

Abstract
The Red Sea sponge Hemimycale arabica afforded the known (Z)-5-(4-hydroxybenzylidene)-hydantoin (1). This natural phenylmethylene hydantoin (PMH) 1 and the synthetic (Z)-5-(4-(ethylthio)benzylidene)-hydantoin (2) showed potent in vitro and in vivo anti-growth and anti-invasive properties against PC-3M prostate cancer cells in MTT, spheroid disaggregation, and in mice models. To explore a possible molecular target of PMHs, the most potent synthetic analogue 2 has been virtually screened against various protein kinases. Molecular modeling study has shown that 2 can be successfully docked within the binding pocket of glycogen synthase kinase-3beta (GSK-3β) similar to the well-known GSK-3β inhibitor I-5. Several PMHs showed potent in vitro GSK-3β inhibitory activity with an IC50 range of 4–20 µM. The most potent analogue 3 showed a significant increase in liver glycogen level at the 5, 15, and 25 mg/kg dose levels, in vivo. Pharmacophore model was built and validated using in-house database of active and inactive GSK-3β inhibitors. The GSK-3β inhibitory activity of PMHs entitles them to be potential leads for the treatment of cancer, Alzheimer’s disease, bipolar disorders, stroke, different tau pathologies, and type-2 diabetes.
1. Introduction
The sponge genus Hemimycale (family Mycalidae) is well known for its bioactive secondary metabolites especially biogenetically complex guanidine alkaloids.1–3 Ptilomycalin A1 has a unique polycyclic guanidine skeleton with a spermidine group linked to a 16-hydroxyhexadecanoic acid moiety.1–3 The ethanolic extract of the abundant shallow water Red Sea sponge H. arabica was targeted because it inhibited the proliferation and invasion of the highly metastatic human prostate cancer PC-3M cell line.4,5 The natural (Z)-5-(4- hydroxybenzylidene)-hydantoin (PMH, 1) and the synthetic (Z)-5-(4-(ethylthio)benzylidene)-hydantoin (2) showed potent in vitro anti-growth and anti-invasive properties against PC-3M prostate cancer cells in MTT, and spheroid disaggregation.4,5 They decreased the orthotopic tumor growth and inhibited the formation of tumor micrometastases in distant organs without apparent cytotoxic effects at the test doses.5
The cyclic imide hydantoins are well investigated for their anticonvulsant activity.6,7 Various pharmacological activities were reported for hydantoins including fungicidal, herbicidal, anti-inflammatory, anti-HIV, analgesic, cannabinoid receptor-1 (CB-1), 5HT, purine P-2X receptor antagonism, platelet aggregation inhibition, anti-arrhythmic and antihypertensive, anti-diabetic, neuroprotective, HDL/cholesterol modulating, antiviral, and growth hormone secretagogue.7
GSK-3β, also called tau phosphorylating kinase I is a serine/threonine kinase implicated in the control of several regulatory proteins.8,9 It was first discovered by virtue of its ability to phosphorylate and inactivate glycogen synthase, the regulatory enzyme of mammalian glycogen synthesis.10 Its pleiotropic but unique activities have made GSK-3β a favorite target for the treatment of several human diseases such as type-2 diabetes,11 Alzheimer’s disease (AD),12 CNS disorders like manic depressive disorder and neurodegenerative diseases,13 and chronic inflammatory disorders.14 The search for GSK-3β inhibitors became a very active research trend for academic centers and pharmaceutical industry. Several structurally diverse compounds were reported to inhibit GSK-3β. Examples of these are thiadiazolidindiones (TDZD), hydantoins, triazoles, thiazoles, maleimides, dithiazolidindiones, and pyrazolepyridines.15–26
Marine natural products from sponges, ascidians, and gastropod mollusks have played a central role in providing novel GSK-3β inhibitors. Examples of these active marine-derived compounds are palinurin, tricantin, hymenialdisine, meridianines, manzamine A, and indirubines.22–24 The marine environment represents an enormous resource for the discovery of potential GSK-3β inhibitors.22–24
The well known protein substrate of GSK-3β is the microtubule-associated protein tau. GSK-3β phosphorylates tau at the specific sites; serine residue 199 and serine residue 396.26 Deregulation of GSK-3β activity is implicated in the pathophysiology of AD by regulating microtubule stability and phosphorylating the microtubule associating protein, tau.27,28 Abnormal hyperphosphorylation of tau and not its aggregation into filaments considered an essential key step in neurofibrillary degeneration.27,28 GSK-3β has been reported to play a role in the toxic effect mediated by β-amyloid, a protein that aggregates as extracellular amyloid plaques in brain of AD patients.29 Exposure of cortical and hippocampal primary neuronal cultures to β-amyloid induces activation of GSK-3β, tau hyperphosphorylation, and cell death.30 Therefore, inhibition of GSK-3β controls tau phosphorylation level in cells overexpressing tau protein in a dose-dependent manner.31 Blockade of GSK-3β expression by antisense oligonucleotides or its activity by lithium addition inhibits β-amyloid-induced neurodegeneration of cortical and hippocampal primary cultures.32–37 Therefore, GSK-3β inhibition is therapeutically important for several neurodegenerative diseases including AD.
In an effort to explore possible molecular target(s) of PMHs, compound 2, the most potent anti-metastatic analogue against PC-3M, has been virtually screened versus several kinases (AKB, CDK1, CDK2, CDK5, PKA, PKC, EGFR, PDGFR, ERK, MEK1, PI-3 kinase, MAPKK, and GSK-3β) available at the RCSB Protein Data Bank (PDB) using Surflex Dock Program implanted in SYBYL 8.0 package. Interestingly, 2 shows structure similarity and binding orientation to I-5 (Figure 1), a potent and selective glycogen synthase kinase-3β inhibitor.38,39 Both I-5 and 2 showed high binding scores at the GSK-3β’s ATP binding site. Therefore, various PMH analogues were selected for in silico screening to explore their possible GSK-3β binding affinity. In vitro and in vivo testing further supported the in silico data. Consequently, a pharmacophore model was established to understand the essential features required for GSK-3β binding and to discover and design new analogues that could be used therapeutically. This study reports the identification and characterization of PMHs as a potent class of GSK-3β inhibitors.
Figure 1.

(A) Molecular Structure of I-5. (B) Compound 2 binds similarly to I-5 into the ATP-binding side of GSK-3β
2. Results and Discussion
2.1. Chemistry
Six known (2–4 and 6–8) and one new (5) PMHs were synthesized using Scheme 1.4,40 This Scheme includes base-catalyzed condensation of hydantoin with substituted benzaldehydes.40 Geometrical isomerism (E/Z isomers) was possible due to the restricted rotation around the exocyclic C=C double bond of the PMHs, however this method regioselectively afforded the Z geometry as confirmed by the analytical data. The IR spectra of PMHs 2–4 and 6–8 showed the stretching vibration bands at a higher frequency region (1660–1675 cm−1) characteristic for Z-oriented C=C bonds, which are distinguishable from those expected for the E isomers (1630–1640 cm−1).40 The chemical shift of the most diagnostic olefinic proton H-6 of 2–4 and 6–8 was downfield shifted in the 1H NMR spectra (δH 6.40–7.00), which further confirmed the Z-orientation of the Δ5,6 system. The expected chemical shift of H-6 in the E isomers should be 6.20–6.30 ppm.40 The downfield shift of H-6 in the Z-isomer is attributed to the anisotropic effect of the spatially nearby C-4 carbonyl group.40 Different electronic (σ) and lipophilc (π) substituents were varied at the aromatic ring of PMH to probe the chemical space around the aromatic ring and its correlation with the activity (Table 1).
Scheme 1.

General synthetic scheme of phenylmethylene hydantoins.40
Table 1.
PMH structures and their in vitro GSK-3β inhibitory activities.
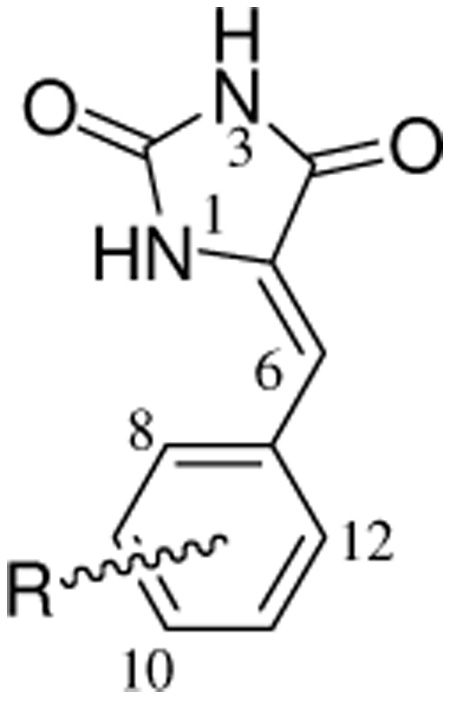 | ||
|---|---|---|
| No. | R | IC50 µM ± SEM |
| 1 | 4-OH | 13.7 ± 1.2 |
| 2 | 4-SCH2CH3 | 7.8 ± 0.7 |
| 3 | 4-N(CH2CH3)2 | 4.2 ± 0.4 |
| 4 | 3-OCH3 | 14.1 ± 1.9 |
| 5 | 4-SCH3 | 6.9 ± 0.9 |
| 6 | 2-thiophene | 13.5 ± 1.1 |
| 7 | 4-thiophene | 6.4 ± 0.6 |
| 8 | 4-Cl | 18.2 ± 2.0 |
| Manzamine Aa | --- | 12.3 ± 1.3 |
Manzamine A was used as a positive control in the in vitro assay.24
The HREIMS data of 5 showed a molecular ion peak at m/z 234.0463, suggesting the molecular formula C11H10N2O2S. Analysis of 1H and 13C NMR data (Experimental Section) indicated that 5 is (Z)-5-(4-(methylthio)benzylidene)-hydantoin. PMH 5 is closely related to the known 2 with the replacement of the S-ethyl group with S-methyl.4,5 The methyl singlet at δ 2.48 was assigned as the S-methyl functionality. This was based on its 3J-HMBC correlation with the quaternary aromatic C-10 (δ 139.7). Meanwhile, the aromatic doublet H-8/H-12 (δ 7.56) showed 3J-HMBC correlations with C-10 and with olefinic methane carbon C-6 (δ 108.7). Detailed 1H, and 13C NMR, and other analytical data for PMHs 1–4 and 6–8 are included in the Supporting Information (Tables S1–S3).
2.2. Molecular Docking Studies
PMHs were docked into the ATP binding site of GSK-3β (PDB code 1q4l) using Surflex Dock interface implemented into SYBYL 8.0.41–43 Surflex is a fully automatic flexible molecular docking algorithm that combines the scoring function from the Hammerhead docking system with a search engine that relies on a surface-based molecular similarity method as a mean to rapidly generate suitable putative poses for molecular fragments.41,42
The corresponding interacting amino acids within the binding site of GSK-3β with structure I-5 are shown in Figure 2A. Compound 3 showed the highest docking score. PMH 3 forms strong interactions with the hinge region of GSK-3β; carbonyl oxygen at position 2 form a H-bonding with backbone nitrogen of Val 135 and the NH at position 3 to the carbonyl oxygen of Asp 133 (Figure 2B). The hydantoin ring was sandwiched between Ala 83, on top, and Leu 188, on the bottom. The aromatic ring is rotated out of plane from the hydantoin plane, allowing extensive interactions with the nucleotide-binding loop. Furthermore, the aniline nitrogen of 3 builds a H-bonding interaction with the guanidine moiety of Arg 141. Interestingly, these interactions are the same hot spots provided by the co-crystallized ligand of I-5, potent and selective inhibitor of GSK-3β (Figures 2A and 2C). Moreover, the phenylmethylene moiety occupies a hydrophobic pocket assembled from Ile 62, Glu 63, and Val 70.
Figure 2.

(A) Detailed view of the co-crystallized structure I-5 and the corresponding interacting amino acids within the binding site of GSK-3β. (B) Detailed view of the docked 3 structure and the corresponding interacting amino-acid moieties within the binding site of GSK-3β. (C) Compound 3 aligned with I-5 in the ATP binding site of GSK-3β. (D) Detailed view of the docked 3 structure in different view angle in the ATP binding site of GSK-3β after the AMBER7 FF02 optimization.
Since the receptor is fixed in docking, the generated PMH 3-GSK-3β complex from docking simulation was pre-minimized using CHARMM. Molecular dynamic simulation using AMBER7 FF02 force field implemented in SYBYL 8.0 was subsequently conducted in the presence of explicit solvent to investigate the stability of H-bonding interactions described above. Figure 2D shows the optimized 3-GSK-3β complex maintaining H-bonding interactions with Asp 133, Val 135, and Arg 141 at optimum distance of 2.11, 1.67, and 3.13 Ǻ, respectively. Although the later H-binding interaction is weaker than the electrostatic interaction created with I-5, targeting Arg 141 is important to improve the activity in the process of designing new derivatives because it is considered the selectivity residue for GSK-3β.44, 45 Many other kinases have either neutral or negatively-charged residues rather than the positively charged guanidine at the same position.45 In CDK2, a kinase that share 33% amino acid identity with GSK-3β, although it bears a positively-charged Lys 86 at that position but it is oriented away from the ligand.46 Accordingly, Arg 141 is unique in GSK-3β and provides an interesting model to explain the high GSK-3β selectivity observed for many inhibitors.46 This unique binding feature is formed in the docked pose of 3, which may justify a possible high PMH selectivity for GSK-3β versus other kinases.
The validation for docking–scoring procedure was performed through employing the same conditions to dock I-5 into the binding pocket of this enzyme (Figure 2A). The docking simulation resulted in a close model to the crystallographic structure (Figure S3, Supporting Information), which highlights the potential and selectivity of PMHs as GSK-3β inhibitors.
2.3. Monitoring GSK-3β Activity Using a Tau [pS396] phosphoELISA™ Kit
A well-known protein substrate of GSK-3β is the microtubule-associated protein tau. GSK-3β phosphorylates tau at the specific sites: serine residue 199 and serine residue 396.26 BioSource has recently introduced phosphoELISA™ kits for monitoring the phosphorylation of tau at serine 396 that have utility in monitoring the activity of GSK-3β (BioSource; KHB7031). The Invitrogen Human Tau [pS396] kit is a solid phase sandwich Enzyme Linked-Immuno-Sorbent Assay (ELISA). To evaluate the inhibitory effect of PMHs against GSK-3β, an in vitro GSK-3β inhibitory assay was conducted. In this inhibitory assay, the concentration of PMH that inhibits 50% of the enzyme, IC50, was measured. Table 1 shows the in vitro GSK-3β inhibitory activities of PMH analogues. Consistent with the in silico studies, PMH 3 shows the most potent inhibition with an IC50 value of 4.2 µM. The validity of the test was established by testing the inhibitory action of the marine-derived GSK-3β inhibitor, manzamine, which showed an IC50 value of 12.3 µM that was comparable to the published data.24 Based on the abovementioned results, PMH 3 was selected for in vivo activity evaluation in rat model for ability to enhance glycogen disposition in liver as a subsequent inhibition of GSK-3β.
2.4. In Vivo Sprague Dawley Rat Model. Determination of Hepatic Glycogen Contents
GSK-3β plays an important role in controlling the hepatic and muscular glycogen content and blood glucose level.35,36 There are different mechanisms by which GSK-3β accomplish this critical function. GSK-3β phosphorylates and inactivates glycogen synthase, the enzyme which converts excess glucose residues to glycogen for storage. Knockdown action, by insulin or small inhibitors, on GSK-3β results in increased glycogen storage in muscle and liver.20–24 A previous study showed that inhibition of GSK-3β selectively reduces glucose-6-posphatase and phosphoenolpyruvate carboxykinase gene expression.47 Consequently, these effects result in the reduction of glucose output and increase of the synthesis of glycogen from d-glucose.47 These findings indicate that GSK-3β inhibitors may have a great therapeutic potential for lowering blood glucose levels.
The most in vitro active PMH 3 was tested in Sprague Dawley rat model to evaluate its in vivo potency by measuring the hepatic glycogen disposition. Figure 3 shows the significant increase in rats’ liver glycogen content at the three dose levels of 3 used in the study (5, 15, and 25 mg/kg) compared to the vehicle control group (P <0.05) in a dose dependent manner.
Figure 3.

Effect of 5, 15, and 25 mg/kg doses of 3 on the liver glycogen storage in Sprague Dawley rats. (n=3 /dose). Error bars indicate the SEM of n=3/dose.
Compound 3 has been previously tested in vivo for its anticonvulsant activity using maximal electroshock seizure (MES) assay for up to 200 mg/kg and did not show CNS-depressant effects.40 In addition, a previous study showed the lack of cytotoxicity of 3 even at 200 µM in vitro against prostate cancer cells.4 These in vitro and in vivo assays provide a preliminary indication of the safety and tolerability of this compound.
2.5. Pharmacophore Model Generation
3D pharmacophore mapping methodology based on distance comparison technique is built for the three most active analogues (2, 3, and 7) using DISCOtech™ module implemented in SYBYL 8.0.48,49 DISCOtech™ is a well established module in designing pharmacophoric map and in the process of virtual screening to discover new leads.50,51 Given a set of molecules that are related by their ability to bind to same protein receptor, DISCOtech™ identifies features that could be elements in a pharmacophore model.48,49 DISCOtech™ operates in distance space and can perform clique detection to generate pharmacophore hypotheses on up to 300 conformers per molecule.48,49 Therefore, DISCOtech™ can be efficiently used with as low as 3–5 compounds to generate validated pharmacophore models.50–53
These diverse conformers are used in DISCOtech’s clique detection routine to find 3D alignments of the pharmacophore features in different molecules.48,49 A clique is a subgraph in which every node is connected to each other’s node.48,49 DISCOtech™ reduces the conformers to sets of pharmacophore features (nodes) and interfeature distances (connections).48,49
The criterion for the validation of the developed pharmacophoric queries was based on in-house database of active and inactive GSK-3β inhibitors (100 each). Representative examples from both sets are included in the supporting information (Figure S1 and S1). Compound is considered active as GSK-3β inhibitor with IC50 ≤ 20 µM.16–21 Model that filters the active and excludes inactive compounds was selected and then optimized. Several initial DISCO runs were performed by varying the tolerance and range of required features. Table 2 shows the results of the initial efforts. Except Run-2(b), all the models turned out to be non-specific for two reasons; either they have few (~4–5) features as in Run-1(a)–Run-1(c) or high tolerance limit as in Run-1(c) and Run-2(c). The highest score query generated from Run-2(b) and named Mod-2(b)-1, which shows a slight selectivity, was selected for further refinement (Figure 4).
Table 2.
Number of models obtained along with the pharmacophoric features and tolerance values for each of the DISCO pharmacophoric run.
| Run Name | Features Range |
Tolerance | Number of Models |
Number of Pharmacophoric Features |
Number of Active GSK-3β Hits* |
Number of Inactive GSK-3β Hits* |
|---|---|---|---|---|---|---|
| Run-1(a) | 3–7 | 0.5 | 164 | 4 | 41 | 22 |
| Run-1(b) | 3–7 | 1 | 297 | 4 | 42 | 27 |
| Run-1(c) | 3–7 | 2 | 497 | 5 | 48 | 21 |
| Run-2(a) | 7–10 | 0.5 | 351 | 9 | 49 | 10 |
| Run-2(b) | 7–10 | 1 | 761 | 9 | 67 | 19 |
| Run-2(c) | 7–10 | 2 | 1011 | 10 | 53 | 22 |
Selected by the pharmacophore model showing the highest score. The highlighted model used for further modification.
Figure 4.

Pharmacophoric features of Mod-2(b)-1and their distance relation generated by DISCOtech™ module. AA–Acceptor atom, DA–Donor atom, HD–Hydrophobic center.
Mod-2(b)-1was optimized by adding and deleting some specific features (Table 3). Adding donor atom feature (DA3) through UNITY interface at the aniline N and removing DA1 and AA2 generated Mod-7 (Figure 5). This query picked more number of GSK-3β inhibitors and only a few numbers of inactive GSK-3β inhibitors. This model is selective enough to pick a complex structure like staurosporine, a potent natural GSK-3β inhibitor (IC50 50 nM), from our in-house database (Figure 5B). Mod-7 has the characteristic features required for an ideal pharmacophoric query, because it possessed the important interactions required for this series of GSK-3β inhibitors, worked consistently with published GSK-3β pharmacophore model, and performed satisfactorily with the in-house database.20,52,53
Table 3.
Models generated by modifying Mod-2(b)-1 along with percentage of hits picked by each model from the in-house database.
| Model no. | Pharmacophoric Featuresa,b |
Number of Active GSK-3β Hits (%) |
Number of Inactive GSK-3β Hits (%) |
|---|---|---|---|
| Mod-1 | DA2, AA1, AA2, | 78 | 11 |
| AA3, HD1, HD2 | |||
| Mod-2 | DA1, DA2, AA2, | 63 | 15 |
| AA3, HD1, HD2 | |||
| Mod-3 | DA1, AA1, AA2, | 61 | 11 |
| AA3, HD1, HD2 | |||
| Mod-4 | DA1, DA2,AA1, | 79 | 10 |
| AA3, HD1, HD2 | |||
| Mod-5 | DA2, AA2, AA3, | 72 | 8 |
| HD1, HD2 | |||
| Mod-6 | AA1, AA2, AA3, | 79 | 8 |
| HD1, HD2 | |||
| Mod-7 | DA2, AA1, AA3, | 89 | 3 |
| HD1, HD2 | |||
| Mod-8 | DA1, AA2, AA3, | 58 | 14 |
| HD1, HD2 |
AA – Acceptor atom, DA – Donor atom, HYD – Hydrophobic center.
The corresponding acceptor and donor sites are not tabulated.
Figure 5.

The Pharmacophoric features of Mod-7 generated by DISCOtech™ module along with PMH 3 (A) and with staurosporine (B), the potent GSK-3β inhibitor. AA–Acceptor atom, DA–Donor atom, HD – Hydrophobic center, DS-Donor site, AS-Acceptor site.
Based on the results above, future design of potent and selective GSK-3β inhibitors should consider the following important hot spots: (i) H-bonding interaction with the hinge region of Asp 133 and Val 135, (ii) targeting Arg 141 and Gln 185 amino acids, and (iii) filling the Val 70, Lys 85 and Cys 99 hydrophobic pocket. For example, keeping the hydantoin ring, and placing carboxylate or other negatively charged moiety at C-9 or C-10 positions, along with benzyl or phenethyl at C-12 can afford potent and selective GSK-3β inhibitors.
3. Conclusion
Experimental in vitro and in vivo GSK-3β inhibitory activities of PMHs were documented. The feasible, cost effective, and regioselective synthesis of this class bode well for their future development as potential therapeutics for cancers, Alzheimer’s disease, bipolar disorders, stroke, different tau pathologies, and type-2 diabetes. The validated pharmacophore model of PMH-derived GSK-3β inhibitors provides a powerful tool to design new leads and to discover virtual screening based-GSK-3β inhibitors from available online databases.
4. Experimental
4.1. General experimental procedures
Melting points were determined using Fisher digital melting point apparatus. IR spectra were recorded on a Varian 800 FT-IR spectrophotometer. Optical rotation measurements were carried out on a Rudolph Research Analytical Autopol III polarimeter. TLC analyses were carried out on precoated silica gel 60 F254 500 µm TLC plates, using MeOH/CHCl3 (1:9) as a developing solvent. The 1H and 13C NMR spectra were recorded in d6-DMSO, using TMS as an internal standard, on a JEOL Eclipse NMR spectrometer operating at 400 MHz for 1H and 100 MHz for 13C. The HREIMS experiments were conducted at the University of Michigan on a Micromass LCT spectrometer.
4.2. Preparation of phenylmethylene hydantoins
Hydantoin (1.0 gm) was dissolved in 10 mL H2O while heating at 70°C on oil bath with continuous stirring (Scheme 1).40 The pH was adjusted to 7.0 using saturated NaHCO3 solution after complete dissolution. The temperature was then raised to 90°C after the addition of 0.9 mL ethanolamine.40 Equimolar quantity of the corresponding aldehyde solution in 2–5 mL EtOH was then added drop-wise with continuous stirring.40 The reaction was kept under reflux for approximately 5–8 h. The reaction was monitored by TLC every hour till a yellow or white precipitate was formed. After complete depletion of the starting aldehyde, the mixture was cooled and the precipitate was filtered and washed with EtOH/H2O (1:5) before recrystallization from EtOH.40 Reaction yield range from 60–90%, based on the nature of the used aldehyde. Generally with few exceptions, it has been noticed that aromatic substitutions with electron donating groups enhance the yield, unlike electron withdrawing functionalities, which decrease the overall yield. Bulk groups at the ortho positions can also diminish the product yield.40
4.2.1. (Z)-5-(4-(methylthio)benzylidene)-hydantoin (5)
Amorphous yellow powder. mp 252–254 °C, 1H NMR (DMSO-d6, 400 MHz) δ 2.48 (s, 3H), 6.38 (s, 1H), 7.25 (d, 2H, J = 8.4), 7.56 (d, 2H, J = 8.1), 10.70 (s, 1H), 11.14 (s, 1H). 13C NMR (DMSO-d6, 100 MHz) δ 14.8 (CH3), 108.7 (CH), 126.2 (2CH), 127.8 (2CH), 129.9 (qC), 130.4 (CH), 139.7 (qC), 156.2 (qC), 166.1 (qC). IR υmax (CHCl3) 3384, 1722, 1648, 1590 cm−1; HREIMS m/z 234.0463 [M]+ (calcd for C11H10N2O2S, 234.0463).
4.3. Molecular Modeling
Three-dimensional structure building and all modeling were performed using the SYBYL program package, version 8.0, installed on DELL desktop workstations equipped with a dual 2.0 GHz Intel® Xeon® processor running the Red Hat Enterprise Linux (version 5) operating system.25 Conformations of each compound were generated using Confort™ conformational analysis. Energy minimizations were performed using the Tripos force field with a distance-dependent dielectric and the Powell conjugate gradient algorithm with a convergence criterion of 0.01 kcal/(mol A).57 Partial atomic charges were calculated using the semiempirical program MOPAC 6.0 and applying the AM1.58
4.3.1. Molecular Docking
Surflex Dock program version 2.0 interfaced with SYBYL 8.0 was used to dock the compounds to the ATP binding site of GSK-3β.43 SurFlex Dock employs an idealized active site ligand (protomol) as a target to generate putative poses of molecules or molecular fragments.59,60 These putative poses were scored using the Hammerhead scoring function.59,60 The 3D structure was taken from the Brookhaven Protein Databank (PDB code: 1q4l).9
4.3.2. Pharmacophore Generation
The three most active analogues 2, 3, and 7, were used to build the pharmacophoric map using DISCOtech™ module. The structure of these compounds were constructed manually using SYBYL 8.0, minimized using the Tripos force field to obtain a local minimum, and partial atomic charges were calculated using the semiempirical program MOPAC 6.0 and applying the AM1. Diverse conformers were generated for each structure using the Confort™ conformational analysis tool in SYBYL. Derivation of the pharmacophore model was undertaken using DISCOtech™. Assignment of the initial pharmacophore features for the DISCO-based pharmacophore mapping was conducted using the following features: aromatic and aliphatic ring centroids as hydrophobic centers, hydrogen bond donors and acceptors, and external site points representing receptor-associated hydrogen bond acceptor sites and donor sites. Validation of the pharmacophoric models was carried out using in-house database search. This database contained a total of 200 compounds including 100 GSK-3β inhibitors and 100 inactive molecules. An acceptable pharmacophoric query should be able to pick up active GSK-3β inhibitors (IC50 ≤20) and should omit the inactive molecules (IC50 >20).
4.4. In vitro GSK-3β Inhibitory Activity Assay Using a Tau [pS396] phosphoELISA™
4.4.1. Preparation of compounds for in vitro enzymatic assay
Each compound was dissolved in DMSO to give a 10 mM solution. Subsequently, 1 µL was transferred to 100 µL HEPES buffer (pH 7.2) to give a final stock solution of 100 µM used for subsequent enzymatic assays.
4.4.2. Determination of the inhibitory IC50 values
Recombinant GSK-3β (Invitrogen) was dissolved in a buffer solution (pH 7.2) containing the following: 40 mM HEPES, 5 mM MgCl2, 5 mM EDTA, 100 µM ATP, and 50 µg/mL heparin to reach a final enzymatic solution of 10 pg/mL. Subsequently, 50 µL aliquots of the enzymatic solution were pipetted into 1.5 mL vials.
Thereafter, appropriate volumes of the compounds’ stock solutions were pipetted into the enzymatic solution to yield 20 µM, 10 µM, 1 µM, and 100 nM of each hit after completion to 75 µL with the buffer solution. The hits were incubated with the enzyme over 30 min at room temperature. Then about 25 µL of 2000 pg/mL tau protein solution in HEPES was added to give a final tau protein concentration of 500 pg/mL. This mixture was incubated over 1 h at room temperature.
The detection of tau phosphorylation was performed as follows: the GSK-3β reaction mixtures were diluted 1:1 with sodium azide aqueous solution (15 mM) to achieve a final tau protein concentration of 250 pg/mL.61 Then 100 µL aliquots of this solution were pipetted into the wells of the tau [pS396] phosphoELISA kit (Invitrogen, Carlsbad, California). Subsequently, the wells were incubated for 2 h at room temperature. Then they were aspirated and washed (with the washing solution provided in the kit). Thereafter, 100 µL aliquots of rabbit detector antibody solution were pipetted in the wells and incubated for 1 h at room temperature. The wells were then aspirated and washed with the washing buffer. About 100 µL aliquots of goat (polyclonal) anti-rabbit IgG-HRP were then added to the wells and incubated for 30 min at room temperature. Subsequently, the wells were aspirated and washed with the washing buffer. Finally, a total of 100 µL of TMB substrate chromogen solution aliquots was added to each well and incubated for 20–30 min. After the termination of the HRP reaction in each well, the solution’s absorbance was measured spectrophotometrically at λ of 450 nm. The marine-derived β-carboline alkaloid GSK-3β inhibitor manzamine A was employed as positive control.24 The IC50 for each compound was calculated using nonlinear regression (curve fit) of log concentration versus percent of inhibition implemented in GraphPad Prism 5.0.
4.5. In Vivo Evaluation
4.5.1. Preparation of Drug Hits
The effect of 3 on liver glycogen content of Sprague Dawley rats was investigated. The sodium salt of 3 was prepared by the addition of equimolar concentration of 1N NaOH solution and the formed salt was then dissolved in PBS (pH 7.2). Three doses of 3 were used; 5, 15 and 25 mg/kg. The drug was dissolved to a concentration (mg/mL) that yielded the desired final dosage (mg/kg) when injected at 0.5 mL/rat.
4.5.2. Determination of Liver Glycogen
Six-week old female Sprague Dawley female rats with average weight of 200 g were used for this investigation. The animals were randomized and fed ad libitum with standard food and water except when fasting was needed in the course of the study. All animals were housed in the same conditions and separated randomly to four groups. Three groups (3 rats/group) used to investigate compound 3 were injected ip with the three escalating doses of PMH 3 (mentioned earlier) and one group was injected ip with PBS as negative control (n=3). On the day of the experiment, food and water were removed 6 h before the injection. The animals were sacrificed by a pentobarbital overdose, and their livers were immediately removed for glycogen determination. Liver glycogen content was determined quantitatively following a reported procedure.62 Briefly, livers were removed immediately after the animals sacrificed and were homogenized using a homogenizer (IKA-T8 Ultra-Turrax, Germany) with appropriate volume of 5% trichloroacetic acid over 5 min.62 The homogenate was centrifuged (Eppendorf centrifuge 5804 R, Germany) at 3000 rpm for 5 min. The supernatant fluid was taken and filtered using acid-washed filter paper, and the residues were homogenized again with another volume of 5% trichloroacetic over 1–3 min to ensure better extraction of glycogen. The glycogen of 1.0 mL of this filtrate was precipitated using ethanol (95%, 5 mL), incubated in water bath at 37–40 °C for 3 h, and centrifuged at 3000 rpm for 15 min. The clear liquid is gently decanted from the packed glycogen, and the tubes were allowed to drain in an inverted position for 10 min. The glycogen was dissolved in distilled water (2 mL) and mixed with 10 mL of the anthrone reagent (0.05% anthrone, 1.0% thiourea in 72% H2SO4). The mixture was incubated in boiling water over 30 min, and subsequently, the absorbance was spectrophotometerically measured at 620 nm by a Spectroscan 80D UV–vis spectrophotometer. Blank and standard solutions were prepared by adding 10 mL of anthrone reagent to 2 mL of water and to 2 mL of glucose solution containing 0.1 mg of glucose in saturated benzoic acid, respectively. The liver glycogen content is estimated using the following formula:
Where DU is the absorbance of the unknown sample and DS is the absorbance of the standard.
Supplementary Material
Acknowledgments
This study was partially supported by the Louisiana Biomedical Research Network.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting Information Available: 1H and 13C NMR, mass, and melting point data of compounds 1–4 and 6–8 (Tables S1–S3) are available. Representative examples for the active and inactive GSK-3β inhibitors used to validate the pharmacophore model in addition to the docking pose of the inhibitor I-5 as produced by docking simulation versus the crystallographic structure of 3 (Figure S1–Figure S3) are also available.
References and notes
- 1.Louwrier S, Ostendorf M, Boom A, Hiemstra H, Speckamp WN. Tetrahedron. 1996;52:2603–2628. [Google Scholar]
- 2.Ohtani I, Kusumi T, Kakisawa H, Kashman Y, Hirsh S. J. Am. Chem. Soc. 1992;114:8472–8479. [Google Scholar]
- 3.Kashman Y, Hirsh S, McConnell OJ, Ohtani I, Kusumi T, Kakisawa H. J. Am. Chem. Soc. 1989;111:8925–8926. [Google Scholar]
- 4.Mudit M, Khanfar M, Muralidharan A, Thomas S, Shah GV, van Soest RWM, El Sayed KA. Bioorg. Med. Chem. 2009;17:1731–1738. doi: 10.1016/j.bmc.2008.12.053. [DOI] [PubMed] [Google Scholar]
- 5.Shah GV, Muralidharan A, Thomas S, Gokulgandhi M, Mudit M, Khanfar M, El Sayed KA. Mol. Cancer Ther. 2009;8:509–520. doi: 10.1158/1535-7163.MCT-08-0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thenmozhiyal JC, Wong PT, Chui WK. J. Med. Chem. 2004;47:1527–1535. doi: 10.1021/jm030450c. [DOI] [PubMed] [Google Scholar]
- 7.Meuesel M, Gutschow M. Org. Prep. & Procedures Int. 2004;36:391–443. [Google Scholar]
- 8.Ishiguro K, Shiratsuchi A, Sato S, Omori A, Arioka M, Kobayashi S, Uchida T, Imahori K. FEBS Lett. 1993;325:167–172. doi: 10.1016/0014-5793(93)81066-9. [DOI] [PubMed] [Google Scholar]
- 9.(a) Lovestone S, Reynolds CH, Latimer D, Davis DR, Anderton BH, Gallo J-M, Hanger D, Mulot S, Marquardt B. Curr. Biol. 1994;4:1077–1086. doi: 10.1016/s0960-9822(00)00246-3. [DOI] [PubMed] [Google Scholar]; (b) Klein PS, Melton DA. Proc. Natl. Acad. Sci. U.S.A. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Pap M, Cooper GM. J. Biol. Chem. 1998;273:19929–19932. doi: 10.1074/jbc.273.32.19929. [DOI] [PubMed] [Google Scholar]
- 10.Welsh GI, Proud CG. Biochem. J. 1993;294:625–629. doi: 10.1042/bj2940625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wagman AS, Johnson KW, Bussiere DE. Curr. Pharm. Des. 2004;10:1105–1137. doi: 10.2174/1381612043452668. [DOI] [PubMed] [Google Scholar]
- 12.Li X, Lu F, Tian Q, Yang Y, Wang Q, Wang JZ. J. Neural Transm. 2006;113:93–102. doi: 10.1007/s00702-005-0303-7. [DOI] [PubMed] [Google Scholar]
- 13.Neary Joseph T, Kang Yuan. J. Neurosci. Res. 2006;84:515–524. doi: 10.1002/jnr.20969. [DOI] [PubMed] [Google Scholar]
- 14.Martinez A, Castro A, Dorronsoro I, Alonso M. Med. Res. Rev. 2002;22:373–384. doi: 10.1002/med.10011. [DOI] [PubMed] [Google Scholar]
- 15.Iqbal K, Grundke-Iqbal I. J. Cell. Mol. Med. 2008;12:38–55. doi: 10.1111/j.1582-4934.2008.00225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinez A, Alonso M, Castro A, Perez C, Moreno FJ. J. Med. Chem. 2002;45:1292–1299. doi: 10.1021/jm011020u. [DOI] [PubMed] [Google Scholar]
- 17.Martinez A, Alonso M, Castro A, Dorronsoro I, Gelpi JL, Luque FJ, Perez C, Moreno FJ. J. Med. Chem. 2005;48:7103–7112. doi: 10.1021/jm040895g. [DOI] [PubMed] [Google Scholar]
- 18.Castro A, Encinas A, Gil C, Brase S, Porcal W, Perez C, Moreno FJ, Martinez A. Bioorg. Med. Chem. 2008;16:495–510. doi: 10.1016/j.bmc.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 19.Naerum L, Norskov-Lauritsen L, Olesen PH. Bioorg. Med. Chem. Lett. 2002;12:1525–1528. doi: 10.1016/s0960-894x(02)00169-5. [DOI] [PubMed] [Google Scholar]
- 20.Witherington J. Glycogen Synthase Kinase 3 (GSK-3) and Its Inhibitors. 2006:281–305. [Google Scholar]
- 21.Taha MO, Bustanji Y, Al-Ghussein MAS, Mohammad M, Zalloum H, Al-Masri IM, Atallah N. J. Med. Chem. 2008;51:2062–2077. doi: 10.1021/jm7009765. [DOI] [PubMed] [Google Scholar]
- 22.Meijer L, Thunnissen AMWH, White AW, Garnier M, Nikolic M, Tsai LH, Walter J, Cleverley KE, Salinas PC, Wu YZ, Biernat J, Mandelkow EM, Kim SH, Pettit GR. Chem. Biol. 2000;7:51–63. doi: 10.1016/s1074-5521(00)00063-6. [DOI] [PubMed] [Google Scholar]
- 23.Alonso GD, Dorronsoro DI, Martinez GA, Panizo dPG, Fuertes HA, Perez Puerto MJ, Martin AE, Perez ND, Medina PM. PCT Int. Appl. 2005:WO 2005054221. [Google Scholar]
- 24.Hamann MT, Alonso D, Martín-Aparicio E, Fuertes A, Pérez-Puerto J, Castro A, Morales S, Navarro ML, del Monte-Millán M, Medina M, Pennaka H, Balaiah A, Peng J, Cook J, Wahyuono S, Martínez A. J. Nat. Prod. 2007;70:1397–1405. doi: 10.1021/np060092r. [DOI] [PubMed] [Google Scholar]
- 25.Dessalew N, Patel DS, Bharatam PV. J. Mol. Graphics Modelling. 2007;25:885–895. doi: 10.1016/j.jmgm.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 26.Godemann R, Biernat J, Mandelkow E, Mandelkow EM. FEBS Lett. 1999;454:157–164. doi: 10.1016/s0014-5793(99)00741-3. [DOI] [PubMed] [Google Scholar]
- 27.Gong CX, Lidsky T, Wegiel J, Zuck L, Grundke-Iqbal I, Iqbal K. J. Biol. Chem. 2000;275:5535–5544. doi: 10.1074/jbc.275.8.5535. [DOI] [PubMed] [Google Scholar]
- 28.Iqbal K, Grundke-Iqbal I. Cur. Drug Target. 2004;5:495–502. doi: 10.2174/1389450043345254. [DOI] [PubMed] [Google Scholar]
- 29.Phiel CJ, Wilson CA, Lee VM-Y, Klein PS. Nature. 2003;423:435–439. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- 30.Takashima A, Noguchi K, Michel G, Mercken M, Hoshi M, Ishiguro K, Imahori K. Neurosci. Lett. 1996;203:33–36. doi: 10.1016/0304-3940(95)12257-5. [DOI] [PubMed] [Google Scholar]
- 31.Culbert AA, Brown MJ, Frame S, Hagen T, Cross DAE, Bax B, Reith AD. FEBS Lett. 2001;507:288–294. doi: 10.1016/s0014-5793(01)02990-8. [DOI] [PubMed] [Google Scholar]
- 32.Takashima A, Noguchi K, Sato K, Hoshino T, Imahori K. Proc. Natl. Acad. Sci. USA. 1993;90:7789–7793. doi: 10.1073/pnas.90.16.7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alvarez G, Munoz-Montano JR, Satrustegui J, Avila J, Bogonez E, Diaz-Nido J. FEBS Lett. 1999;453:260–264. doi: 10.1016/s0014-5793(99)00685-7. [DOI] [PubMed] [Google Scholar]
- 34.Hoshi M, Takashima A, Noguchi K, Murayama M, Sato M, Kondo S, Saitoh Y, Ishiguro K, Hoshino T, Imahori K. Proc. Natl. Acad. Sci. USA. 1996;93:2719–2723. doi: 10.1073/pnas.93.7.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.MacAulay K, Woodgett JR. Expert Opin. Therap. Targets. 2008;12:1265–1274. doi: 10.1517/14728222.12.10.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu S, Shen Z. Yaoxue Xuebao. 2007;42:1227–1231. [Google Scholar]
- 37.Medina M, Castro A. Current Opin. Drug Discovery Devel. 2008;11:533–543. [PubMed] [Google Scholar]
- 38.Smith DG, Buffet M, Fenwick AE, Haigh D, Ife RJ, Saunders M, Slingsby BP, Stacey R, Ward RW. Bioorg. Med. Chem. Lett. 2001;11:635–639. doi: 10.1016/s0960-894x(00)00721-6. [DOI] [PubMed] [Google Scholar]
- 39.Bertrand JA, Thieffin eS, Vulpetti A, Cristiani C, Valsasina B, Knapp S, Kalisz HM, Flocco M. J. Mol. Biol. 2003;333:393–407. doi: 10.1016/j.jmb.2003.08.031. [DOI] [PubMed] [Google Scholar]
- 40.Thenmozhiyal JC, Wong PT, Chui WK. J. Med. Chem. 2004;47:1527–1535. doi: 10.1021/jm030450c. [DOI] [PubMed] [Google Scholar]
- 41.Jain AN. J. Med. Chem. 2003;46:499–511. doi: 10.1021/jm020406h. [DOI] [PubMed] [Google Scholar]
- 42.Kellenberger E, Rodrigo J, Muller P, Rognan D. Proteins. 2004;57:225–242. doi: 10.1002/prot.20149. [DOI] [PubMed] [Google Scholar]
- 43.Tripos Associates. SYBYL Molecular Modeling Software. St. Louis, MO: Tripos Associates; 2007. version 8.0 http://www.tripos.com. [Google Scholar]
- 44.Kuo GH, Prouty C, DeAngelis A, Shen L, O'Neill DJ, Shah C, Connolly PJ, Murray WV, Conway BR, Cheung P, Westover L, Xu JZ, Look RA, Demarest KT, Emanuel S, Middleton SA, Jolliffe L, Beavers MP, Chen X. J. Med. Chem. 2003;46:4021–4031. doi: 10.1021/jm030115o. [DOI] [PubMed] [Google Scholar]
- 45.Hanks S, Quinn AM. Methods Enzymol. 1991;200:38–62. doi: 10.1016/0076-6879(91)00126-h. [DOI] [PubMed] [Google Scholar]
- 46.Lawrie AM, Noble ME, Tunnah P, Brown NR, Johnson LN, Endicott JA. Nat. Struct. Mol. Biol. 1997;4:796–801. doi: 10.1038/nsb1097-796. [DOI] [PubMed] [Google Scholar]
- 47.Lochhead PA, Coghlan M, Rice SQJ, Sutherland C. Diabetes. 2001;50:937–946. doi: 10.2337/diabetes.50.5.937. [DOI] [PubMed] [Google Scholar]
- 48.Martin YC, Bures MG, Danaher EA, DeLazzer J, Lico I, Pavlik PA. J. Comp.-Aided Mol. Design. 1993;7:83–102. doi: 10.1007/BF00141577. [DOI] [PubMed] [Google Scholar]
- 49.Clark RD. J. Chem. Info. Comp. Sci. 1997;37:1181–1188. [Google Scholar]
- 50.Marriott DP, Dougall IG, Meghani P, Liu Y-J, Flower DR. J. Med. Chem. 1999;42:3210–3216. doi: 10.1021/jm980409n. [DOI] [PubMed] [Google Scholar]
- 51.Spadoni G, Balsamini C, Diamantini G, Di Giacomo B, Tarzia G, Mor M, Plazzi PV, Rivara S, Lucini V, Nonno R, Pannacci M, Fraschini F, Stankov BM. J. Med. Chem. 1997;40:1990–2002. doi: 10.1021/jm960651z. [DOI] [PubMed] [Google Scholar]
- 52.Kim H-J, Choo H, Cho YS, No KTi, Pae AN. Bioorg. Med. Chem. 2008;16:636–643. doi: 10.1016/j.bmc.2007.10.047. [DOI] [PubMed] [Google Scholar]
- 53.Patel DS, Bharatam PV. J. Comp-Aided Mol. Design. 2006;20:55–66. doi: 10.1007/s10822-006-9036-4. [DOI] [PubMed] [Google Scholar]
- 54.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv. Drug Del. Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 55.Jones G, Willett P, Glen RC. J. Mol. Biol. 1995;245:43–53. doi: 10.1016/s0022-2836(95)80037-9. [DOI] [PubMed] [Google Scholar]
- 56.Jones G, Willett P, Glen RC, Leach AR, Taylor R. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 57.Clark M, Cramer RD, III, van Opdenbosch N. J. Comput. Chem. 1989;10:982–1012. [Google Scholar]
- 58.Stewart JJ. J. Comput. Aided Mol. Des. 1990;4:1–103. doi: 10.1007/BF00128336. [DOI] [PubMed] [Google Scholar]
- 59.Welch W, Ruppert J, Jain AN. Chem. Biol. 1996;3:449–462. doi: 10.1016/s1074-5521(96)90093-9. [DOI] [PubMed] [Google Scholar]
- 60.Ruppert J, Welch W, Jain AN. Protein Sci. 1997;6:524–533. doi: 10.1002/pro.5560060302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cho JH, Johnson GV. J. Biol. Chem. 2003;278:187–193. doi: 10.1074/jbc.M206236200. [DOI] [PubMed] [Google Scholar]
- 62.Carroll NV, Longley RW, Roe JH. J. Biol. Chem. 1956;220:583–593. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.