

Abstract
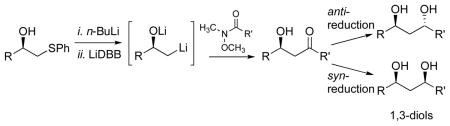
Synthesis of ketone aldol products using a non-aldol route was developed. The β-phenylthio alcohols were prepared from optically pure oxiranes. Deprotonation and reductive lithiation generated the key intermediate, a β-oxyanionic alkyllithium reagent. Addition to a Weinreb amide produced the β-hydroxy ketone in >90% yield using only 1.5 equivalents of the phenylthio alcohol. Stereoselective reduction of the ketone led to either the syn- or anti-1,3-diol. This simple, convergent sequence was used to prepare aculeatins A, B, and D from a common intermediate.
Ketone aldol reactions are very useful for the construction of polyacetate and polypropionate natural products.1 They allow carbon chains to be joined together to make much larger structures, and are often the key disconnection in the retrosynthesis.2 We recently reported an alternative sequence (Scheme 1) in which a β-oxyanionic alkyllithium reagent (2) was prepared and added to a Weinreb amide (3) to produce a β-hydroxy ketone (4). The sequence was used to prepare most of the carbon skeleton of amphidinol 3.3 This strategy nicely complements the traditional aldol sequence, and offers several advantages, including the direct introduction of the stereogenic center from a chiral building block. Other groups have begun to adopt this method.4 One limitation is the use of a large excess of the alkyllithium reagent (3–4 equiv). In this communication, we have reinvestigated the generation of β-oxyanionic alkyllithium reagents and demonstrated that with normal precautions to exclude moisture, as little as 1.5 equivalents of the alkyllithium precursor results in excellent yield of the coupled product. The utility of the improved procedure was demonstrated in a unified synthetic approach to aculeatins A, B and D.
Scheme 1.

Aldol Product from β-Oxyanionic Alkyllithium Reagents3
The β-oxyanionic alkyllithium reagents have been prepared by a number of methods that include mercury-lithium exchange,5 reductive lithiation of chlorohydrins,6 oxiranes,7 and β-phenylthio alcohols.8 A few complex β-oxyanionic alkyllithium reagents have been prepared by reduction of oxiranes,7,9 but these reagents have not been used widely in synthesis. The β-phenylthio alcohols are typically prepared by nucleophilic opening of oxiranes; thus the most direct route to the alkyllithium reagents would be by direct reduction of oxiranes.
Reductive lithiation and coupling of a model oxirane is presented in Scheme 2. The reductive lithiation of oxirane 5 generated β-oxyanionic alkyllithium 6, which coupled only poorly with the Weinreb amide 7 to give the expected product in 34% yield. Both Cohen7b and Yus7c reported good yields for reductive lithiation of oxiranes followed by addition to aldehydes. Presumably the issue is not with the generation of the anion, but rather with the coupling efficiency with the much less reactive Weinreb amide under these conditions.10 We considered the possibility that added thiophenol might catalyze the reduction of the oxirane or stabilize the alkyllithium agent. The addition of 20 mol % of thiophenol to the reaction did neither, and actually resulted in a lower yield of product. Although oxiranes have been reduced and added to aldehydes with excellent results,7 in our hands they did not add to a Weinreb amide efficiently.
Scheme 2.

Aldol Products Formed by Reductive Lithiation of Oxiranes
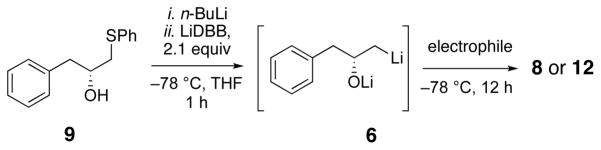
Lithiation of β-phenylthio alcohols was much more effective. The model phenylthio alcohol 9 was prepared from oxirane 5 using catalytic LiClO4.11 The alcohol 9 was dissolved in THF with 1,10-phenanthroline and titrated with n-BuLi to prepare the alkoxide and to remove all traces of water. The reductive lithiation was carried out with LiDBB (lithium 4,4′-di-tert-butylbiphenylide, Freeman’s reagent)12 at −78 °C in THF. The first entries show the addition to Weinreb amide 7. With 1.5 equivalents of 9, the product 8 was isolated in excellent yield. Reducing the ratio of phenylthio alcohol 9 and Weinreb amide 7 to 1:1 gave 68–85% of the product, a surprisingly good outcome.13 One reaction using the phenylthio alcohol 9 as the limiting reagent only gave 56%, but this result could certainly be improved upon. The use of a morpholine amide 10 was also effective, but resulted in slightly diminished yields compared with the Weinreb amide. Aldehydes are much better electrophiles, and generated the 1,3-diol 12 in 92% yield using phenylthio alcohol 9 as the limiting reagent. As expected, the 1,3-diol was produced as a 1:1 mixture of diastereomers, underscoring the need for an alternative strategy to introduce the second stereogenic center. Using a modest excess (1.5 equiv) of the phenylthio alcohol led to >90% yields of the desired aldol product.
The aculeatins A–D have attracted significant attention from synthetic chemists. They were isolated by Heilmann from the rhizomes of Amomum aculeatum, and were reported to have significant antiprotozoal and antiplasmodial activity.14 In addition, they show low to sub-micromolar activity against KB cell lines, and aculeatin A was found to be active against MCF-7 (human breast cancer cells) using an in vivo hollow fiber assay.15 The synthesis of racemic aculeatin A and B was first reported by Wong,16 followed by enantioselective syntheses by Marco.17 Synthesis of aculeatin D was first reported by Baldwin.18 A number of other syntheses have followed, all of which use a final phenol oxidation to assemble the spirocyclic system.19 The aculeatins are interesting synthetic targets and promising lead compounds in a number of important therapeutic areas.
The retrosynthetic analysis of the aculeatins is presented in Figure 1. Aculeatin A and B are epimeric at the acetal center, with A having the thermodynamically favored configuration.17b Similarly, aculeatin D is thermodynamically disfavored with respect to its C6 epimer. The final step is a biomimetic cyclization using a dithiane in place of the C6 ketone.16 syn-1,3-Diol 13 is the precursor to A and B, whereas anti-1,3-diol 14 is the precursor to aculeatin D. Both diols share a common 2R-configuration (aculeatin numbering), and they will be derived from the common intermediate 15 by stereoselective reduction. The key building blocks for the planned syntheses are Weinreb amide 16 and the optically pure phenylthio alcohol 17.
Figure 1.

The structures and unified retrosynthetic analyses of aculeatins A, B and D.
Synthesis of the building blocks and an initial attempt at the coupling reaction are shown in Scheme 3. Dithiane 18 was prepared by Wong’s procedure.19d,16 Formation of the Weinreb amide20 and protection of the phenol led to the coupling partner 16. The optically pure hydroxy phenylthio alcohol 17 was prepared by epoxidation of 1-pentadecene, followed by Jacobsen kinetic resolution.21 The thiophenol addition was catalyzed by LiClO4 to give optically pure22 phenylthio alcohol 17 in 34% overall yield. The initial coupling experiments, using the conditions reported in Table 1, gave none of the desired aldol product and returned both the Weinreb amide 16 and the phenylthio alcohol 17. Recovery of 17 indicated that the reductive lithiation reaction had failed.
Scheme 3.

Synthesis of Aculeatin Precursors and an Attempted Coupling Reaction
Table 1.
Optimized Coupling Between β-Oxyanionic Alkyllithium and Electrophiles
 | ||||
|---|---|---|---|---|
| 9: equiv | electrophile | equiv | product | yield |
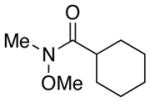
| 1.5 |
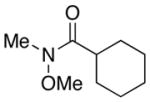 7 |
1.0 |
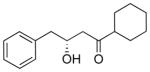 8 |
92–94% |
| 1.0 |
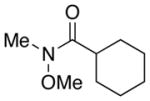 7 |
1.0 |
 8 |
68–85% |
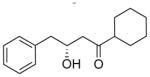
| 1.0 |
 7 |
1.1 |
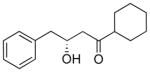 8 |
56% |
| 1.0 |
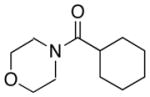 10 |
1.0 |
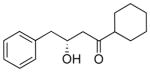 8 |
52–60% |
| 1.0 |
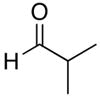 11 |
1.5 |
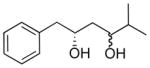 12 |
92%b,c |
Compound 9 (100 mg) was titrated with n-BuLi with 1,10-phenanthroline as an indicator at 0 °C. The alkoxide was reduced with LiDBB at −78 °C for 1 h, followed by addition of the electrophile and stirring for 12 h. Isolated yields are reported and ranges are given for multiple runs.
The mixture was stirred for 2 h after addition of the electrophile.
The syn- and anti-diols were isolated as a 47:53 mixture of diastereomers.
The failure of the lithiation reaction led us to reoptimize the conditions for the coupling. The obvious difference between the reactive phenylthio alcohol 9 and the unreactive 17 was the long alkyl chain in 17. We hypothesized that the lipophilic alkoxide formed by deprotonation of 17 was not available for reduction, possibly because it separated into a different phase or perhaps formed micelles in the THF solution. In an effort to avoid the hypothesized phase separation, we investigated different solvents and additives.23 The lithiation was successful when the reaction was conducted in a 1:1 mixture of THF and hexanes, Scheme 4. Addition of Weinreb amide 16 in THF resulted in a final solvent mixture of 70:30 THF/hexanes, and delivered the aldol adduct 15 in 60–79% yield using 1.5 equivalents of phenylthio alcohol 17.24 Ketone 15 is the common intermediate for all three aculeatins. Anti reduction using Evans’ method,25 and deprotection led to anti-1,3-diol 14 as a 10:1 dr in very good yield. The final oxidative cyclization was conducted under citrate-buffered conditions19g to generate aculeatin D in 30% yield, accompanied by the separable C6 epimer in 35% yield. Without the added buffer, only the C6 epimer was produced.19g Aculeatin D was prepared in 7 steps from 1-pentadodecane.
Scheme 4.

Synthesis of Aculeatin D
The synthesis of aculeatin A and B is shown in Scheme 5. Attempts to reduce 15 to the syn diol using Narasaka’s conditions (EtOBEt2, NaBH4)26 led to very slow and unselective reduction of the ketone. The steric crowding from the dithiane may slow the addition, and competing boron coordination with the sulfur atoms may reduce the selectivity. Eventually we found that Evans’ catecholborane procedure27 was effective on the deprotected phenol, and generated the syn-1,3-diol 13 with 5:1 dr. Oxidative deprotection and cyclization using Wu’s buffered conditions gave aculeatin A (38%) and aculeatin B (20%).19g The small amount of aculeatin D and its epimer were separated by chromatography at this stage. The NMR data for synthetic aculeatins A, B and D were all identical to the literature data, and the optical rotations were consistent with the assigned configurations.
Scheme 5.

Synthesis of Aculeatin A and B
The β-oxyanionic alkyllithium addition to a Weinreb amide is a practical segment-coupling reaction to assemble β-hydroxy ketones. The aculeatins A, B and D were prepared from a common intermediate using this method. This coupling reaction enables convergent synthesis of polyol chains and will be a valuable tool in natural product synthesis.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of General Medicine (GM-043854) and by a generous gift from the Schering-Plough Research Institute. The S. T. Li foundation also provided financial support. VM thanks the Department of Education (GAANN) for a predoctoral fellowship.
Footnotes
Supporting Information Available: Characterization data and experimental procedures for all compounds described are included. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Yeung KS, Paterson I. Chem Rev. 2005;105:4237–4313. doi: 10.1021/cr040614c. [DOI] [PubMed] [Google Scholar]; (b) Schetter B, Mahrwald R. Angew Chem, Int Ed. 2006;45:7506–7525. doi: 10.1002/anie.200602780. [DOI] [PubMed] [Google Scholar]
- 2.Recent examples of strategic aldol reactions in synthesis, see: Evans DA, Kvaerno L, Dunn TB, Beauchemin A, Raymer B, Mulder JA, Olhava EJ, Juhl M, Kagechika K, Favor DA. J Am Chem Soc. 2008;130:16295–16309. doi: 10.1021/ja804659n.Dunetz JR, Julian LD, Newcom JS, Roush WR. J Am Chem Soc. 2008;130:16407–16416. doi: 10.1021/ja8063205.Bock M, Dehn R, Kirschning A. Angew Chem, Int Ed. 2008;47:9134–9137. doi: 10.1002/anie.200803271.Lu L, Zhang W, Carter RG. J Am Chem Soc. 2008;130:7253–7255. doi: 10.1021/ja803012n.Lanners S, Norouzi-Arasi H, Salom-Roig XJ, Hanquet G. Angew Chem, Int Ed. 2007;46:7086–7089. doi: 10.1002/anie.200701749.
- 3.Huckins JR, De Vicente J, Rychnovsky SD. Org Lett. 2007;9:4757–4760. doi: 10.1021/ol7020934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kartika R, Gruffi TR, Taylor RE. Org Lett. 2008;10:5047–5050. doi: 10.1021/ol802254z. [DOI] [PubMed] [Google Scholar]
- 5.(a) Barluenga J, Fananas FJ, Yus M. J Org Chem. 1979;44:4798–4801. [Google Scholar]; (b) Barluenga J, Fananas FJ, Yus M. J Org Chem. 1981;46:1281–1283. [Google Scholar]
- 6.(a) Barluenga J, Florez J, Yus M. J Chem Soc, Chem Commun. 1982:1153–1154. [Google Scholar]; (b) Barluenga J, Florez J, Yus M. J Chem Soc, Perkin 1. 1983:3019–3026. [Google Scholar]; (c) Najera C, Yus M, Seebach D. Helv Chim Acta. 1984;67:289–300. [Google Scholar]
- 7.(a) Bartmann E. Angew Chem, Int Ed. 1986;25:653–655. [Google Scholar]; (b) Cohen T, Jeong IH, Mudryk B, Bhupathy M, Awad MMA. J Org Chem. 1990;55:1528–1536. [Google Scholar]; (c) Bachki A, Foubelo F, Yus M. Tetrahedron: Asymmetry. 1996;7:2997–3008. [Google Scholar]; (d) Dorigo AE, Houk KN, Cohen T. J Am Chem Soc. 2002;111:8976–8978. [Google Scholar]; (e) Grobelny Z. Eur J Org Chem. 2004;2004:2973–2982. [Google Scholar]
- 8.(a) Foubelo F, Gutierrez A, Yus M. Tetrahedron Lett. 1997;38:4837–4840. [Google Scholar]; (b) Foubelo F, Gutierrez A, Yus M. Synthesis. 1999:503–514. [Google Scholar]
- 9.(a) Soler T, Bachki A, Falvello LR, Foubelo F, Yus M. Tetrahedron: Asymmetry. 2000;11:493–517. [Google Scholar]; (b) Falvello LR, Foubelo F, Soler T, Yus M. Tetrahedron: Asymmetry. 2000;11:2063–2066. [Google Scholar]; (c) Yus M, Soler T, Foubelo F. Tetrahedron: Asymmetry. 2001;12:801–810. [Google Scholar]; (d) Yus M, Maci B, Gûmez C, Soler T, Falvello LR, Fanwick PE. Tetrahedron. 2005;61:3865–3871. [Google Scholar]
- 10.Reductive lithiation of oxirane 5 with LiDBB and quenching with 1.5 equiv of isobutyraldehyde resulted in 59% of compound 12. In a side-by-side reaction, quenching the alkyllithium with 1.0 equiv of Weinreb amide resulted in <20% of ketone 8, accompanied by a significant amount of allylbenzene.
- 11.Azizi N, Saidi MR. Catal Commun. 2006;7:224–227. [Google Scholar]
- 12.Freeman PK, Hutchinson LL. J Org Chem. 1980;45:1924–1930. [Google Scholar]
- 13.The reaction was quenched with NH4Cl (aq) and did not show any indication of racemization or retro-aldol side reactions. When the reaction was quenched with MeOH-d4, we observed partial deuteration at the positions adjacent to the ketone, but not at the benzylic position. Traces of retro-aldol product may be present, but apparently the forward aldol reaction is unfavorable. In cases where the yields of 8 were modest, most of the unreacted Weinreb amide 7 was recovered.
- 14.(a) Heilmann J, Mayr S, Brun R, Rali T, Sticher O. Helv Chim Acta. 2000;83:2939–2945. [Google Scholar]; (b) Heilmann J, Brun R, Mayr S, Rali T, Sticher O. Phytochemistry. 2001;57:1281–1285. doi: 10.1016/s0031-9422(01)00174-1. [DOI] [PubMed] [Google Scholar]; (c) Salim AA, Su BN, Chai HB, Riswan S, Kardono LBS, Ruskandi A, Farnsworth NR, Swanson SM, Kinghorn AD. Tetrahedron Lett. 2007;48:1849–1853. doi: 10.1016/j.tetlet.2007.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chin YW, Salim AA, Su BN, Mi Q, Chai HB, Riswan S, Kardono LBS, Ruskandi A, Farnsworth NR, Swanson SM, Kinghorn AD. J Nat Prod. 2008;71:390–395. doi: 10.1021/np070584j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong YS. Chem Commun. 2002:686–687. doi: 10.1039/b200740a. [DOI] [PubMed] [Google Scholar]
- 17.(a) Falomir E, Alvarez-Bercedo P, Carda M, Marco JA. Tetrahedron Lett. 2005;46:8407–8410. [Google Scholar]; (b) Alvarez-Bercedo P, Falomir E, Carda M, Marco JA. Tetrahedron. 2006;62:9641–9649. [Google Scholar]
- 18.Baldwin JE, Adlington RM, Sham VWW, Marquez R, Bulger PG. Tetrahedron. 2005;61:2353–2363. [Google Scholar]
- 19.(a) Chandrasekhar S, Rambabu C, Shyamsunder T. Tetrahedron Lett. 2007;48:4683–4685. [Google Scholar]; (b) Peuchmaur M, Wong YS. Synlett. 2007:2902–2906. [Google Scholar]; (c) Peuchmaur M, Wong YS. J Org Chem. 2007;72:5374–5379. doi: 10.1021/jo0707986. [DOI] [PubMed] [Google Scholar]; (d) Peuchmaur M, Saidani N, Botte C, Marechal E, Vial H, Wong YS. J Med Chem. 2008;51:4870–4873. doi: 10.1021/jm8007322. [DOI] [PubMed] [Google Scholar]; (e) Ramana CV, Srinivas B. J Org Chem. 2008;73:3915–3918. doi: 10.1021/jo800026n. [DOI] [PubMed] [Google Scholar]; (f) Suresh V, Selvam JJP, Rajesh K, Venkateswarlu Y. Tetrahedron: Asymmetry. 2008;19:1509–1513. [Google Scholar]; (g) Zhen ZB, Gao J, Wu Y. J Org Chem. 2008;73:7310–7316. doi: 10.1021/jo801296x. [DOI] [PubMed] [Google Scholar]
- 20.(a) Nahm S, Weinreb SM. Tetrahedron Lett. 1981;22:3815–3818. [Google Scholar]; (b) Williams JM, Jobson RB, Yasuda N, Marchesini G, Dolling UH, Grabowski EJJ. Tetrahedron Lett. 1995;36:5461–5464. [Google Scholar]
- 21.(a) Tokunaga M, Larrow JF, Kakiuchi F, Jacobsen EN. Science. 1997;277:936–938. doi: 10.1126/science.277.5328.936. [DOI] [PubMed] [Google Scholar]; (b) Schaus SE, Brandes BD, Larrow JF, Tokunaga M, Hansen KB, Gould AE, Furrow ME, Jacobsen EN. J Am Chem Soc. 2002;124:1307–1315. doi: 10.1021/ja016737l. [DOI] [PubMed] [Google Scholar]
- 22.Compound 17 {[α] 24D –27.2 (c 0.99, CHCl3)} was found to be a single enantiomer (>98% ee) by 1H NMR analysis of the R and S Mosher’s ester derivatives.
- 23.Toluene/THF mixtures were also effective. Other additives (LiCl, HMPA) did not lead to any coupled product.
- 24.The coupling of Weinreb amide 16 was attempted without the TBS protecting group, but neither the phenol nor phenoxide was sufficiently soluble in the THF/hexanes mixture to react with the alkyllithium reagent.
- 25.Evans DA, Chapman KT, Carreira EM. J Am Chem Soc. 1988;110:3560–3578. [Google Scholar]
- 26.(a) Narasaka K, Pai FC. Tetrahedron. 1984;40:2233–2338. [Google Scholar]; (b) Chen KM, Hardtman GE, Prasad K, Repic O, Shapiro MJ. Tetrahedron Lett. 1987;28:155–158. [Google Scholar]
- 27.Evans DA, Hoveyda AH. J Org Chem. 1990;55:5190–5192. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.