37) used Sephadex G-25 beads for calibration and could emulate cells well. They found that fluorescein bounded to Sephadex beads had no significant difference in excitation spectra compared to free fluorescein. The wavelength shift caused by linkage was not greater than ∼10 nm higher.
The use of Superdex 200 particles also eliminated the need for exogenous cell permeabilizing agents for in-situ calibration. Other researchers have used the antifungal drug amphotericin B (35, 36) or nigericin, an antibiotic (8, 39), to permeabilize the cell membrane to facilitate dye uptake for flow cytometry calibration. In these instances, cells had to be treated with these substances in high K+ buffers to equilibrate internal and external pH by changing the internal K+ with external H+. Beyond the obvious potential artifacts of using cell permeabilizing agents, the use of strains other than that ultimately being examined may cause artifacts.
Upon comparing the results between photometry and flow cytometry, it was apparent that higher ICP values were achieved with flow cytometry. Our photometric data display a range between 5.7 and 6.2. It should be mentioned here that glycolytic enzymes usually have their working optimum at higher ICP values of around pH 7 (23). Low photometric ICP values could be an artifact resulting from movement of the dye from within the cells to the pH 3 buffer used during measurement. It is known that fluorescent dye is often released from cells into external buffer (5-7). Consequently, one part of the dye is located in the buffer external to the yeast cells, whereas the other part is within the yeast cells. Photometry captures the complete fluorescence of yeast and buffer, producing a composite value of both. The CF dye in a low-pH 3 buffer will decrease the actual ICP value since it has low fluorescence in a low-pH environment. Since flow cytometry measures only the fluorescence of the yeast cells, the resulting ICP is not confounded by the fluorescent signal of low pH buffer. This is one of the main differences between photometry and cytometry for ICP measurements.
Breeuwer et al. (5) reported that CF is well retained in cells of S. cerevisiae, which are stored on ice. At higher temperatures (30°C) the efflux of the stain increases rapidly. The efflux is also dependent on the external pH: the lower the pH, the faster it leaves the cell. In later experiments (7), these authors reported that in cells where glucose was added to the extracellular medium CF was completely translocated to the vacuoles in about 20 min, and in about 1 h it was exported to the extracellular medium. Imai and Ohno (21), which used the same CF stain, reported that this compound cannot easily enter the cell organelles and proved this with fluorescence microscopy. Based on these two published finding, we feel there was little risk for the dye to enter the yeasts' vacuoles and since there is also no carbon source in the buffer used for flow cytometry. The efflux of CF was reduced, because we conducted the sample preparation at very low temperatures to enhance the stain retention. Centrifugation was performed at 4°C, and during further sample preparation time yeast was kept on ice or ice-cold buffer was used. Nevertheless, ICP measurements were performed at room temperature, where stain extrusion rose. A quick detection of sample fluorescence was important to avoid errors, our measurements were completed within one minute introducing the cells to the cytometer.
A final important aspect that must be considered when comparing the two ICP measurement techniques are the device settings of the flow cytometer. All ICP values measured with flow cytometry are not absolute because the settings of the signal amplification must be adjusted to achieve the best signal. The combination of the voltage signal amplification for each wavelength determines the location of the maximum peak, and this can be dramatically shifted by adjusting these settings. Furthermore, the signal-to-noise ratio differs for each and every flow cytometer, and thus device settings are machine-specific. We tested the optimal settings with different stress experiments and in combination with comparative photometer measurements to find a voltage signal amplification setting with a strong signal-to-noise ratio.
Stress and storage experiments.
Stress experiments were performed only to show the range of detectable ICP values measurable by flow cytometry. Nonvital yeast populations were produced by different stressing conditions, and their ICP was measured by flow cytometry. With yeast populations having high vitality, the range of ICP values obtained from flow cytometry ranged from pH 6.2 to 7.2. With stress populations, the range expanded such that values as low as pH 5.2 were obtained. This broadening of the range of ICP values within the population was only observable with the flow cytometer and not with the fluorescence photometer.
A range of ICP values was produced with the various stressing conditions. Yeast stored for 45 h in saline buffer had a significant loss of vitality. Imai proposed a maximum storing time of 24 h in saline solution at low temperature to minimize the loss of vitality. Storage for nearly twice as long resulted in a significant loss of vitality, which can be explained by the lack of nutrition, slowing of yeast metabolism, and lowering ATPase activity and thus lower ICP (1). With additional heat treatment vitality was not significantly lower. The light heat shock for 3 h to 35°C was meant to initiate a heat stress response (11, 34) and thus expected to produce a more significant loss of vitality. Storage in ethanol affected yeast vitality to a greater extent. Ethanol is a cytotoxin which permeabilizes the cell membrane and disturbs metabolic transport systems (25, 26). A high sorbitol concentration in extracellular medium provoked osmotic pressure (2, 28, 29) and caused the greatest loss of vitality and death. This may be due to the longer exposure to stress in comparison to other stress events.
In conclusion, the transfer of an ICP method for determining yeast vitality from fluorescence photometry to flow cytometry was implemented successfully. Differentiation between exponential and stationary phases during propagations could be achieved with cytometric measurements but not with photometric measurements. Moreover, we demonstrated the ability to detect less vital yeast subpopulations in the stationary phase with cytometry that were not observable with photometry. Since the time for sample preparation and measurement was the same for both detection systems, the more detailed information about the distribution of ICP values within a population coupled with greater precision makes flow cytometry a more useful tool for assessing the health of a yeast population.
Acknowledgments
We thank Silke Jährig, Saskia Henniger, and Angelika Gebauer from the Berlin Sugar Institute for their help with the laboratory work and allocation of the photometer and cytometer.
This study was supported by the Bundesministerium für Wirtschaft und Technologie, Inno-Watt, grant IW061204.
Footnotes
Published ahead of print on 6 July 2009.
REFERENCES
- 1.Back, W., T. Imai, C. Forster, and L. Narziss. 1998. Hefevitalität und Bierqualität. Monatsschrift. Brauwissenschaft. 11/12:189-195. [Google Scholar]
- 2.Blomberg, A. 2000. Metabolic surprises in Saccharomyces cerevisiae during adaptation to saline conditions: questions, some answers and a model. FEMS Microbiol. Lett. 182:1-8. [DOI] [PubMed] [Google Scholar]
- 3.Boulton, C., W. Box, D. Quain, and S. Molzahn. 2001. Vicinal diketone reduction as a measure of yeast vitality. Tech. Q. Master Brew. Assoc. Am. 2:89-93. [Google Scholar]
- 4.Bracey, D., C. D. Holyoak, G. Nebe-von Caron, and P. J. Coote. 1998. Determination of the intracellular pH (pHi) of growing cells of Saccharomyces cerevisiae: the effect of reduced-expression of the membrane H+-ATPase. J. Microbiol. Methods 31:113-125. [Google Scholar]
- 5.Breeuwer, P., J. L. Drocourt, F. M. Rombouts, and T. Abee. 1994. Energy-dependent, carrier-mediated extrusion of carboxyfluorescein from Saccharomyces cerevisiae allows rapid assessment of cell viability by flow cytometry. Appl. Environ. Microbiol. 60:1467-1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Breeuwer, P., J. L. Drocourt, N. Bunschoten, M. H. Zwietering, F. M. Rombouts, and T. Abee. 1995. Characterization of uptake and hydrolysis of fluorescein diacetate and carboxyfluorescein diacetate by intracellular esterases in Saccharomyces cerevisiae, which result in accumulation of fluorescent product. Appl. Environ. Microbiol. 61:1614-1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Breeuwer, P., and T. Abee. 2000. Assessment of the intracellular pH of immobilized and continuously perfused yeast cells employing fluorescence ratio imaging analysis. J. Microbiol. Methods 39:253-264. [DOI] [PubMed] [Google Scholar]
- 8.Chow, S., D. Hedley, and I. Tannock. 1996. Flow cytometric calibration of intracellular pH measurements in viable cells using mixtures of weak acids and bases. Cytometry A 24:360-367. [DOI] [PubMed] [Google Scholar]
- 9.Cimprich, P., J. Slavík, and A. Kotyk. 1995. Distribution of individual cytoplasmic pH values in a population of the yeast Saccharomyces cerevisiae. FEMS Microbiol. Lett. 130:245-251. [DOI] [PubMed] [Google Scholar]
- 10.Dinsdale, M. G., D. Lloyd, P. McIntyre, and B. Jarvis. 1999. Yeast vitality during cider fermentation: assessment by energy metabolism. Yeast 15:285-293. [DOI] [PubMed] [Google Scholar]
- 11.Ferguson, S. B., E. S. Anderson, R. B. Harshaw, T. Thate, N. L. Craig, and H. C. M. Nelson. 2005. Protein kinase A regulates constitutive expression of small heat-shock genes in an Msn2/4p-independent and Hsf1p-dependent manner in Saccharomyces cerevisiae. Genetics 169:1203-1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fernández, S., G. González, and A. Sierra. 1991. The acidification power test and the behavior of yeast in brewery fermentations. Tech. Q. Master Brew. Assoc. Am. 28:89-95. [Google Scholar]
- 13.Fritsch, M., J. Starruß, A. Loesche, S. Mueller, and T. Bley. 2005. Cell cycle synchronization of Cupriavidus necator by continuous phasing measured via flow cytometry. Biotechnol. Bioeng. 92:635-642. [DOI] [PubMed] [Google Scholar]
- 14.Hewitt, C. J., and G. Nebe-Von-Caron. 2001. An industrial application of multiparameter flow cytometry: assessment of cell physiological state and its application to the study of microbial fermentations. Cytometry 44:179-187. [DOI] [PubMed] [Google Scholar]
- 15.Hutter, K.-J. 2002. Flow cytometry: a new tool for direct control of fermentation processes. J. Inst. Brew. 108:48-51. [Google Scholar]
- 16.Hutter, K. 2002. Flow cytometric determinations of glycogen content of yeast during fermentation. J. Inst. Brew. 108:52-53. [Google Scholar]
- 17.Hutter, K., and F. Nitzsche. 2002. Untersuchungen über die Alterung der Bierhefen mit Hilfe der flusszytometrischen Analyse. Monatsschrift. Brauwissenschaft. 9/10:196-199. [Google Scholar]
- 18.Hutter, K., C. Kliem, F. Nitzsche, and M. Wießler. 2003. Biomonitoring der Betriebshefen in praxi mit fluoreszenzoptischen Verfahren. IX. Mitteilung: Trehalose-Stressprotektant der Saccharomyces-Hefen. Monatsschrift. Brauwissenschaft. 7/8:121-125. [Google Scholar]
- 19.Imai, T., I. Nakajima, and T. Ohno. 1994. Development of a new method for evaluation of yeast vitality by measuring intracellular pH. J. Am. Soc. Brew. Chem. 52:5-8. [Google Scholar]
- 20.Imai, T., and T. Ohno. 1995. Measurement of yeast intracellular pH by image processing and the change it undergoes during growth phase. J. Biotechnol. 38:165-172. [DOI] [PubMed] [Google Scholar]
- 21.Imai, T., and T. Ohno. 1995. The relationship between viability and intracellular pH in the yeast Saccharomyces cerevisiae. Appl. Environ. Microbiol. 61:3604-3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kara, B. V., W. Simpson, and J. R. M. Hammond. 1988. Prediction of the fermentation performance of brewing yeast with the acidification power test. J. Inst. Brew. 94:153-158. [Google Scholar]
- 23.Madshus, I. H. 1988. Regulation of intracellular pH in eukaryotic cells. Biochem. J. 250:1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Müller, S., K.-J. Hutter, T. Bley, L. Petzold, and W. Babel. 1997. Dynamics of yeast cell states during proliferation and nonproliferation periods in a brewing reactor monitored by multidimensional flow cytometry. Bioprocess Biosyst. Eng. 17:287-293. [Google Scholar]
- 25.Nagar-Legmann, R., and P. Margalith. 1986. A comparative study of the lipid composition of yeasts with different fermentative capacities. Appl. Environ. Microbiol. 26:49-54. [Google Scholar]
- 26.Nagodawithana, T. W., and K. H. Steinkraus. 1976. Influence of the rate of ethanol production and accumulation on the viability of Saccharomyces cerevisiae in “rapid fermentation.” Appl. Environ. Microbiol. 31:158-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peddie, F. L., W. J. Simpson, B. V. Kara, S. C. Robertson, and J. R. M. Hammond. 1991. Measurement of endogenous oxygen uptake rates of brewers' yeast. J. Inst. Brew. 97:21-25. [Google Scholar]
- 28.Reed, R. H., J. A. Chudek, R. Foster, and G. M. Gadd. 1987. Osmotic significance of glycerol accumulation in exponentially growing yeasts. Appl. Environ. Microbiol. 53:2119-2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rep, M., M. Krantz, J. M. Thevelein, and S. Hohmann. 2000. The transcriptional response of Saccharomyces cerevisiae to osmotic shock. J. Biol. Chem. 275:8290-8300. [DOI] [PubMed] [Google Scholar]
- 30.Rodrigues, P. G., A. A. Barros, J. A. Rodrigues, A. A. Ferreira, C. Gonçalves, and J. R. M. Hammond. 2004. Vital titration: a new method for assessment of yeast vitality. Tech. Q. Master Brew. Assoc. Am. 41:277-281. [Google Scholar]
- 31.Slavík, J. 1982. Intracellular pH of yeast cells measured with fluorescent probes. FEBS Lett. 140:22-25. [DOI] [PubMed] [Google Scholar]
- 32.Smart, K. 2003. Brewing yeast fermentation performance, 2nd ed. Blackwell Publishers, Oxford, United Kingdom.
- 33.Thiele, F., and W. Back. 2005. Measurement of yeast vitality using a modified version of the intracellular pH measurement (ICP). Monatsschrift. Brauwissenschaft. 1/2:2-5. [Google Scholar]
- 34.Trotter, E. W., L. Berenfeld, S. A. Krause, G. A. Petsko, and J. V. Gray. 2001. Protein misfolding and temperature up-shift cause G1 arrest via a common mechanism dependent on heat shock factor in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 98:7313-7318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Valli, M., M. Sauer, P. Branduardi, N. Borth, D. Porro, and D. Mattanovich. 2005. Intracellular pH distribution in Saccharomyces cerevisiae cell populations, analyzed by flow cytometry. Appl. Environ. Microbiol. 71:1515-1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Valli, M., M. Sauer, P. Branduardi, N. Borth, D. Porro, and D. Mattanovich. 2006. Improvement of lactic acid production in Saccharomyces cerevisiae by cell sorting for high intracellular pH. Appl. Environ. Microbiol. 72:5492-5499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Visser, J. M., A. A. M. Jongeling, and H. J. Tanke. 1979. Intracellular pH-determination by fluorescence measurements. J. Histochem. Cytochem. 27:32-35. [DOI] [PubMed] [Google Scholar]
- 38.Wheatcroft, R., Y. H. Lim, D. B. Hawthorne, B. J. Clarke, and T. E. Kavanagh. 1988. An assessment of the use of specific oxygen uptake measurements to predict the fermentation performance of brewing yeast. Proc. Int. Conv. Inst. Brew. 20:193-199. [Google Scholar]
- 39.Wieder, E. D., H. Hang, and M. H. Fox. 1993. Measurement of intracellular pH using flow cytometry with carboxy-SNARF-1. Cytometry Part A 14:916-921. [DOI] [PubMed] [Google Scholar]