
Fig. 2.
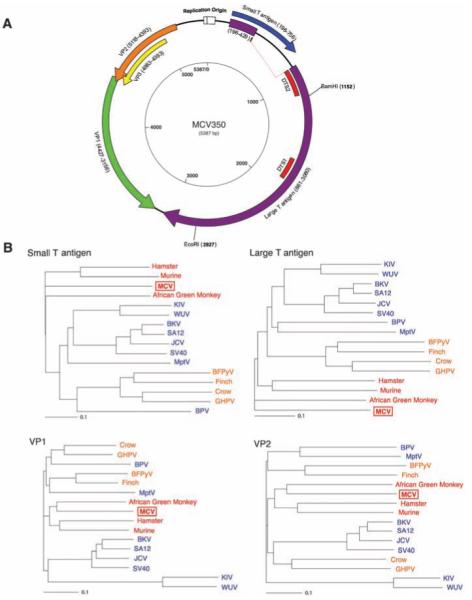
(A) Schematic of MCV genome. Genome walking was used to clone the full MCV genome from tumor MCC350. The genome encodes typical features of a polyomavirus, including large T (purple) and small T (blue) open reading frames. Also shown are predicted VP1 (green) and overlapping VP2 (orange) and VP3 (yellow) genes. DTS1 and DTS2 (red) represent cDNA fragments originally identified by DTS screening. The former was used to identify MCV, and the latter is a spliced transcript with no homology to known polyomavirus sequences. (B) Neighbor-joining trees for putative MCV large T, small T, VP1, and VP2 proteins. The four known human polyomaviruses (BKV, JCV, KIV, and WUV) cluster together in the SV40 subgroup (blue), whereas MCV is most closely related to MuPyV subgroup viruses (red). Both subgroups are distinct from the avian polyomavirus subgroup (orange). Scale bars indicate an evolutionary distance of 0.1 amino acid substitutions per position in the sequence.