

Abstract
Fluorescence microscopy and intensity histogram analysis techniques were used to monitor spatially-resolved albumin adsorption kinetics to model heterogeneous surfaces on sub-μm scales. Several distinct protein subpopulations were resolved, each represented by a normal distribution of adsorption densities on the adsorbent surface. Histogram analyses provided dynamic information of mean adsorption density, spread in adsorption density, and surface area coverage for each distinct protein subpopulation. A simple adsorption model is proposed in which individual protein binding events are predicted by the summation of multiple protein's surface sub-site interactions with different binding energy sub-sites on adsorbent surfaces. This model is predictive of the albumin adsorption on the patterns produced by one step μ-contact printing (μCP) of octadecyltrichlorosilane (OTS) on glass but fails to describe adsorption once the same patterns are altered by a thermal annealing step.
Keywords: adsorption kinetics, albumin, surface heterogeneity, fluorescence imaging
Introduction
Proteins are surface-active molecules, which concentrate at interfaces by so-called »non-specific« adsorption. This general observation is supported by a large body of experiments and thousands of published papers.1–5 Almost any material, when exposed to a physiological, protein-containing solution, becomes coated with proteins within seconds. For example, plasma protein adsorption on surfaces of artificial materials is considered to be the principal means by which a material becomes thrombogenic.6 Biomaterial's interface with blood is the locality where a number of processes concurrently proceeds including surface–proteins–cells interactions. From an experimentalist's point of view, one is faced with a problem of defining the most important processes and finding methods to study them. Many past studies were limited to one protein or a smaller selection of plasma proteins adsorbing from (diluted) plasma onto a »model« biomaterial.3,4,7–13 In addition, a number of assumptions, such as monolayer adsorption coverage, uniform surface energy and protein-surface affinity, and protein–protein surface exchange, were typically introduced to manage the experimental complexity of such systems.
In contrast to the »nonspecific« protein-surface interactions, interactions between cells and proteins or surface-adsorbed proteins primarily proceed via specific molecular recognition mechanisms. Molecular recognition is one of the basic tenets of biology. The unique combination of salt bridges, hydrogen bonds, hydrophobic associations, and other interactions, coupled with the complementary size and shape of the interacting molecules (the so-called »lock-and-key« mechanism14,15) assure that the binding partners, ligands and receptors, will be bound for some required duration. One can imagine a ligand molecule that encounters a protein receptor in a cell membrane; a number of spatially discrete interactions are presented to the incoming molecule almost like a molecular tapestry full of fine details. When these interactions are complementary to the incoming molecule »a binding event« will occur until it is overcome and disrupted by thermal forces.
A majority of the protein binding experiments are typically made in a macroscopic fashion. Techniques such as ellipsometry, total internal reflection spectroscopy, surface plasmon resonance, quartz crystal microbalance, and Fourier transform infrared attenuated internal reflection have an inherent inability to resolve the protein adsorption at a sub-μm scale and will provide only an averaged measure of protein adsorption.16–19 As a consequence, in the cases where a rich local surface micro-structure affects protein adsorption, such macroscopic approaches will »smear-out« and/or overlook these discrete effects. Hence, the transition between nonspecific and specific protein-surface interaction can be quite elusive.
We have previously used so-called »gradient surfaces« as tools to study the effects of surface chemistry spatial distribution on protein adsorption kinetics. Based on the idea originally presented by Elwing,20 we have utilized three different surface gradients to study protein adsorption: octadecyldimethylsilyl (C18),21–23 polyethylene oxide (PEO),24 and a positively charged quaternary amine gradient.25 Using octadecyltrichlorosilane (OTS) gradient surfaces, we have previously shown that albumin adsorption increases with increasing OTS surface coverage only up to a certain limit: any increase in OTS surface coverage above 42 % neither affected albumin adsorbed amount nor its adsorption kinetics from a dilute solution.21 However, these gradients surfaces were typically 10–15 mm long and the spatial resolution information was limited to > 50 μm length scales. In an effort to increase the spatial resolution with which OTS molecules are placed on a surface, we have used in this study a μ-contact printing (μCP) technique to create spatially controlled OTS surface patterns for protein binding. By manipulating the OTS patterns in a post-stamping thermal annealing step, we have previously shown that we could control the OTS density both in each pattern and also in the gradient regions between the stamped and nonstamped areas.26 In this paper, we present the results of spatially-resolved human serum albumin (HSA) adsorption kinetics on such patterns as measured with fluorescence microscopy (FM) and the statistical analysis of binding events from the molecular recognition point of view.
Generic Model for Protein–Binding Site Recognition on Heterogeneous Surfaces
We assume that each protein binding site on a nonuniform, heterogeneous adsorbent surface may contain a discrete distribution of different molecular species (in the case of the OTS-stamped surface patterns on glass these may contain nonpolar OTS chains or negatively charged surface silanol groups). Furthermore, any local combination of surface species can, in principle, result in an average effect of attractive or repulsive interactions with the adsorbing protein molecule. Similarly, nonuniform sites are presented on a heterogeneous outside protein surface containing an arrangement of molecular moieties such as different side chains or exposed polypetide chain segments. This spatial arrangement may or may not match the character and the discrete distribution of the molecular species on the underlying adsorbent site. The extent of spatial complementarity between the discrete molecular adsorbent characteristics and the respective binding regions on the protein molecule will determine the ultimate free energy change upon binding and thus the affinity of protein for each binding site and each protein orientation. For example, albumin has several fatty acid binding regions that may also bind OTS alkyl chains.27 The exact spatial complementarity of OTS chains surface distribution to the distribution of albumin's fatty acid binding regions can result in a high affinity and strong binding (Figure 1A). These hydrophobic interactions are superimposed on all other contributions to the free energy of adsorption: repulsive or attractive electrostatic interactions, van der Waals interactions, and any steric hindrance to adsorption. In the case of a spatial mismatch between the surface OTS chains and albumin's fatty acid binding regions, binding affinity may be different. Even if the number of OTS chains at the adsorbent binding site exceeds the number of fatty acid binding regions on the albumin molecule, the resulting binding may be weaker due to steric hindrance to OTS–fatty acid binding pocket and the proximity of albumin charged residues to nonpolar OTS clusters (Figure 1B).
Figure 1.
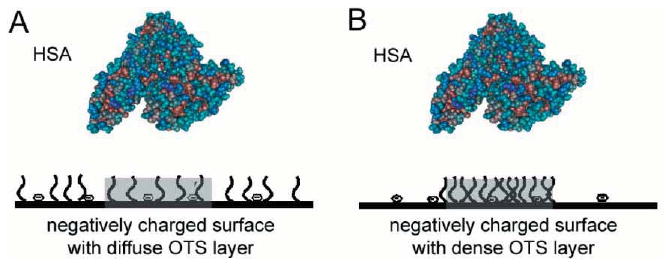
Different arrangements of surface-attached OTS chains result in different affinities for single albumin molecules. A – A spatial match of the surface-attached OTS chains and negative surface charges to the fatty acid binding regions in albumin will result in a better binding affinity. B – Clustered OTS molecules prevent the individual alkyl chains from binding into the fatty acid binding pockets in the albumin molecule. Thus, the scenario in B better mimics albumin binding to a hydrophobic, nonpolar surface.
Protein binding to a 2-D array of hydrophobic sites has been extensively studied and reviewed by Jennissen.28 Other mechanistic models for nonspecific protein adsorption have also been proposed in the literature.29–33 Our approach here is analogous to the probability model developed by Lancet et al. for molecular recognition events in biological receptor repertoires.34 The main difference is that Lancet et al. have in their model only allowed for the attractive interactions, which were assumed to be of the same magnitude, while our approach includes both positive and negative interactions thus leading to a more realistic distribution of adsorption free energies. The analogy between ligand binding to a repertoire of biological receptors as described by Lancet et al.34 and the protein binding to a nonuniform surface with heterogeneous binding sites we undertake here is obvious: a heterogeneous surface displays a repertoire of potential binding sites for protein molecule (Figure 2). Both the heterogeneous surface and the hypothetical protein are assumed to contain a set of sub-sites, denoted by symbols, »x«, »y«, and »o«, each representing a particular type of molecular interactions. If the hypothetical protein, portrayed in Figure 2 as a cube with 3 × 3 × 3 sub-sites is rotated by 180 degrees around its vertical axis and placed with its shaded face on the unshaded area in the middle of the surface (Figure 2, left panel shows a schematic of a 12 by 12 sub-site surface area), the following interacting sub-sites will be paired: xx, oy, yo, yx, yy, ox, xx, xx, yx (counting from upper left to lower right). Assuming that the interactions are additive, the local free energy of adsorption, ΔGloc is:
Figure 2.
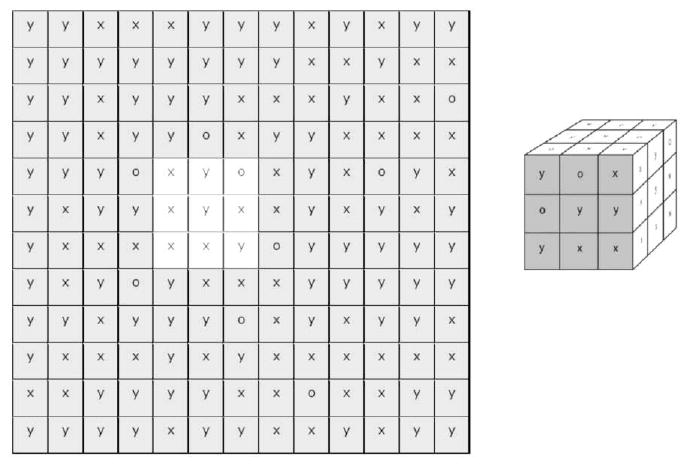
A 12 × 12 array of surface binding sub-sites (left panel) each with its discrete type of interactions (x, y, o) is sampled by a 3 × 3 array of discrete binding sub-sites on the protein molecule (right panel). The shaded 3 × 3 array on the right represents one of the several protein orientations at the interface.
| (1) |
where gi is the free energy contribution originating from the individual sub-site – sub-site interactions and m is the total number of interacting sub-site pairs. In addition to the additivity of the sub-site interactions, it is also assumed that each sub-site interaction is independent of the neighboring protein-surface paired sub-sites. Thus, for the example shown in Figure 2 ΔGloc = – (gxx + goy + gyo + gyx + gyy + gox + gxx + gxx + gyx).
To calculate the magnitude of ΔGloc, one needs to specify the rules for the interactions between x, y, and o sub-site types. One possible example of these rules is given below; other scenarios may also be envisioned. Let us assume that x, valued at 2, represents a nonpolar, hydrophobic sub-site; y, valued at −1, is a generic electrically charged sub-site; and the o site will make no final free energy contribution with any other site (i.e., its value is 0). The so-called »value« of each sub-site is in free energy units; its magnitude could, for example, amount to a kBT. In such a case, the sub-site – sub-site interaction xx, with its interaction contribution of 4 (i.e., 2 × 2), may represent the spatial complementarity of two hydrophobic sub-sites and will result in a free energy contribution leading to an attractive interaction. Similarly, xy and yx, with their interaction values of −2, may represent spatial interaction mismatch in which a charged site (positive or negative) is brought in the neighborhood of a nonpolar hydrophobic sub-site, thus leading to repulsion. Finally, the yy combination, with its interaction value of 1, represents attractive electrostatic contribution to ΔGloc.
In order to calculate the distribution of all potential local values of ΔGs and thus the distribution of potential affinities for a particular surface and a given protein orientation, one needs to compute each local ΔG by sampling all possible surface sub-sites with an array of protein sub-sites. The process of sampling each surface sub-site and its (m − 1) neighbors by an array of protein sub-sites is equivalent to a discrete convolution between an adsorbent surface array and protein surface array, with the resulting value of ΔGloc being assigned to the position of the central sub-site.35 The process is then repeated for the next sub-site position until the entire adsorbent surface array is sampled. For example, the sampling of all potential interactions on a 12 × 12 array of surface sub-sites in Figure 2 by a 3 × 3 array of protein sub-sites will give 121 potential ΔGloc values [i.e., (12 − 1) × (12 − 1)].
The Sampling of Potential ΔGloc on Simulated OTS Patterns
An example of the surface-protein sub-site array convolution that is more pertinent to HSA adsorption onto μCP OTS patterns is shown in Figure 3. In the preceding paper, we measured the average fraction of OTS surface coverage in stamped and nonstamped regions before and after an annealing step.26 The OTS surface coverage in the nonannealed μCP pattern was 14 % in the nonstamped areas and 44 % in the stamped lanes. In the annealed μCP sample, the average OTS surface coverage increased to 25 % and 74 % for the nonstamped and stamped areas, respectively.26 To simulate the heterogeneity of these surface regions, we generated a randomly distributed 256 × 256 array of binding sub-sites that, on average, had the desired surface fractions of OTS sub-sites. Figure 3A (top panels) represents a random distribution of sub-sites in a simulated nonannealed OTS μCP pattern. The top left image in Figure 3A is a randomly generated array representing the nonstamped, nonannealed area with 14 % OTS sub-sites (represented by white pixels, x = 2), 76 % of charged glass sites (represented by black pixels, y = −1) and 10 % of inert sites (represented by gray pixels, o = 0), respectively. The top right image in Figure 3A is a random array representing the stamped, nonannealed band containing 44 % OTS sub-sites (x = 2, white pixels), 46 % charged glass sites (y = −1, black pixels), and 10 % inert sites (o = 0, gray pixels). The single protein orientation is represented by a 9 × 9 array (middle panel, magnified fourfold for details) containing 23 nonpolar sub-sites (white, x = 2), 8 charged sub-sites (black, y = −1) and 50 gray sub-sites (o = 0).
Figure 3.
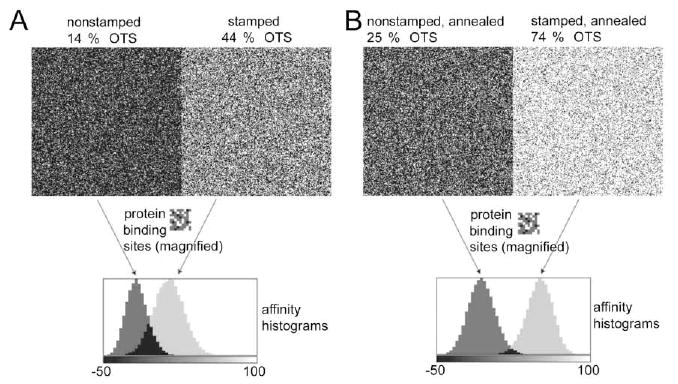
Convolution of random sub-site distributions in the simulated OTS patterns and hypothetical protein sub-sites array. A – Top left image is a randomly generated array representing a nonstamped, nonannealed OTS area. Top right image in A is a random array representing a stamped, nonannealed OTS band. B – Top left image is a random array representing a nonstamped, annealed OTS area. Top right image in B is a random array representing a stamped, nonannealed OTS band. The hypothetical protein orientation is represented by a 9 × 9 array (middle panels in A & B, magnified 4× for details). Normalized histograms show the distribution of potential −ΔGloc values resulting from the convolution for each of the four simulated OTS areas with single protein orientation shown in middle panels.
After convolving these surface and protein sub-site arrays, the local −ΔGloc values from the middle area (200 × 200 pixels) were ranked in the form of normalized histograms (lower panel, Figure 3A). The distribution of the potential −ΔGloc values ranged from −50 to 76 interaction units (Table I). Note that in this example the negative values indicate positive free energy change and consequently should result in no protein binding. One also finds an overlap between the two histograms; some binding sites in the nonstamped and stamped areas are likely to produce identical affinities for this single hypothetical protein orientation.
TABLE I.
ΔGloc distribution results
| Area | Min | Max | Mean | Mode | FWHM |
|---|---|---|---|---|---|
| »Stamped, nonannealed« | −45 | 76 | 13.0 | 14.5 | 34.5 |
| »Nonstamped, nonannealed« | −50 | 35 | −18.1 | −18.5 | 25.0 |
| »Stamped, annealed« | −15 | 94 | 49.6 | 50.5 | 29.3 |
| »Nonstamped, annealed« | −50 | 48 | −6.8 | −6.5 | 30.2 |
It is instructive to compare the nonannealed OTS simulation results with results for the annealed OTS patterns simulation (Figure 3B). The −ΔGloc histograms for the identical protein array and simulated annealed OTS μCP patterns were different than for the nonannealed patterns both in the position and in width, as well as in the extent of overlap between the simulated nonstamped (Figure 3B, upper left) and stamped (Figure 3B, upper rigth) OTS regions.
The Implication of −ΔGloc Distribution on Protein Adsorption Kinetics
The model that relates the distribution of all potential binding free energies to HSA adsorption kinetics depends on how −ΔGloc is related to the adsorption and desorption rate constants. Namely, −ΔGloc is related to the local association constant, Kloc:
| (2) |
which, in turn, is defined by the local adsorption and desorption rate constants:
| (3) |
Although the 256 × 256 array of sub-sites contains 2552 potential binding sites, the actual protein binding capacity of the same array for the hypothetical 9 × 9 array protein is much less since only up to 282 molecules can bind there in a close-packed monolayer. In reality, this number will be further diminished due to steric exclusion, jamming, and other limiting factors.
For the comparison with the experimental results we recognize that 282 closed-packed albumin molecules with an average size of 16 nm2, will fit onto the area of 111 by 111 nm2 which is, as it will be shown below, approximately the size of the surface area imaged by a single CCD camera element (so-called »pixel«) in the protein adsorption experiments.
The adsorption rate observed at any position by a single CCD camera pixel, dΓpix / dt, is assumed to follow simple binding model:13
| (4) |
where c(0,t) is the protein solution concentration adjacent to the area of the adsorbent surface observed by a single pixel of CCD camera, kon,pix and koff,pix are the average adsorption and desorption rate constants for the same area, Γpix is the adsorbed amount per pixel area and is the maximal adsorption per pixel area thus making the , average surface coverage of adsorbed protein in the area observed by the pixel of CCD camera.
In principle, kon,pix and koff,pix parameters are experimentally accessible for each pixel position and c(0,t) can be related to the bulk protein concentration by accounting for the transport of the protein molecules through the concentration boundary layer (so-called »unstirred« layer).8,13 However, interpreting the experimental adsorption rate constants in terms of actual −ΔGloc distribution remains a challenge. One may assume that the distribution of −ΔGloc, taken over the area observed by a pixel of CCD camera for all possible protein orientations, is representative of the distribution of kBT ln kon,loc for protein binding at low-to-intermediate surface coverages. This leads to a general conclusion that the local surface areas with higher affinity binding sub-sites will adsorb protein faster than the areas with predominantly lower affinity sub-sites.
The sub-site convolution and histogram approach do not account for the blocking of neighboring sub-sites by the adsorbed protein. Namely, −ΔGloc distributions shown above are computed for each and every sub-site. Once the protein molecule binds to a higher affinity binding site, it will not occupy only the central sub-site (for which the −ΔGloc was computed) but also numerous outside sub-sites. For example, for the model in Figure 3 protein binds to 1 central and 80 outside sub-sites. Accordingly, when the two high affinity binding sites are separated by less than sub-sites, occupation of one site will prevent the second binding site to be filled. Hence, not all high −ΔGloc values can be realized in experiments. Another limitation may be due to slow desorption: adsorption to any site may result in a transient coverage possessing sufficiently long lifetime that will effectively prevent binding to any site less than sub-sites away. In other words, adsorption history matters as it influences the adsorption outcome.
Experimental
Methods and protocols used to prepare and characterize the μCP OTS patterns have been fully described in the preceding paper.26 Here, we repeat only the most important points and focus on protein adsorption methods.
Microcontact Printing
Poly(dimethylsiloxane) (PDMS) stamps containing a 3 × 3 mm2 area patterned with 5 μm wide, 1540 nm high, 3 mm long mesas separated by 5 μm wide troughs, and stamp pads were used to produce OTS patterns through micro-contact printing.36 Upon stamping the glass coverslip (thickness #1), the OTS patterns were either immediately rinsed with hexane to remove any unreacted OTS and then sonicated in an ethanol-double-distilled water (1:1 v/v, mole fraction) mixture, or first annealed followed by hexane rinsing and an ethanol-water sonication step. Positive controls of both nonannealed and annealed OTS films were produced using smooth, planar stamps. Clean glass cover-slip surfaces served as negative controls.
OTS Pattern Characterization
Advancing water contact angles were measured on the OTS-patterned substrates using the sessile drop method to ascertain the average OTS coverage for each of the micro-patterned areas.26 OTS coverages for patterned areas not directly accessible to the sessile drop method were calculated using the Cassie equation.37 An SFM instrument (Explorer, Topometrix) was used to map friction forces using lateral force microscopy mode (LFM). An OTS-modified SFM tip was employed in all measurements.
Protein Adsorption Experiments
Human serum albumin (HSA; ICN, Fraction V Fatty Acid Free) was labeled with Alexa Fluor 488 (AF488, Molecular Probes, Eugene, OR) and then isolated by gel permeation chromatography and purified through dialysis. UV/Vis spec-trophotometry was used to determine the degree of labeling and protein concentration in the final AF488-HSA solution.17 Fluorophore conjugation reaction resulted in a degree of labeling of 0.83 mol AF488 per mol of HSA. The final AF488-HSA solution was diluted to a concentration of 5 μg/mL in 10 mmol dm–3 phosphate buffered saline (PBS, pH = 7.4, ionic strength 0.165 mol dm–3), divided into small vials, stored at −20 °C and thawed immediately prior to each adsorption experiment. The same 5 μg/mL AF488--HSA stock solution was used for all adsorption experiments.
The adsorption flow cell was designed to enable realtime fluorescence imaging of protein adsorption onto OTS patterned glass coverslips from a flowing AF488-HSA solution. The flow cell contained a microscope objective accessible opening on the side of the sample to enable imaging of the adsorbed layer. Experiments were conducted on the stage of an upright fluorescence microscope (Nikon Eclipse E400) at room temperature.
The flow cell was initially filled with PBS to acquire a background image of the sample surface. PBS in the inflow tubing was replaced with AF488-HSA solution immediately prior to beginning each adsorption experiment. A fast flow rate (2 mL/min) of protein solution (or PBS in the desorption segment of each experiment) was used during the first 30 seconds to bring the solution to the observation area and to limit mixing and dilution effects. The flow was reduced to 0.1 mL/min thereafter.
Fluorescence images of adsorbed AF488-HSA on the OTS-modified and control coverslips were recorded using a 100×, 1.25 N.A., oil immersion objective (Leitz) in the epi-fluorescence configuration and a 12-bit Peltier-cooled CCD camera (MicroMax, Princeton Instruments). Neutral density filters served to attenuate the excitation light in order to minimize photobleaching of the AF488 fluorophore and an exposure time of 4 seconds was used to reduce the short term effects of temporal and spatial fluctuations in the excitation light source (75 W Xenon lamp). An appropriate set of excitation band-pass, dichroic mirror and emission band-pass filters (Omega) for the AF488 fluorophore was used to separate the excitation and emission photons.
During each adsorption experiment, the fluorescence intensity at any given pixel position was composed of in-focus fluorescence emission from the adsorbed layer, IAds, and out-of-focus fluorescence emanating from protein solution in the flow cell, ISol. The surface concentration of surface bound AF488-HSA, ΓHSA, was calculated by finding the optical relationship between the two fluorescence components, IAds and ISol. The conversion from IAds and ISol to ΓHSA used here parallels the quantification of protein adsorbed amount in total internal fluorescence spectroscopy13,21 and is outlined in the preceding paper.26 Each fluorescence image was also corrected for long-term variations in the excitation light intensity caused by Xenon arc-lamp aging and possible variations in the microscope optical alignment, nonuniformity in the excitation illumination, and for fluorescence background originating from solution fluorescence, scattered light, and electronic CCD readout.26 The error in the measurement of the adsorbed HSA surface density as represented by pixel intensities was ± 3.5 % as estimated from the standard deviations of protein fluorescence intensity. A linear relationship between the measured fluorescence intensity and the adsorbed HSA surface density was assumed and not checked experimentally. This assumption was justified based on the previous quantification of fluorescein-labeled HSA adsorption using radiography and mass transfer-limited adsorption processes.21,22
A multipeak fitting software (Igor Pro v4.05A, Wave-metrics) was used to deconvolve HSA surface density (ΓHSA) histograms for the various OTS patterns and controls into normal distributions.
Results
Protein Adsorption
Monitoring protein adsorption as a function of time using fluorescence imaging provided a wealth of information about the adsorption process and how it relates to microheterogeneities in the OTS films. Figure 4 displays time-sequence fluorescence images of AF488-HSA accumulation on typical nonannealed (Figure 4A) and annealed (Figure 4B) OTS patterns. AF488-HSA adsorption proceeded at a faster rate and reached steady state sooner on stamped bands than on the nonstamped areas for both the annealed and nonannealed patterns. As expected, AF488-HSA adsorbed at a higher adsorption density on the more hydrophobic areas in the nonannealed pattern. Somewhat surprisingly, however, the adsorption process resulted in a final higher surface density of AF488-HSA on the less hydrophobic nonstamped bands on the annealed pattern. The contrast in the fluorescence images for the annealed pattern switched at about 12 minutes into the adsorption process due to the capacity of the nonstamped bands to adsorb more protein than the stamped bands.
Figure 4.

Time sequence fluorescence image montage of AF488-HSA adsorption onto OTS Patterns. A – Nonannealed pattern. The images display the same surface area (33.6 by 30.8 μm2) imaged up to 4800 seconds of adsorption. The stamped OTS band is located at the top of each image. Adsorption time in seconds is in each image. B – Same for the annealed pattern.
Figures 5A and 5B show the AF488-HSA adsorption density distribution evolution for the time-sampled images presented in Figures 4A and 4B. Each image pixel intensity was first converted into adsorbed amount, and then the adsorbed amount histograms were constructed and plotted in 3D plots (% coverage vs. time vs. adsorbed amount). Histogram analysis showed that the temporal distribution of protein surface density on patterned substrates could be fitted to the sum of several adsorbed protein subpopulations each described with a normal distribution. Furthermore, each normal distribution possessed a unique mean density, spread in adsorption density range (defined as full width half maximum, FWHM), and subpopulation fractional area coverage (integrated area under normal distribution divided by the total histogram area). The relation between the typical protein subpopulations and their spatial positions on the pattern is represented by Figure 5C where fitted normal distributions are superimposed on histograms. Experimental distribution (Figure 5C, upper panel) was fitted well by the histograms each representing the stamped and nonstamped areas (Figure 5C, lower panel), while the fits to the histograms representing gradient areas between the stamped and nonstamped regions were less accurate.
Figure 5.
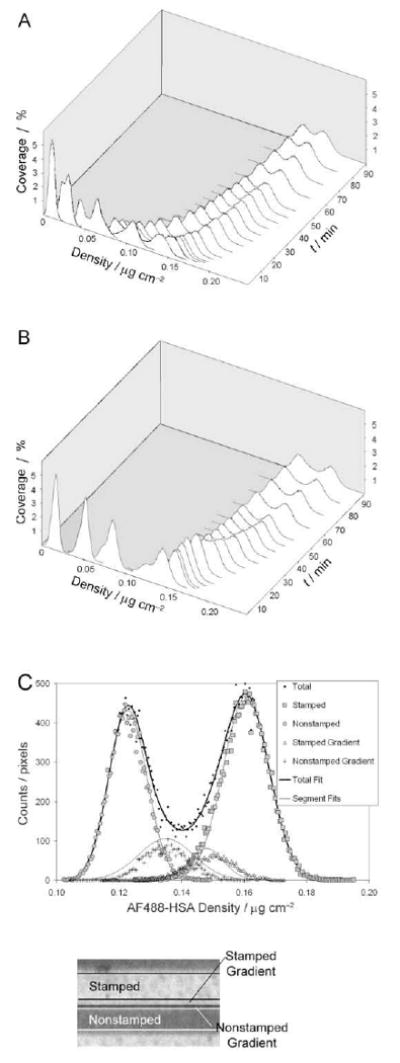
Adsorption density histogram evolution and normal distribution deconvolution. A – Evolution of the AF488-HSA density distribution on the nonannealed OTS pattern plotted as a function of time. B – Evolution of the AF488-HSA density distribution on the annealed OTS pattern plotted as a function of time. C – Deconvolution of fluorescence intensity histogram into four normal distributions. The upper panel shows the intensity histogram for the area shown in the lower panel. Solid lines in the graph represent the four normal distributions deconvolved from the total histogram data and resulting total fit using multipeak fitting software. Fitted normal distributions coincided well with the stamped and nonstamped area histograms, while the fitting algorithm was less accurate for the smaller gradient areas.
The results of the histogram analyses are displayed in graphical form for the patterned surfaces and controls in Figures 6 through 8, and 9, respectively. Each protein subpopulation exhibited unique adsorption kinetics as represented by changes in mean AF488-HSA adsorption density over time (Figure 6). The adsorption density spread (i.e., FWHM for each protein subpopulation) (Figure 7) and the fractional area of each subpopulation coverage (Figure 8) also changed with adsorption time.
Figure 6.
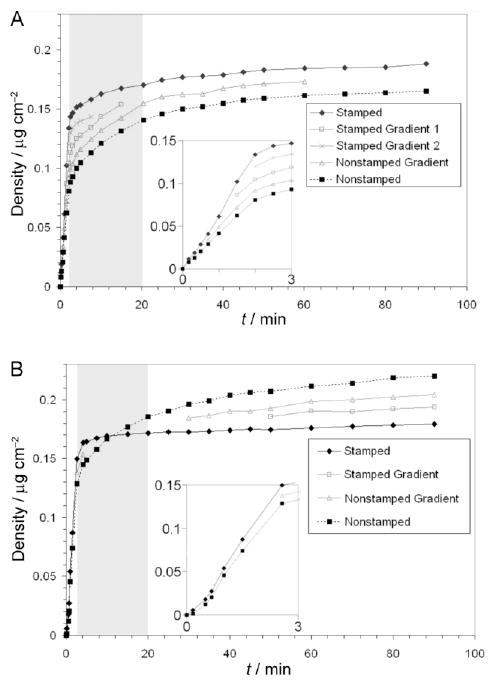
Mean AF488-HSA adsorption density kinetics on OTS patterns. A – Mean adsorption density kinetics on the nonannealed OTS pattern. B – Mean adsorption density kinetics on the annealed OTS pattern. The first 3 minutes of adsorption kinetics are inset for each sample. The grey boxed areas demark the time period during which adsorption to intermediate energy binding sites predominated.
Figure 8.
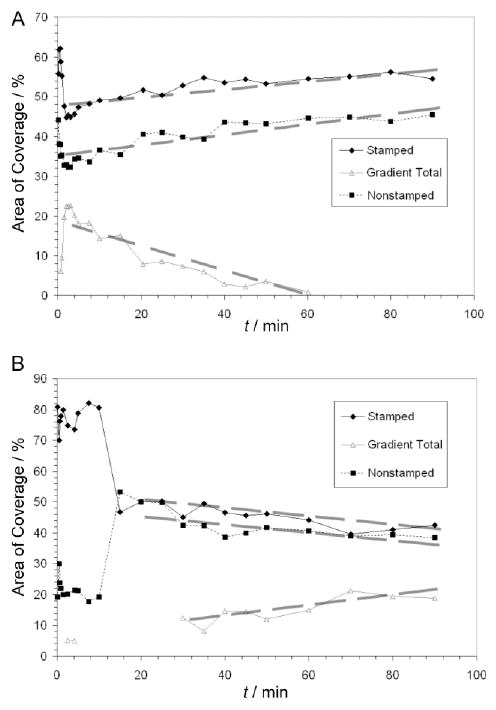
AF488-HSA subpopulation fractional coverages on OTS patterns. A – Changes in the fractional coverage of each protein subpopulation on the nonannealed OTS pattern. B – Changes in the fractional coverage of each protein subpopulation on the annealed OTS pattern.
Figure 9.
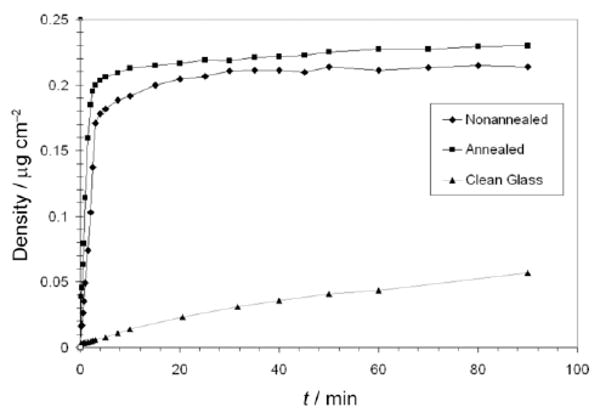
Mean AF488-HSA adsorption density kinetics on control surfaces.
Figure 7.
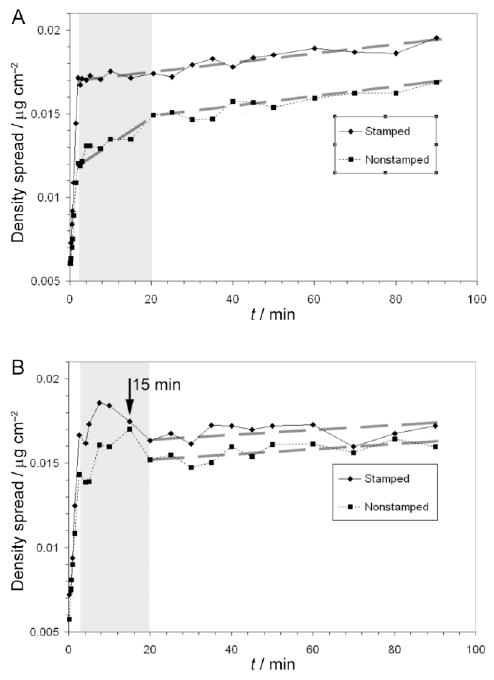
Spread of AF488-HSA adsorption density on OTS patterns. A – Changes in the spread of adsorption densities for the sub-populations on the nonannealed pattern. B – Changes in the spread of adsorption densities for the sub-populations on the annealed pattern. The grey boxed areas demark the time period during which adsorption to intermediate energy binding sites predominated.
The AF488-HSA surface densities after 90 minutes of adsorption agreed well with the literature,21 and approached the theoretical density for a full HSA monolayer of side-on orientation adsorption calculated to be 0.25 μg/cm.2,38 The fitted normal distributions were not only representative of AF488-HSA distribution throughout the whole pattern, but also accounted for the majority of fluorescence signal found in a spatially well-defined area within a given OTS pattern. Each of the stamped and nonstamped bands was represented by one protein subpopulation while the edges of stamped and nonstamped bands were represented by one or more populations depending on the type of pattern and progression of protein adsorption. As the mean density of surface AF488-HSA subpopulations increased over adsorption time, so did the adsorption density spread (Figure 6 vs. Figure 7). In the case of nonannealed OTS patterns (Figure 8A), the fractional area for the two major protein subpopulations grew at the expense of the gradient subpopulations after the initial decrease, however, in the case of annealed OTS patterns the fractional area changes were opposite (Figure 8B); the gradient subpopulations became more and more distinguishable over time.
Although each sample was subjected to 90 minutes of desorption, only a small fraction of AF488-HSA molecules desorbed (data not shown). The average desorbed amount for the OTS modified substrates was 4 % of the total adsorbed amount and was never greater than 6 % for any of the sample areas monitored. The control glass substrate, however, experienced a 10 % reduction in protein coverage over 90 minutes of desorption. Adsorption and desorption results for the control OTS surfaces were similar to those for the respective stamped areas of the patterned surfaces while the adsorbed amount on the clean glass surface reached a lower average density of 0.057 μg/cm2 after 90 minutes of adsorption (Figure 9).
The morphology of the OTS distribution on both nonannealed and annealed samples as detected by using LFM is shown in Figure 10. The histogram to the left of each image shows the distribution of friction forces for that area. Each area contained at least two distinct friction force populations that corresponded to the local areas of high and low OTS surface density. The annealed stamped lanes presented the most homogeneous OTS layer, while both nonstamped areas were more heterogeneous. The area percentages with high and low OTS surface coverages are listed as well as the maximum average AF488-HSA adsorbed density and the average OTS coverage as determined by water contact angle measurements in the previous study.26
Figure 10.
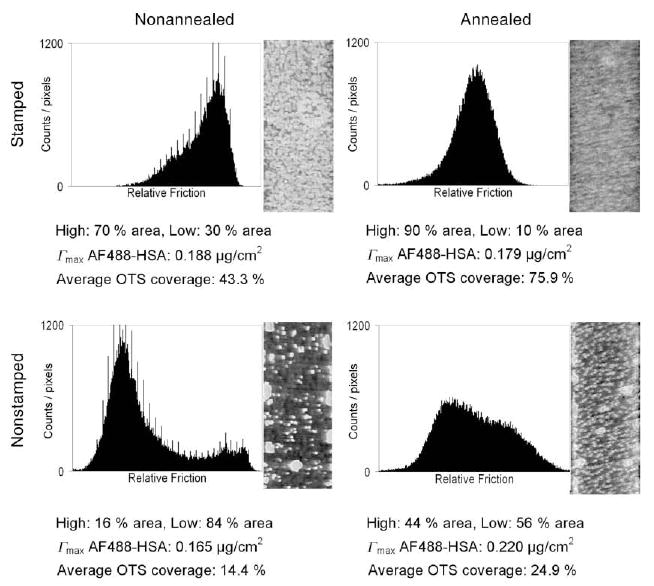
LFM images of the areas in the OTS patterns. The percent area with high and low OTS coverage are listed as well as the maximum mean AF488-HSA adsorbed density and the average OTS coverage as determined by water contact angle measurements in the previous study.26
Discussion
The OTS patterns produced in this study contained both largely homogeneous areas, chemical heterogeneities on a μm and sub-μm scales, and hydrophobicity gradients less than 2 μm in width. We have already demonstrated in the preceding paper,26 that each pattern contained several discrete areas each of which represents a unique combination of low and high energy binding sites and differentially affects albumin adsorption. Here, we proposed a generic adsorption model and histogram analysis method that can provide additional information about the dynamics of the adsorption processes at each of these distinct surface areas. Deviations in the experimental data from the model predictions should also provide insights into processes occurring during adsorption, if it can be determined which assumptions used in the model were not satisfied. Before we critically approach the differences between the model and experimental results, we first examine how the present results compare with the past reports on albumin adsorption on nonuniform surfaces.
Although it has been shown in the past that HSA preferentially adsorbs to substrates of greater hydrophobicity,21,39–41 there is also strong evidence that HSA will bind more readily and strongly to mixed surfaces containing both hydrophobic and hydrophilic sites, and at amphiphilic boundaries, than to hydrophobic substrates with densely packed hydrophobic molecules. It has been shown that the wetting transition zone of a carefully prepared hydrophobicity gradient will adsorb greater HSA densities than the more hydrophobic portions of the gradient.42,43 It has also been shown that HSA molecules will preferentially adsorb to defect boundaries in organosilane films created on hydrophilic substrates.42,44 Several studies have shown that organosilane films of intermediate surface coverage irreversibly bind HSA with a higher affinity than more hydrophobic or hydrophilic areas indicating more favorable interactions between the segregated alkyl chains and the HSA molecules.41,43,45 It is known that delipidized HSA possesses binding sites that interact with alkyl chains46 and that albumin denaturation disrupts fatty acid binding.47 Tilton et al. also recently provided strong evidence that HSA specifically and preferentially binds to intermediate coverages of surface bound nonpolar hydrocarbon chains.48 Consistent with previous findings,43 we observed in this study that the highest maximal HSA adsorption on areas containing intermediate average OTS fractional coverages of 0.25 and 0.43 on the nonstamped annealed areas and stamped nonannealed lanes, respectively. The friction maps obtained by LFM measurements, however, provide more compelling evidence that increased HSA adsorption is due to surface heterogeneities in areas containing relatively high fractions of normally low adsorbing sites (Figure 10). Histogram analysis of the high and low OTS density coverage sites for the different areas in the annealed and nonannealed patterns show that HSA adsorbed in greater amounts to areas that contained distributions of high to low OTS coverage areas approaching a 1:1 ratio despite large differences in average OTS coverage for the various areas (Figure 8). In particular, HSA consistently adsorbed more readily to the nonstamped areas of the annealed patterns than to the stamped bands despite that the stamped bands contained an average OTS surface coverage nearly three times greater than the nonstamped areas. We propose therefore that the microheterogeneities in the OTS patterns prepared in this study had an important impact on protein adsorption and that areas of intermediate coverage facilitated increased HSA adsorption when compared to densely packed hydrophobic organosilane films.
Mean HSA Surface Density
The mean protein surface density provided the first indication that surface heterogeneities affect protein adsorption kinetics. Each distinct area in the annealed and nonannealed patterns adsorbed albumin at a rapid rate for the first 2 to 3 minutes of adsorption and then continued to adsorb protein at varying slower rates over the remainder of the 90 minutes of adsorption (Figures 6A and B), which indicated that the majority of the highest energy binding sites are rapidly filled at the rates limited by protein transport to the interface. Once the majority of surface sites are occupied, the transport-limited rate switches to the adsorption-limited rate because the diffusion through the unstirred layer replenishes protein concentration next to the interface faster than what the process of adsorption removes from the adjacent solution. In this regime, lower energy binding sites continued to adsorb protein from the solution and true adsorption rate constants could be ascertained.
Even the observed transport-limited kinetics revealed information about the nature of heterogeneities in the various surfaces: the average adsorbed amount at the end of the transport-limited process, when expressed as a fraction of the average final adsorbed amount, is a direct measure of the fraction of high energy binding sites on the surface (Table II). Because adsorption to lower affinity sites is much slower and the transport-limited adsorption prevails for only short initial periods of time, it is assumed that the adsorbed amount at the end of the transport-limited regime is representative of only the high affinity sites.
TABLE II.
AF488-HSA absolute and fractional coverages sorted by binding site energy and type of OTS pattern. Absolute coverages are in ng/cm2 and fractional coverages in paretheses ().Bold typed values demark the area with the greatest absolute coverage for each binding energy
| Area | High | Intermediate | Low | |||
|---|---|---|---|---|---|---|
| Stamped, nonannealed | 134 | (0.71) | 36 | (0.19) | 18 | (0.10) |
| Nonstamped, nonannealed | 81.0 | (0.49) | 60 | (0.36) | 24 | (0.15) |
| Stamped, annealed | 150 | (0.84) | 22 | (0.12) | 7.0 | (0.04) |
| Nonstamped, annealed | 129 | (0.59) | 57 | (0.26) | 34 | (0.15) |
For instance, at the end of the transport-limited regime, the stamped bands for the annealed and nonannealed samples had adsorbed 84 % and 71 % of final adsorbed amounts while the nonstamped areas for the annealed and nonannealed samples had only adsorbed 59 % and 49 % of the final adsorbed amounts, respectively. These values (84 % vs. 71 %, and 59 % vs. 49 %) indicate that both the stamped and nonstamped areas in the annealed pattern contained larger fractions of high energy binding sites than the corresponding areas in the nonannealed pattern, and that the stamped areas in each pattern, as expected, contained a higher fraction of high energy binding sites than the nonstamped areas.
It is interesting, however, that although the nonstamped areas in the annealed pattern contain a lower average OTS coverage (24.9 %) than the stamped lanes in the nonannealed pattern (43.3 %), these areas adsorbed nearly equal amounts of protein during the transport-limited process (0.134 μg/cm2 for the nonannealed, stamped bands compared to 0.129 μg/cm2 for the annealed, nonstamped areas). This finding indicates that the two areas contained nearly equal numbers of protein-occupied high energy binding sites. This is also supported by LFM measurements (Figure 10) that demonstrated the formation of small OTS islands in the non-stamped areas during the annealing process. Furthermore, the annealed, nonstamped areas adsorbed an additional 0.091 μg/cm2 of HSA during the adsorption-limited regime, while the nonannealed, stamped bands only adsorbed an additional 0.054 μg/cm2 of HSA. The conclusion is that, although each area contained equal numbers of high affinity binding sites, the annealed, nonstamped areas contained local arrangements of OTS molecules at lower average surface densities. These local areas provided more favorable interactions for protein binding than the areas of greater average OTS coverage found in the nonannealed, stamped lanes.
We also observed that the initial adsorption rate for all distinct protein populations increases with increased hydrophobicity (Figures 6 and 7), which agrees with HSA adsorption trends reported elsewhere.49 It has been shown by others that HSA and bovine serum albumin denature to a greater extent on more hydrophobic surfaces50,51 and that initial binding rates increase with the ability of HSA to denature upon adsorption.52 Thus, we postulate that strong hydrophobic interactions between a more densely packed OTS film cause HSA molecules to denature upon adsorption in such a way that multiple surface binding sites are filled and that an adsorption maximum is reached more quickly because of the rapid depletion of available sites.
HSA Subpopulation Adsorption Density Spread
The protein density distribution within a given subpopulation also changes over time. The adsorption density spread changes over time serve as an indicator of the spatial distribution of high, intermediate, and low energy binding sites available to the proteins. Based on this hypothesis it is proposed that high energy sites bind protein first at times when adsorption to lower energy sites is still negligible. Thus the adsorption density spread transition from fast initial increase to a slower change with time (Figure 7) can be used a marker of the completion of adsorption to the highest energy binding sites as well as an indicator of the spatial distribution of these high energy sites. Any further changes in adsorption density spread with time are indicative of the distribution of lower energy binding sites which continue to adsorb protein at much slower rates. In principle, changes in adsorption density spread over time can be divided into 3 cases: (1) short term attainment of a steady state level of adsorption density spread indicates a surface with homogeneous spatial distribution of sites, (2) long term increase in adsorption density spread indicates a surface with heterogeneous distribution of binding sites, and (3) a constant level of adsorption density spread over time where the mean adsorbed amount continues to increase indicates that all resolvable sites adsorb protein at equal rates.
The spread in AF488-HSA adsorption densities for all protein populations increased quickly during the first 2 to 3 minutes of adsorption (Figures 7A and B). The nonannealed, stamped bands reached the spread in HSA adsorption density of 17.1 ng/cm2 compared to a value of 16.6 ng/cm2 on the annealed, stamped bands even though the annealed bands had adsorbed 11.0 ng/cm2 more protein than the nonannealed bands. The nonstamped annealed areas, however, exhibited a greater spread in HSA densities at 14.3 ng/cm2 than the nonannealed, nonstamped areas at 12.0 ng/cm2, although some of this difference may be attributable to a greater adsorbed amount on the annealed, nonstamped areas. Based on the assumption that greater spreads in densities at the end of the transport-limited adsorption regime correlate with greater spatial heterogeneity in high energy binding sites, these findings indicate that the annealing step decreased the spatial heterogeneity of the high energy binding sites in the stamped bands but increased spatial heterogeneity of the high energy sites in the nonstamped areas. This is further supported by LFM measurements showing that annealing reduced OTS film defects in the stamped bands but increased OTS island formation in the nonstamped areas (Figure 10).
At longer adsorption times, HSA adsorption density spread changes provided information about the heterogeneity for the remaining intermediate and low energy binding sites not filled during initial adsorption periods. In the nonannealed sample, density spread in the non-stamped area population continued to increase at a different rate until ≈20 minutes, then proceeded at a slower rate through the remainder of the experiment (Figure 7A). In the annealed pattern, a transient maximum in adsorption density spread was observed for both the stamped and nonstamped areas (Figure 7B). It is believed that the transient maxima in adsorption density spread are not the result of an experimental artifact as no noticeable variations in absolute intensities, average densities, or image focus were observed at these time points. Instead, we propose that areas containing already low HSA densities adsorbed protein at somewhat faster rates than high HSA density areas (i.e., the lowest pixel intensity values were replaced by brighter pixels, while the brightest pixels did not increase in intensity).
Based on the optics used in this study, each theoretically resolvable area of 222 × 222 nm2 was imaged by 2 × 2 CCD camera pixels. Since the optical point-spread-function of a point source of fluorescence is larger than the pixel size, some contribution in observed adsorption density spread must also be due to the optics of the system. Many thousands of OTS molecules and hundreds of HSA molecules can occupy sites in the area imaged by a single CCD pixel. As a result, data captured by each pixel present on average possibly hundreds of adsorption events on a sub-μm area. Consequently, the adsorption density spread is a measure of heterogeneity on a scale of 0.222 μm and little knowledge can be gained of the distribution of individual protein molecules within the areas imaged by each pixel using the FM imaging technique presented in this study.
Lateral force microscopy (LFM) was used to assess the local distribution of surface heterogeneity on the OTS patterns (Figure 10). Frictional force between the LMF probe and surface was a measure of interactions between the OTS molecules on the SFM tip and molecular species on the sample. Although probing these interactions allowed us to measure the spatial arrangements of OTS molecules in the patterns, the OTS-modified SFM tip did not fully sample all types of interactions that occurred between HSA molecules and sample surface binding sites. Thus, it is tempting to also consider the use of FM and protein adsorption as a means to probe surfaces for heterogeneities. Namely, the changes in adsorption density spread data suggest additional information about the distribution and variety of protein binding sites not measurable by the LFM imaging made in this study. In the LFM images shown in Figure 10, each pixel represents an area of 30 × 30 nm2. However, if each HSA molecule is considered acting as a discrete probe then each LFM pixel is reporting to an equivalent of ca. 56 HSA molecules area. Thus, if it were possible to monitor the position and occurrence of each HSA binding event, the HSA adsorption itself would report on a more highly resolved surface site heterogeneity than LFM.
HSA Subpopulation Fractional Area Coverage
Comparison of the areas below each AF488-HSA adsorption density distribution revealed that the fractional coverage of each subpopulation also changes with time (Figure 8). During the adsorption on the nonannealed pattern (Figure 8A), fractional coverage of stamped and nonstamped subpopulations initially decreased while the coverage of combined gradient areas initially increased. After 2 minutes of adsorption, fractional coverage of stamped and nonstamped subpopulations began again to increase while the fractional coverage of combined gradient population could not be distinguished in the histograms. These data indicate that during the transport-limited adsorption process, high binding energy sites in the stamped and nonstamped areas quickly filled while the high energy sites in the gradient areas lagged in binding rate. Thus, the fractional area coverage analysis points to two distinct energy binding site subtypes within the high energy binding site populations for the nonannealed patterns; higher affinity sites were located in the stamped and nonstamped areas and lower affinity sites were found in the gradient areas.
In the annealed OTS pattern (Figure 8B), the subpopulation of initially high HSA fractional coverage comprised nearly 80 % of the surface area at early times then gradually decreased in favor of the subpopulation of initially low fractional coverage. At 12 minutes of adsorption, the two subpopulations started to comprise nearly equal fractions of the sample surface. After 25 minutes of adsorption, a new subpopulation of intermediate HSA coverage was resolved in the histograms: it began to appear at the boundary between high and low coverage populations and continued to grow in area throughout the rest of the adsorption process. These data, in conjunction with mean adsorbed amount and adsorption density spread kinetics (Figures 6B and 7B), indicate that the population in the nonstamped area decreased in size most likely due to continued adsorption to the center of that area at a rate faster than adsorption at the edges. Additionally, adsorption at the edges caused the gradient areas to grow in size because of a faster adsorption rate than at the centers of the stamped lanes. Thus, for these data, area analysis enabled us to distinguish adsorption rates by pattern area. Area analysis data demonstrate the transient nature of discrete protein subpopulations, as defined by the histogram analysis method, and the histogram analysis method provides a way to monitor protein density changes even at gradient boundaries less than 2 μm wide. These results also indicate that small variations in OTS distribution cause significant changes in local adsorption kinetics.
Together, the FM adsorption data suggested that up to three levels of binding site energy heterogeneity existed in each of the patterned areas, each of which could be fitted to a single discrete normal distribution (Table II). The first level was comprised of high energy binding sites, which rapidly adsorbed protein from solution during the transport-limited regime. The fractional protein coverage deposited during this process indicates the fraction of high energy binding sites, while the absolute adsorbed amount indicates the total number of high energy sites, and the adsorption density spread at the completion of the transport-limited process is an indication of the spatial distribution of the high energy binding sites. The second and third levels of heterogeneity were comprised of the distribution of intermediate and low energy binding sites, respectively. The absolute additional protein coverage deposited during the first ≈17 minutes of the adsorption-limited process is an indication of the total number of intermediate energy binding sites, while the corresponding change in adsorption density spread suggests the spatial distribution of intermediate sites. Similarly, any additional changes in average adsorbed amount and density distribution after ≈20 minutes of adsorption provides information about the numbers and spatial distribution of the low energy sites. Thus, in these experiments, adsorption proceeded in as many as three temporally discrete groups in which information about the heterogeneity of high, intermediate, and low energy binding sites could be determined separately by tracking the evolving normal distribution in time representing a well-defined area of the sample.
Model Correlation to Experimental Data
The experimental results observed in this study both support and contradict the proposed generic model for adsorption kinetics on a heterogeneous surface. The adsorption model predicted that final HSA coverage increases with OTS coverage, which was clearly supported by adsorption kinetics to the stamped and nonstamped areas of the nonannealed pattern. However HSA adsorption on the annealed pattern did not follow the model since the area of lowest OTS coverage produced the greatest adsorbed amount for any of the patterned surfaces. Additionally, the annealed, stamped bands adsorbed slightly less protein than the nonannealed, stamped bands despite of greater OTS coverage.
We propose three major violations of the assumptions used in developing the generic adsorption model that may explain why the model could not predict the adsorbed amount and coverage distributions on all OTS patterns produced in this study. The first violation was due to the area scale mismatch. In the model, a distribution of binding site energies is formed by sampling all possible interactions on a surface area corresponding to the size of one CCD pixel used in this study. More importantly, the model describes interactions at a much higher resolution than could be monitored by optical imaging. Namely, the model as presented here describes a distribution function for a single CCD pixel area, while in the experiments the same area is observed as a single intensity (i.e. proportional to the adsorbed amount) value completely devoid of distribution information. The second violation of the model concerns the nature of OTS heterogeneities. We found through LFM and adsorption histogram analysis that each pattern area, modeled as a random distribution of a particular OTS coverage, was in fact comprised of sub-μm scale OTS density heterogeneities that were non-randomly distributed within the modeled area. In the case where a critical density of OTS molecules is required for adsorption to occur, a surface with a majority of the OTS molecules segregated into densely packed sites will adsorb more protein than a diffuse, randomly populated OTS film that is on average below the critical coverage at which adsorption begins to occur. Thus the proposed adsorption model most likely underestimated the adsorbed amount for the OTS surfaces used in this study. The third violated assumption concerns the rigid nature of the modeled protein sub-sites and the effectiveness of protein packing in the adsorbent layer. The adsorption model does not account for eventual change of protein conformation, which would result in an exchange of protein sub-sites or even the addition or deletion of sub-site interactions. We believe that the model correctly predicts that adsorbed protein amount should increase with hydrophobic binding sites but it does not account for the fact that strong protein-surface hydrophobic interactions can lead to protein denaturation and spreading. In such a scenario, the model fails to predict the depletion of available binding sites due to a denaturing event that increases the footprint of each HSA molecule and the incomplete filling of the all binding sites because of effect of preceding adsorption history.
Overall, this study provided the evidence that the energy barrier for nonspecific adsorption is lower for HSA on alkyl chain grafted surfaces than for specific molecular recognition events between alkyl chains and HSA fatty acid binding pockets. This is evidenced by slower adsorption rates and higher maximal HSA coverages for low coverage OTS films and fast adsorption rates and denaturation resulting in lower maximal HSA coverages on high OTS coverage areas. Both of these findings demonstrate how important the balance of interaction energies is in biological systems and illustrate how foreign materials can easily disrupt this balance, cause protein denaturation and thus elicit undesired host immune response.
Conclusions
In this study we have shown that fluorescence microscopy imaging is a viable technique to monitor protein adsorption kinetics and protein temporal-spatial changes down to a sub-μm length scale. The FM technique used to monitor protein adsorption kinetics in this study can easily be extended to monitor other biological processes that exhibit evolving spatial distributions of fluorescently labeled species over time. Additionally, histogram analysis of spatially resolved adsorption kinetics revealed new information about the nature of protein adsorption to heterogeneous surfaces. By correlating FM data with OTS patterned substrate properties, we have shown that HSA initial binding rates increase with substrate hydrophobicity and that heterogeneities on the μm and sub-μm length scale in OTS patterns have an important effect on protein adsorption kinetics, adsorbed amount, and surface distribution. We have also demonstrated that the effects caused by the heterogeneity within high, intermediate, and low energy binding sites can be separated by monitoring adsorption characteristics during both transport-limited and adsorption-limited protein binding regimes. Although the presented model accurately describes protein densities at low OTS coverages, it fails when strong hydrophobic interactions deform the normal albumin structure and when sub-μm heterogeneities predominate over randomly distributed nanoscale heterogeneities. Based on these conclusions, we propose that surface heterogeneity effects are strong predictors of temporal-spatial adsorption kinetics and that mere average properties such as average surface coverage of hydrophobic moiety is insufficient to describe complex biomolecular binding events at interfaces.
Acknowledgments
The authors acknowledge the discussions with Drs. N. Kallay, H.P. Jennissen, W. M. Reichert and P. A. Tresco. This work has been supported by the grants EB00278 and HL084586.
Footnotes
Dedicated to Professor Nikola Kallay on the occasion of his 65th birthday
References
- 1.Andrade JD, editor. Protein Adsorption. Vol. 2. Plenum Press; New York: 1985. Surface and Interfacial Aspects of Biomaterials. [Google Scholar]
- 2.Horbett TA, Brash JL, editors. Proteins at Interfaces II. ACS; Washington D.C.: 1995. [Google Scholar]
- 3.Leonard EF, Turrito VT, Vroman L, editors. Ann N Y Acad Sci. Vol. 516. N.Y. Acad. Sci.; New York: 1987. Blood in Contact with Natural and Artificial Surfaces. [PubMed] [Google Scholar]
- 4.Brash JL, Wojciechowski PW, editors. Interfacial Behavior of Bioproducts. M. Dekker; New York: 1996. [Google Scholar]
- 5.Malmsten M, editor. Biopolymers at Interfaces. 2nd revised. M. Dekker; New York: 2001. [Google Scholar]
- 6.Horbett TA. Cardiovasc Path. 1993;2(3):137S–148S. [Google Scholar]
- 7.Vroman L, Adams AL. J Biomed Mater Res. 1996;3:43–67. doi: 10.1002/jbm.820030106. [DOI] [PubMed] [Google Scholar]
- 8.Corsel JW, Willems GM, Kop JMM, Cuypers PA, Hermens WT. J Colloid Interface Sci. 1986;111:544–554. [Google Scholar]
- 9.Slack SM, Horbett TA. J Colloid Interface Sci. 1989;133:148–165. [Google Scholar]
- 10.Brash JL, Horbett TA, editors. Proteins at Interfaces Physicochemical and Biochemical Studies. ACS; Washington D.C.: 1987. [Google Scholar]
- 11.Brash JL, Ten Hove P. J Biomater Sci, Polym Ed. 1993;4:591–99. doi: 10.1163/156856293x00230. [DOI] [PubMed] [Google Scholar]
- 12.Andrade JD, Hlady V. Ann N Y Acad Sci. 1987;516:158–172. doi: 10.1111/j.1749-6632.1987.tb33038.x. [DOI] [PubMed] [Google Scholar]
- 13.Hlady V, Ho CH, Britt DW. Quantitative Analysis of Protein Adsorption Kinetics. In: Kallay N, editor. Interfacial Dynamics. Vol. 88. M. Dekker; New York: 2000. pp. 405–418. (Surfactant Science). [Google Scholar]
- 14.Lancet D, Horovitz A, Katchalski-Katzir E. Molecular Recognition in Biology: Models for Analysis of Protein-Ligand Interactions. In: Behr JP, editor. The Lock-and-Key Principle: The State of the Art – 100 Years On, Perspectives in Supramolecular Chemistry. J Wiley; New York: 1994. pp. 25–71. [Google Scholar]
- 15.Lauffenburger DA, Linderman JL. Receptors: Models for Binding, Trafficking and Signaling. Oxford University Press; New York: 1993. pp. 236–359. [Google Scholar]
- 16.Kallay N, Hlady V, Jednačak-Bišćan J, Milonjić S. Techniques for the Study of Adsorption from Solution. In: Rossiter BW, Baetzold R, editors. Investigation of Surfaces and Interfaces – Part A. 2nd. IX. John Wiley & Sons; New York: 1993. pp. 73–140. (Physical Methods in Chemistry). [Google Scholar]
- 17.Hlady V, Buijs J, Jennissen HP. Methods for studying protein adsorption. In: Wetzel R, editor. Amyloid, Prions, and Other Protein Aggregates, Methods Enzymol. Vol. 309. 1999. pp. 402–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tengvall P, Lundstrom I, Liedberg B. Biomaterials. 1998;19:407–422. doi: 10.1016/s0142-9612(97)00110-5. [DOI] [PubMed] [Google Scholar]
- 19.Chittur KK. Biomaterials. 1998;19:357–69. doi: 10.1016/s0142-9612(97)00223-8. [DOI] [PubMed] [Google Scholar]
- 20.Elwing H, Nilsson B, Svensson KE, Askendahl A, Nilsson UR, Lundström I. J Colloid Interface Sci. 1988;125:139–147. [Google Scholar]
- 21.Lin YS, Hlady V. Colloids Surf, B: Biointerfaces. 1994;2:481–491. doi: 10.1016/0927-7765(94)80056-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hlady V, Ho CH. Mat-wiss Werkstofftech. 2001;32:185–192. doi: 10.1002/1521-4052(200102)32:2<185::AID-MAWE185>3.0.CO;2-W. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hlady V. Adsorption kinetics of low density lipoprotein onto a hydrophobic-hydrophilic gradient surface. In: Pi-fat-Mrzljak G, editor. Supramolecular Structure and Function. Vol. 7. Kluwer Academic; New York: 2001. pp. 45–61. [Google Scholar]
- 24.Lin YS, Hlady V, Gölander CG. Colloids Surf, B: Biointerfaces. 1994;3:49–62. doi: 10.1016/0927-7765(93)01114-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin YS, Hlady V. Colloids Surf, B: Biointerfaces. 1995;4:65–75. doi: 10.1016/0927-7765(94)01150-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hodgkinson G, Hlady V. J Adhes Sci Technol. 2005;19:235–255. doi: 10.1163/1568561054352612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Curry S, Mandelkow H, Brick P, Franks N. Nat Struct Mol Biol. 1998;5:827–835. doi: 10.1038/1869. [DOI] [PubMed] [Google Scholar]
- 28.Jennissen HP. J Colloid Interface Sci. 1986;111:570–586. [Google Scholar]
- 29.Kulik EA, Kalinin ID, Sevastianov VI. Artif Organs. 1991;15:386–391. doi: 10.1111/j.1525-1594.1991.tb00748.x. [DOI] [PubMed] [Google Scholar]
- 30.Minton AP. Biophys J. 1999;76:176–187. doi: 10.1016/S0006-3495(99)77187-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szleifer I. Biophys J. 1997;72:595–612. doi: 10.1016/s0006-3495(97)78698-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Minton AP. Biophys Chem. 2000;86:239–247. doi: 10.1016/s0301-4622(00)00151-4. [DOI] [PubMed] [Google Scholar]
- 33.Minton AP. Biophys J. 2001;80:1641–1648. doi: 10.1016/S0006-3495(01)76136-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lancet D, Sadovsky E, Seidemann E. Proc Natl Acad Sci USA. 1993;90:3715–3719. doi: 10.1073/pnas.90.8.3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jähne B. Practical handbook on image processing for scientific applications. Boca Raton: CRC Press; 1997. [Google Scholar]
- 36.Pompe T, Fery A, Herminghaus S. Langmuir. 1999;15:2398–2401. [Google Scholar]
- 37.Cassie ABD. Discuss Faraday Soc. 1952;75:5041. [Google Scholar]
- 38.Baszkin A, Lyman DJ. J Biomed Mater Res. 1980;14:393–403. doi: 10.1002/jbm.820140406. [DOI] [PubMed] [Google Scholar]
- 39.Van Dulm P, Norde W. J Colloid Interface Sci. 1983;91:248–255. [Google Scholar]
- 40.Goodman SL, Simmons SR, Cooper SL, Albrecht RM. J Colloid Interface Sci. 1990;139:561–570. [Google Scholar]
- 41.Gölander CG, Pitt WG. Biomaterials. 1990;11:32–35. doi: 10.1016/0142-9612(90)90048-u. [DOI] [PubMed] [Google Scholar]
- 42.Ho CH, Britt DW, Hlady V. J Mol Recognit. 1996;9:444–455. doi: 10.1002/(sici)1099-1352(199634/12)9:5/6<444::aid-jmr281>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 43.Gölander CG, Lin YS, Hlady V, Andrade JD. Colloids Surf. 1990;49:289–302. [Google Scholar]
- 44.Fang J, Knobler CM. Langmuir. 1996;12:1368–1374. [Google Scholar]
- 45.Petrash S, Sheller NB, Dando W, Foster MD. Langmuir. 1997;13:1881–1883. [Google Scholar]
- 46.Boulkanz L, Vidal-Madjar C, Balcar N, Baron MH. J Colloid Interface Sci. 1997;188:58–67. [Google Scholar]
- 47.Davis BD, Dubos RJ. J Exp Med. 1947;86:215–228. doi: 10.1084/jem.86.3.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choi EJ, Foster MD, Daly S, Tilton R, Przybycien T, Marjkrzak CF, Witte P, Menzel H. Langmuir. 2003;19:5464–5474. [Google Scholar]
- 49.Young BR, Pitt WG, Cooper SL. J Colloid Interface Sci. 1988;125:246–260. [Google Scholar]
- 50.Ge S, Kojio K, Takahara A, Kajiyama T. J Biomater Sci, Polym Ed. 1998;9:131–150. doi: 10.1163/156856298x00479. [DOI] [PubMed] [Google Scholar]
- 51.Noinville S, Revault M, Baron MH, Tiss A, Yapou-djian S, Ivanova M, Verger R. Biophys J. 2002;82:2709–2719. doi: 10.1016/S0006-3495(02)75612-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pitt WG, Grasel TG, Cooper SL. Biomaterials. 1988;9:36–46. doi: 10.1016/0142-9612(88)90067-1. [DOI] [PubMed] [Google Scholar]