

Abstract
Purpose
Lens epithelium–derived growth factor (LEDGF) is upregulated in response to stress and enhances the survival of neurons in the retina and optic nerve, as well as a wide range of other cells, such as fibroblasts and keratinocytes. Photoreceptor protection was investigated in the RCS rat retinal degeneration model after Ledgf delivery with an adeno-associated virus (AAV) and the mechanism of protection explored.
Methods
Thirty-six RCS and nine P23H rats had bilateral subretinal injections of AAV-Ledgf in one eye and buffer in the contralateral eye as the control. Retinal function was evaluated 8 weeks later by the electroretinogram and compared with photoreceptor cell layer count. LEDGF mRNA and protein levels and mRNA levels of known stress-related factors were compared in treated and control retinas to explore the mechanism of LEDGF protection. Nine RCS rats were treated with adenovirus-heat shock protein 27 (Ad-HSP27) and examined for protection.
Results
Significant photoreceptor protection was evident functionally and morphologically in 65% to 100% of the RCS rats treated at early ages of up to 7 weeks. Cell protection was more prominent in the superior retinal hemisphere which has a slower natural degeneration rate in untreated eyes. Although many of the heat shock proteins and other stress-related genes showed significant elevation in the AAV-Ledgf–treated eyes, all increases were approximately twofold or less. Transduction of retinal cells with Ad-HSP27 also resulted in photoreceptor protection. AAV-Ledgf elicited no photoreceptor functional protection in P23H rhodopsin transgenic rat retina.
Conclusions
Chronic LEDGF treatment via AAV-Ledgf administration gave successful protection of photoreceptors in the RCS rat retina and retarded cell death by about 2 weeks. Induction of heat shock proteins also gave photoreceptor protection. However, compelling evidence was not found that LEDGF protection was associated with upregulation of heat shock proteins.
Sequence changes in more than 50 genes have been identified to date as causing retinitis pigmentosa (RP),1 a progressive photoreceptor degeneration leading to blindness.2 These genes are involved in diverse functions, from the phototransduction cascade to protein folding, cell–cell interactions, intracellular trafficking, RNA splicing, proteasomal degradation, cell cycle control, and more.2 Despite this genetic heterogeneity, many forms of RP converge to a common mechanism of apoptosis in human diseases and in animal models.3,4 Therefore, a propitious treatment strategy for drug development for RP would be to target the apoptotic cascade generally rather than each specific causal mutation.5,6
Lens epithelium–derived growth factor (LEDGF), also known as PC4 and SFRS1 interacting protein 1 (Psip1), is a survival factor of the hepatoma-derived growth factor protein family,7 which becomes less sensitive to heat and proteolytic degradation by binding to heparin.8 LEDGF is a secreted protein that is naturally induced by stress signals, localizes to the nucleus, and, in turn, activates stress-related genes.9,10 Enhancing endogenous LEDGF supply, as well as exogenous administration of LEDGF, enhances the survival of retinal neurons under in vitro and in vivo conditions.11
LEDGF binds to promoter elements of many stress-related genes, including those for heat shock proteins (HSPs).11 HSPs are a group of proteins that are upregulated in response to stress and protect cells from damage subsequent to exposure to heat, ischemia, or toxins.12–14 Several groups have found that HSPs, including HSP25 and αβ-crystallin, exert a direct and negative effect on apoptosis.15,16
We previously reported that intravitreous injection of LEDGF promotes photoreceptor survival in light-damaged and Royal College of Surgeons (RCS) rats.17 The RCS rat is a model of progressive photoreceptor degeneration over the first few months of life.18 It harbors a mutation in the receptor tyrosine kinase Mertk gene19 that leads to photoreceptor cell loss from defective phagocytosis by the retinal pigment epithelium (RPE) and causes an abnormal accumulation of debris between the RPE and the neural retina. In this study, we asked whether the effectiveness of LEDGF could be increased by chronic expression in genetic animal models of retinal degeneration. For this purpose, we introduced the Ledgf gene to the retina of RCS rats via an adeno-associated virus (AAV) construct. In addition, conclusions drawn in our previous study were limited by use of the fusion protein GST-LEDGF, which complicated the interpretation, even though we found no effect of GST alone. We have circumvented this problem by using AAV-Ledgf. We found that LEDGF delivery via subretinal injection of the AAV-Ledgf construct was successful in preserving photoreceptors and retinal function in the RCS rat retina but not in P23H rhodopsin transgenic rats. The protection in RCS rats was not associated with substantial overexpression of small HSPs. However, providing the HSP27 gene (human orthologue of rat Hsp25) via an adenovirus vector to the RCS rat retina produced protection similar to that of AAV-Ledgf.
Methods
Isolation of the Rat Ledgf Clone
The rat Ledgf open reading frame (ORF) was cloned by RT-PCR. The first strand of rat Ledgf cDNA was synthesized by using random primers and the total rat retina RNA as template. For amplifying rat Ledgf, the Ledgf gene–specific primers were designed based on the sequences of human, bovine, and mouse genes. The PCR product was cloned into a vector (pCRII-Blunt; Invitrogen, San Diego, CA) for sequencing analysis (pLedgf). A genetic analysis system (CEQ 8000; Beckman Coulter, Fullerton, CA) was used for DNA sequencing, with analysis by a commercial computer program (DNAstar; Madison, WI).
Introducing a Silent Mutation and a Kozac Site into the Rat Ledgf
Site-directed mutagenesis of the pLedgf plasmid DNA was performed in a standard protocol (QuikChange kit; Stratagene, La Jolla, CA) with a pair of oligonucleotides (5′-CATGAGAAAGAAGCgGCAGATCGGAAACGC-3′ and 5′-GCGTTTCCGATCTGCcGCTTCTTTCTCATG-3′). The new plasmid, pMLedgf, was confirmed to carry the silent mutation by sequencing. Two oligonucleotides (5′-CTAGCTAGCGCCACCATGACTCGCGATTTCA AACCTG-3′ and 5′-GCCGGATTTTCTTCAGGGTTGT-3′) were synthesized as PCR primers to introduce an NheI restriction site and a Kozac site of GCCACC in front of the ATG start codon with pMLedgf as the template.
Recombinant AAV-Ledgf Plasmid Construction, Production, and Purification
The Cis AAV-Ledgf vector was made by inserting the NheI and EcoRI cut fragment from the pMLedgf (Koz) plasmid into the same restriction enzyme site of pZac2.1 (University of Pennsylvania Medical School, Philadelphia, PA), in which the rat Ledgf is driven by a CMV promoter. The AAV2/5-Ledgf construct was produced and purified as described elsewhere.20,21 Briefly, AAV2/5-Ledgf was produced by triple transfection of 293 cells. The first plasmid encoded the rat Ledgf expression cassette packaged between the AAV2 internal terminal repeats; the second plasmid encoded the rep and AAV5 cap genes, and the third encoded the adenoviral helper function genes. The virus was purified on a CsCl gradient. The virus physical titer was assessed by real-time PCR, and prep infectivity was assessed by infectious center assay.22 The ratio of genomic copy number (GC) to infectious center assay (ICA) was 650.
Recombinant Ad-CMV-HSP27 Generation
The plasmid DNA of pcDNA-HSP27 was a gift from Karl Csaky (Department of Ophthalmology, Duke University Medical Center, Durham, NC) and the adenoviral shuttle vector was purchased from ViraQuest Inc. (pAdCMV; North Liberty, IA). The open reading frame of human HSP27 (NCBI accession number AB020027; National Center for Biotechnology Information, Bethesda, MD; http://www.ncbi.nlm.nih.gov/ Genbank) was released from the plasmid of pcDNA-HSP27 by XbaI and EcoRI digestion and inserted into the same restriction sites of the pAdCMV vector. Recombinant Ad-CMV-HSP27 was produced and purified with a rapid adenovirus production system (RAPA; ViraQuest Inc.) at 3.0 pfu/mL.
AAV-Ledgf Delivery to the Rat Retina
Dystrophic RCS (RCS-p+/Lav) and heterozygous P23H rhodopsin transgenic line 1 (P23H) were used in this study. The animals were anesthetized by intraperitoneal injection of ketamine (80 mg/kg body weight) and xylazine (5 mg/kg body weight) and subretinal injection was performed.23 Briefly, an incision was made in the temporal conjunctiva 2 to 3 mm from the limbus, and a 30-gauge needle was used to puncture the exposed sclera. A blunt 33-gauge needle was inserted through the puncture and positioned to penetrate the retina, and the solution was injected into the subretinal space with a syringe (Hamilton, Reno, NV). One eye of each animal was injected with 2 μL of AAV-Ledgf solution, and the fellow eye was injected with PBS (pH 7.4) as the control. Animal studies were conducted in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research, and the protocols were approved by the Animal Care and Use Committee of the National Eye Institute of the National Institutes of Health.
ERG Recording
Retinal function was examined 3 to 9 weeks after AAV-Ledgf administration. Animals were dark-adapted overnight and handled under dim red light. Pupils were fully dilated with topical corneal 0.5% tropicamide and 2.5% phenylephrine HCl. ERGs were recorded from both eyes simultaneously by placing a gold-wire loop electrode on each cornea after topical 0.5% proparacaine HCl corneal anesthesia. A gold-wire reference electrode touched the sclera near the limbus of each eye, and a neutral electrode was clipped to the ear. ERGs were elicited with xenon white photostrobe 10-μs flashes delivered into a globe to provide a full-field stimulus. Dark-adapted responses were recorded across a 7-log-unit range of stimulus intensity in 1-log-unit steps beginning just below normal response threshold up to maximum intensity of 0.6 log cd · s/m2. Light-adapted responses were recorded against a constant white background light of 34 cd/m2 that suppresses rod function. ERG responses were amplified and filtered (5000 gain, 0.1–1000 Hz). Small amplitude differences may exist between eyes regardless of treatment. We established a strict protection criterion of b-wave amplitude at the maximum stimulus intensity as ≥100% larger in the AAV-Ledgf–treated eye versus the contralateral control eye injected with PBS or empty viral vector. After the ERG recordings, retinas were removed for protein and RNA analyses or histology.
mRNA Isolation and RT-PCR
After the electroretinogram (ERG) was recorded to evaluate retinal function, the animals were euthanatized by CO2 inhalation, and the eyes were enucleated and retinas removed. The RNA was isolated and purified (TRIzol reagent; Invitrogen), and random primers and M-MLV reverse transcriptase (Invitrogen) were used for first-strand synthesis. The exogenous Ledgf expression was tested by PCR-RFLP, with the oligonucleotide primers 5′-AAAAGGGGCCAGAGGAAAAACC-3′ and 5′-GCCGGA TTTTCTTCAGGGTTG T-3′. The PstI site was used to distinguish exogenous Ledgf from endogenous Ledgf.
Expression of Hsp25 and αβ-crystallin was determined in retinal extracts from treated animals. The primers for amplifying Hsp25 were 5′-CACAGCCGCCTC TTCGAT CAA-3′ and 5′-CGGGCAGAGGGCGCA-CATA-3′. αβ-crystallin primers were 5′-CTTCGGCCACCCTCCT-TCCT-3′ and 5′-CGTCCT GGCGCTCTTCGTG-3′. The reaction conditions were 95°C 30 seconds (denaturation); 57 to 65°C (depending on primer Tm), 30 seconds (annealing); 72°C, 30 seconds (primer extension). PCR products were separated on the agarose gel and visualized by adding ethidium bromide.
Real-Time Quantitative PCR
The same first-strand cDNA was used for real-time PCR of Ledgf, Hsp25, αβ-crystallin, and Bcl2. Equal amounts of cDNA were used in triplicate and amplified with PCR supermix (Platinum SYBR Green qPCR SuperMix-UDS; Invitrogen). The PCR conditions were set at 50°C for 2 minutes, 95°C for 3 minutes followed by 40 cycles of 95°C for 30 seconds, 57°C to 65°C (depending on each primer's Tm) for 30 seconds, 72°C for 30 seconds. Additional reactions were performed on known copy numbers of plasmid DNA corresponding to each gene as a PCR template to construct a standard curve relating threshold cycle to template copy number. Amplification efficiencies were validated and then normalized against the housekeeping gene glyceraldehyde-3-phyosphate dehydrogenase (GAPDH). Primers for real-time PCR of Ledgf were 5′-AGGCCAAAAGGAAGAAGAGAAG-3′ and 5′-TGTGAGCAGCC TGGAAGTTT-3′. The primers for real-time PCR of the remaining genes were the same as for the nonquantitative RT-PCR.
Apoptosis Pathway and Stress and Toxicity Pathway Array Analysis
Neural retinas were collected from three treated RCS rats for which the ERG had shown protection and analyzed individually. Total RNA was isolated from each retina (TRIzol reagent; Invitrogen), and purified (RNeasy Mini kit; Qiagen, Valencia, CA) according to the manufacturer's instruction. Two pathway array analyses were conducted by an apoptosis PCR profiler array (RT2) and stress and toxicity array systems (all from SuperArray Biosciences; Frederick, MD). Equal amounts of RNA (800 ng) were taken for all samples, and reverse transcription was performed (RT2 First-Strand Kit; SuperArray), according to the manufacturer's instruction. Total reaction volume was 20 μL diluted to 100 μL. PCR reactions were performed using PCR profiler arrays of the rat stress and toxicity pathway and rat apoptosis pathway (SuperArray Biosciences) on a thermocycler (iCycler; Bio-Rad, Hercules, CA) and real-time PCR master mix (RT2 SYBR Green PCR master mix; SuperArray Biosciences) with 25 μL total PCR reaction volume. Thermocycler parameters were 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. Relative changes in gene expression were calculated with the ΔΔCt (threshold cycle) method. The PCR array results were validated for 17 genes individually by real-time PCR run in triplicate.
Western Blot Analysis
Neural retinas were homogenized individually and 30 μg of protein from each sample was separated on 10% SDS PAGE gel, transferred onto nitrocellulose membranes (Hybond; Hyclone, Logan, UT), and incubated with antibody (anti-LEDGF, anti-HSP25 or anti-αβ-crystallin). For LEDGF detection, a rabbit polyclonal LEDGF antibody against the C-terminal of LEDGF (amino acid residues 420-438) was generated. The protein was extracted from rat retina, and its concentration was determined with a bicinchoninic acid (BCA) protein assay reagent kit (Pierce, Rockford, IL). Proteins were detected with Western blot detection reagent (ECL; GE Healthcare, Piscataway, NJ). Band intensities were quantified (Kodak Image Station system; Eastman Kodak Co., Rochester, NY), and the intensity ratio of the protein over β-actin in each blot was used to evaluate protein expression level.
Histology
Eyes from a group of five rats showing protection in the AAV-Ledgf–treated eye 8 weeks after injection were enucleated after the ERG recording for evaluation of the distribution of photoreceptor loss. This time point was chosen based on the number of animals showing protection and the time course of AAV gene expression in the rat. Tissue was fixed overnight in 2.5% glutaraldehyde and 2.0% paraformaldehyde, rinsed in 1× PBS and embedded in Araldite. Sections 0.5 μm thick were stained with toluidine blue. As photoreceptor cell protection may be uneven across the retina, we evaluated the retina methodically by counting photoreceptor nuclei at 200-μm intervals in serial vertical sections from the nasal to the temporal side. Each section was sampled every 300 μm from the superior edge to the inferior edge. The blocks were slightly turned continuously, and retinal morphology was monitored to ensure that sections were radial. Inner nuclear layer (INL) and inner plexiform layer (IPL) thicknesses, as well as Müller cell processes crossing the IPL continuously in the plane of section, were used to judge that the sections were not oblique. Generally, sections at the far periphery on both nasal and temporal sides did not meet these criteria and were excluded. Statistical analysis was performed with two-way ANOVA (variable 1, AAV-Ledgf vs. PBS; variable 2, retinal site). In addition, each section was scanned entirely from the superior edge to the inferior edge, and the minimum and maximum number of photoreceptor cell nuclei in the superior and inferior hemispheres of each section was noted.
Results
Functional Protection by AAV-Ledgf
Forty-two animals were treated by subretinal injection of AAV-Ledgf in one eye. PBS buffer in the contralateral eye served as the control. Since functional protection was determined by interocular comparison, six animals were excluded because of lens or corneal opacity in either eye. Results of the remaining 36 animals are summarized in Table 1. Two thirds of the cases showed protection in rats injected at age 5 to 7 weeks and examined 5 weeks later. We found protection in all treated animals with longer expression time of the virus in animals treated at 5 to 7 weeks and examined 8 to 9 weeks later. We then examined the protection when treating at an earlier stage of degeneration. Animals treated at 2.5 to 3 weeks and examined 8 to 9 weeks after injection showed protection in 63% of the cases. We found no evidence of protection in two animals treated at an advanced stage of degeneration.
Table 2.
ONL Cell Count in AAV-Ledgf–Treated versus Control Retinas
| Inferior Hemisphere | Superior Hemisphere | |||||||
|---|---|---|---|---|---|---|---|---|
| AAV-Ledgf | PBS | AAV-Ledgf | PBS | |||||
| Rat No. | Average | Range | Average | Range | Average | Range | Average | Range |
| 1 | 0.44 ± 0.31 | 0 to 1.11 ± 0.89 | 0.23 ± 0.17 | 0 to 0.83 ± 0.39 | 2.49 ± 0.75 | 2.26 ± 1.37 to 4.00 ± 1.41 | 0.93 ± 0.51 | 0.20 ± 0.56 to 2.13 ± 0.66 |
| 2 | 2.48 ± 1.37 | 1.33 ± 1.05 to 3.83 ± 2.21 | 1.60 ± 0.42 | 0.40 ± 0.72 to 3.57 ± 1.98 | 3.18 ± 0.81 | 2.26 ± 0.61 to 5.94 ± 1.80 | 1.66 ± 0.57 | 1.05 ± 0.63 to 4.27 ± 1.05 |
| 3 | 0.91 ± 0.41 | 0.07 ± 0.23 to 2.54 ± 0.56 | 1.26 ± 1.08 | 0.55 ± 1.03 to 3.55 ± 1.91 | 2.36 ± 0.64 | 1.74 ± 0.45 to 4.61 ± 1.08 | 1.64 ± 1.06 | 0.80 ± 0.85 to 4.16 ± 1.15 |
| 4 | 0.62 ± 0.39 | 0 to 1.68 ± 0.73 | 0.63 ± 0.40 | 0 to 1.84 ± 0.74 | 0.70 ± 0.49 | 0 to 3.18 ± 1.37 | 0.77 ± 0.35 | 0 to 2.23 ± 0.59 |
| 5 | 1.24 ± 0.68 | 0.07 ± 0.26 to 2.83 ± 0.64 | – | – | 2.36 ± 1.07 | 0.77 ± 0.94 to 4.87 ± 0.55 | – | – |
AAV-Ledgf construct was delivered at 3 weeks of age, and the morphology was evaluated 8 weeks later. Serial sections, 200 μm apart, were evaluated for each retina. For each hemisphere in each section, the average number of photoreceptor cells, as well as the range (minimum and maximum cell count) was recorded. Since two-way ANOVA showed no nasal–temporal differences, counts in all retinal slices per hemisphere per eye were averaged. Data are represented as the mean ± SD. Tissue from the control eye of animal 5 was damaged in preparation.
A representative example of functional protection is seen in Figure 1A. In the dark-adapted ERG, the b-wave response of the PBS treated eye is seen for only the two brightest stimuli, whereas in the AAV-Ledgf–treated eye, the b-wave is evident for stimuli 3 log units dimmer at −3.4 log cd · s/m2. A population of 12 animals injected at 2 to 3 weeks and examined at 11 to 12 weeks of age showed protection of b-wave amplitude in both dark- and light-adapted responses, and for both conditions the amplitudes were consistently two to four times higher in AAV-Ledgf–treated eyes than in control eyes (Fig. 1B).
Figure 1.
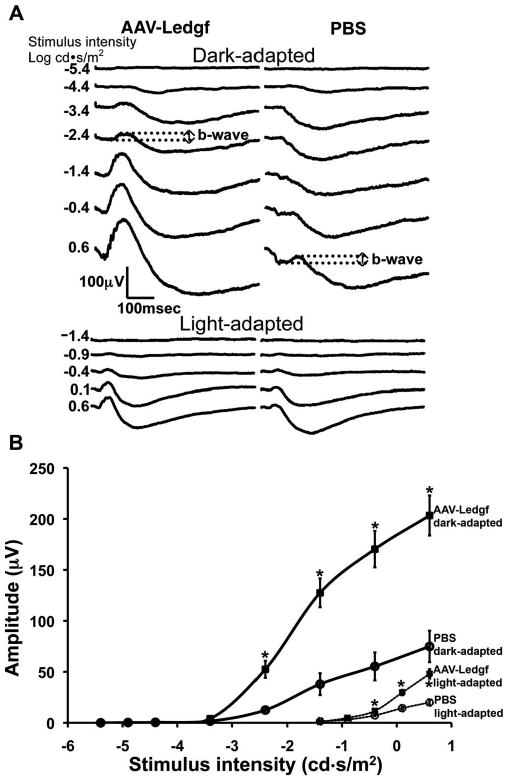
Functional evidence of protection. (A) Representative ERG responses of AAV-Ledgf–treated eyes versus PBS-treated contralateral control eyes show larger amplitudes and a 3-log-unit difference in stimulus intensity to reach equivalent response amplitude (dotted lines demonstrate that b-wave amplitude to maximum stimulus intensity in the control eye is equivalent to b-wave amplitude in response to a stimulus intensity that is 3 log units lower in the AAV-Ledgf–treated eye). (B) Dark- and light-adapted ERG intensity response functions from RCS rats treated with subretinal injection of AAV-Ledgf in one eye and PBS in the contralateral eye. The eyes were treated at 2.5 to 3 weeks of age and examined at 11 to 12 weeks. Bars, mean ± SE. *Amplitudes different at P < 0.01 level by paired Student's t-test (n = 12).
We examined the association between Ledgf mRNA level and functional protection. A silent mutation was introduced into the Ledgf open reading frame (T927G) to eliminate the unique PstI restriction site. This allows distinction between endogenous mRNA and exogenous mRNA introduced by the AAV-Ledgf construct, as the endogenous mRNA is cut by PstI whereas the exogenous mRNA remains uncut (Fig. 2A). Nine animals were injected at 5 to 7 weeks of age with AAV-Ledgf in one eye and PBS in fellow eyes. ERGs were recorded 5 weeks later and the eyes removed for PCR analysis. ERG showed that six of the nine AAV-Ledgf–treated eyes had functional protection; they also had exogenous Ledgf mRNA. One eye showed no protection and no exogenous Ledgf mRNA, and the two remaining did not show protection but did demonstrate exogenous Ledgf. Thus, seven of the nine treated eyes were concordant for functional protection and exogenous Ledgf message. Real-time PCR was performed in the six animals showing protection to compare total Ledgf RNA message levels between the AAV-Ledgf eyes and PBS eyes. The amplification efficiency was validated and normalized by calculating the copy number ratio of Ledgf to the Gapdh housekeeping gene (Fig. 2B). This ratio was 2.1 ± 0.2 times higher in the AAV-Ledgf eyes versus PBS eyes (mean ± SD, n = 6, P = 0.002 by paired t-test). Retinas from a second group of four animals treated at 3 weeks and showing protection by ERG 8 weeks later had a statistically significant yet only slight increase in LEDGF protein in AAV-Ledgf–treated eyes by Western blot analysis (mean ± SD of the ratio LEDGF:b-actin was 1.03 ± 0.01 in the AAV-Ledgf–treated eyes and 0.98 ± 0.01 in control eyes, P < 0.05, Fig. 2C). It is possible that LEDGF protein levels were higher at earlier time points; therefore a larger number of animals and examination at different time points after injection is required to substantiate this effect.
Figure 2.
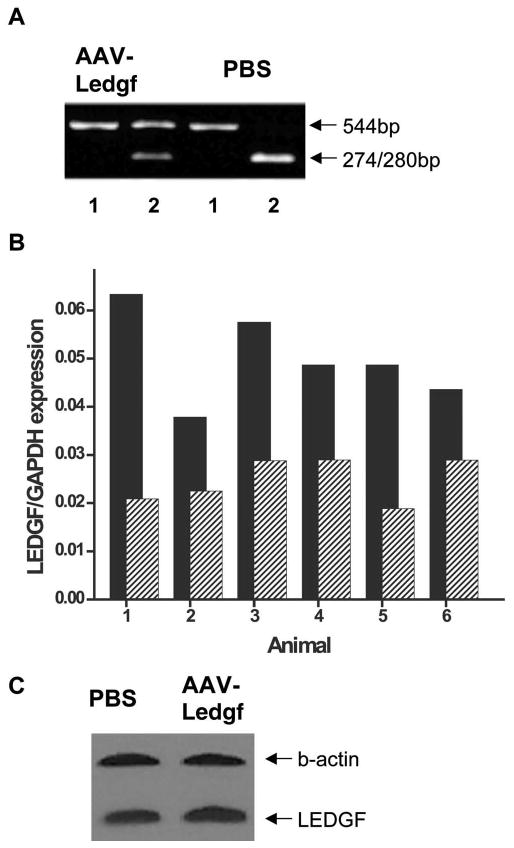
Real-time PCR and RFLP of Ledgf. (A) RT-PCR product from AAV-Ledgf–treated and contralateral PBS–treated retina 8 weeks after injection. RT-PCR product after incubation with buffer only (lane 1) or with PstI which cuts the endogenous Ledgf, but not the exogenous protein from AAV-Ledgf expression, into 274- and 280-bp bands (lane 2). (B) Transcription levels of Ledgf by quantitative real-time PCR, normalized by Gapdh. RNA was isolated from six RCS rat retinas and amplified with Ledgf-specific primers. Values were 2.1 ± 0.2 times higher in the AAV-Ledgf eyes (■) versus the PBS control eyes ( ) (mean ± SE, n = 6, P = 0.002 by paired t-test). (C) A representative example of Western blot analysis of LEDGF protein in AAV-Ledgf–injected and control eyes.
) (mean ± SE, n = 6, P = 0.002 by paired t-test). (C) A representative example of Western blot analysis of LEDGF protein in AAV-Ledgf–injected and control eyes.
Correlation between Functional and Morphologic Protection
To describe the magnitude of protection, we plotted the degree of functional and morphologic degeneration of RCS retinas in untouched eyes as a function of time (Fig. 3). Figure 3B shows functional changes and the average range of photoreceptor cell count in each hemisphere in sections through the optic nerve from four retinas at each age. Photoreceptor cell loss was faster in the inferior retina, with a rate of approximately two layers of cells per week from 4 to 7 weeks and approximately 1 cell width per week from 7 to 11 weeks. The superior retina steadily lost approximately one photoreceptor cell layer a week from 4 to ∼ 11 to 12 weeks. The b-wave declined linearly until it was no longer evident at 13 weeks, corresponding with the sharp decline in photoreceptor count during this time period. We also measured the change in the electronegative potential after the b-wave, which in the responses of the dystrophic RCS rat persist in advanced stages of retinal degeneration when the b-wave was no longer detected (Fig. 3B, dashed lined with open circles). The change in the electronegative potential lagged behind changes in the b-wave, maintaining constant amplitude until loss of several cell layers by 6 weeks. It then decreased for approximately 4 weeks, and the reduction in amplitude tapered off by 10.5 weeks and beyond, coinciding with the slower rate of photoreceptor cell loss.
Figure 3.
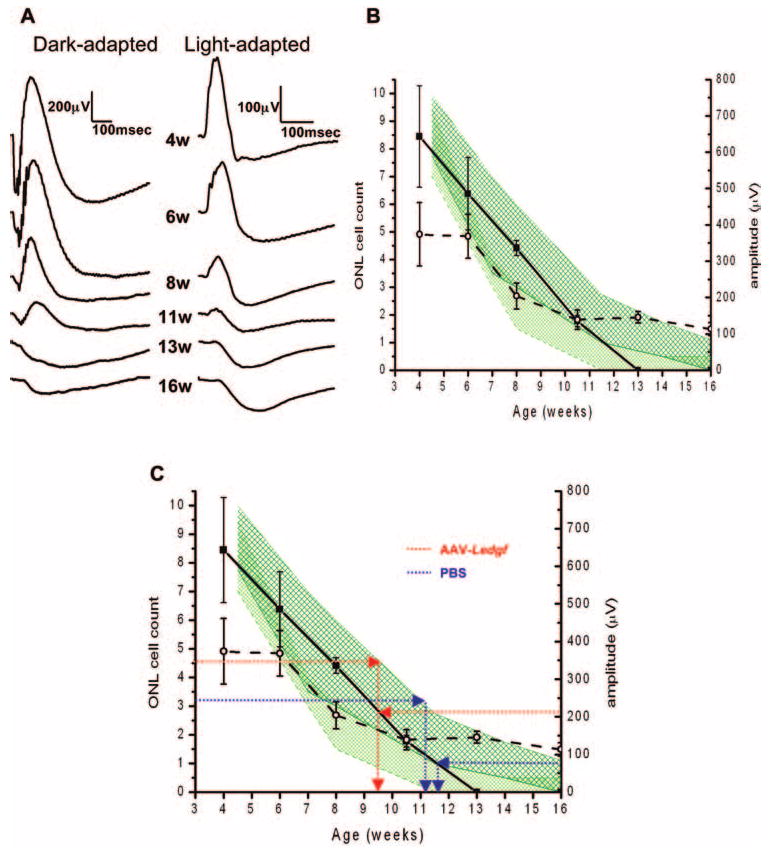
Degeneration rate in AAV-Ledgf and control RCS eyes. (A) Representative dark- and light-adapted ERG responses of RCS rats at 4 to 16 weeks of age, for 0.6 cd · s/m2 stimulus intensity. (B) Plot of dark-adapted ERG b-wave amplitude (solid line, squares) and amplitude of electronegative potential (dashed line, open circles) with range of ONL width in the superior (dark green area) and inferior (light green area) hemispheres in sections through the optic nerve. Mean ± SE (n = 4). (C) Functional and morphologic state of AAV-Ledgf– and PBS-treated eyes indicates a slowing of degeneration equivalent to a 2-week age difference in AAV-Ledgf–treated retina.
Combining the data in Figures 1B and 3B demonstrates the extent of protection by AAV-Ledgf (Fig. 3C). PBS and untouched eyes have comparable b-wave amplitudes at 11 weeks of age of roughly 75 μV for the maximum stimulus (dotted blue line, Fig. 3C), whereas the b-wave of the AAV-Ledgf–treated eyes was roughly 210 μV at the same age (dotted red line, Fig. 3C), which corresponds to b-wave amplitudes of untouched RCS eyes approximately 2 weeks younger. Retinal morphology also indicated a delay in degeneration of about 2 weeks. Maximum photoreceptor layer cell count in the PBS-treated eyes in the superior hemisphere averaged 3.2 cells (Table 2), which corresponds to an untouched retina of 11weeks (Fig. 3C, dotted blue line). Maximum photoreceptor count in the AAV-Ledgf–treated eyes averaged 4.5 cell layers, which corresponds to approximately 9 weeks of age in the untouched eyes (dotted red line in Fig. 3C).
Retinal Distribution of Degeneration and Protection
Cell protection was not homogeneous and was especially evident in the superior retina. When each retinal hemisphere was considered separately, the superior retina had a significantly higher number of cells in the AAV-Ledgf–treated eye than in the contralateral control eye in three of four animals (P < 0.01). Outer nuclear layer (ONL) width in cell number in this hemisphere was up to 10-fold greater in the AAV-Ledgf–treated eyes than in control eyes and the maximum cell count was nearly twice that in the control retinas (Table 2).
Subretinal injections were given in the temporal retina near the equator, although the protection from the secreted LEDGF could extend to areas distant from the injection site. To learn the extent of the effect of LEDGF across the retina, we analyzed the ONL width in vertical sections taken at 200-μm increments along the nasal–temporal axis in animals that showed functional protection. Using the average ONL width in the superior or inferior half of sections at each nasal–temporal position, two-way ANOVA analysis revealed significantly higher photoreceptor cell counts in the AAV-Ledgf–treated eyes than in the control eyes (P = 0.0002) in both superior and inferior hemisphere (Fig. 4A).
Figure 4.
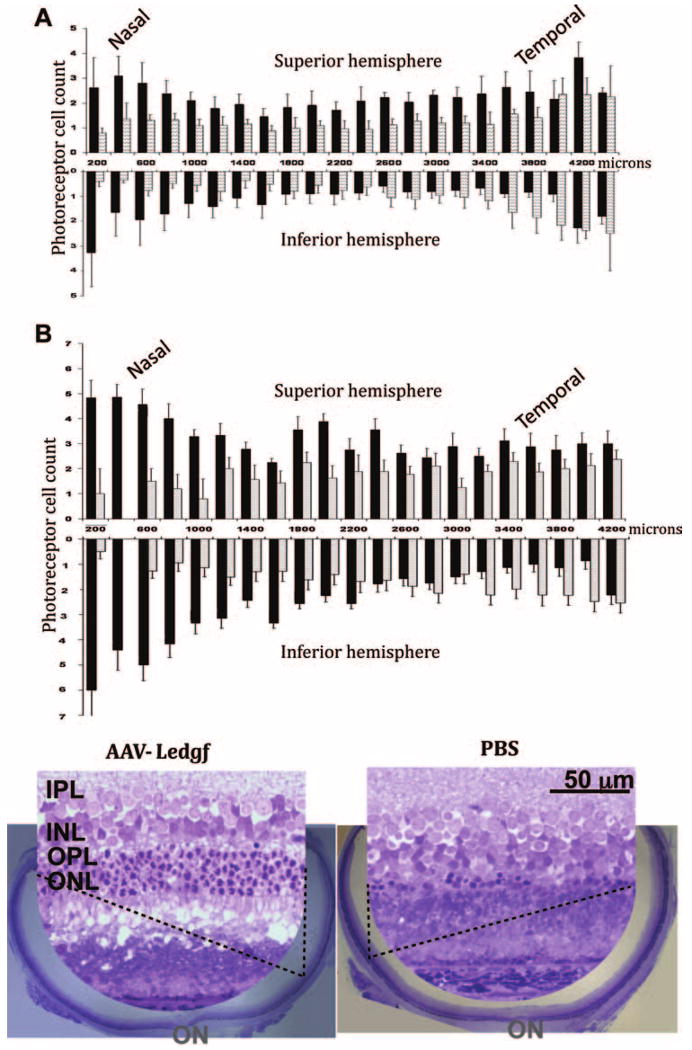
Morphologic evidence of photoreceptor protection. (A) Average photoreceptor cell counts in the superior and inferior halves of radial retinal sections taken every 200 μm perpendicular to the nasal–temporal plane. AAV-Ledgf–treated (n = 5, ■) and PBS treated (n = 4,  ) rats were injected at 3 weeks of age and evaluated 8 weeks later. Standard error bars are shown. (B) Photoreceptor cell count in a representative AAV-Ledgf–treated eye (■) from the group presented in (A) and its contralateral PBS–treated eye () across the retina, and the corresponding retinal morphology in sections through the optic nerve. Insets are from the superior hemisphere. OPL, outer plexiform layer; ON, optic nerve head.
) rats were injected at 3 weeks of age and evaluated 8 weeks later. Standard error bars are shown. (B) Photoreceptor cell count in a representative AAV-Ledgf–treated eye (■) from the group presented in (A) and its contralateral PBS–treated eye () across the retina, and the corresponding retinal morphology in sections through the optic nerve. Insets are from the superior hemisphere. OPL, outer plexiform layer; ON, optic nerve head.
Since animals vary in degree of degeneration and in precise position of vector delivery, positional effects on the degree of protection may be lost in averaging across animals. An example of morphologic protection in a single animal injected at 3 weeks and examined 8 weeks later is shown in Figure 4B. The control eye showed massive cell loss in the inferior retina. The nasal side of this hemisphere had counts between zero and two layers of cells, and in the temporal side there were no more than four layers of cells. The AAV-Ledgf–treated eye had a higher number of surviving cells in the inferior retina. No gaps devoid of photoreceptor cells were observed across most of this hemisphere which maintained a minimum of one- to two-cell thickness at all points. The superior retina showed homogenous cell loss in the PBS control eye, with zero- to four-cell thicknesses remaining. Conversely, the AAV-Ledgf–treated eye showed cell protection particularly in the superior retina, with up to six layers of cells remaining in this hemisphere.
HSP Involvement in LEDGF-Induced Protection
Our previous work,17 as well as other studies,24,25 suggests that HSPs may be involved in LEDGF-induced protection. We used a quantitative real time RT-PCR analysis to examine mRNA levels of αβ-crystallin and Hsp25, relative to Gapdh, in six animals that showed functional protection. Message levels of αβ-crystallin were not detectably different between AAV-Ledgf–injected and control retinas (αβ-crystallin/Gapdh ratio was 0.50 ± 0.13; mean ± SD) in AAV-Ledgf retinas and 0.33 ± 0.12 in control retinas; P = 0.44). Nor was the Hsp25 mRNA level detectably different (0.0075 ± 0.0031 in AAV-Ledgf retinas and 0.0067 ± 0.003 in control retinas; P = 0.87). With a second set of five animals, we examined protein expression in the retina by Western blot analysis and no detectable difference was found in αβ-crystallin or HSP25 protein levels between AAV-Ledgf–treated and control eyes (data not shown).
We extended our search for genes that may be upregulated in response to Ledgf by performing mRNA array analysis of genes involved in apoptosis, stress, and toxicity. Animals were treated with AAV-Ledgf at 3 weeks of age and retinas were removed for analysis at 11 weeks of age; results of three animals are shown in Table 3. Many of these genes were significantly upregulated in the AAV-Ledgf–treated eyes, but only a few exceeded a twofold increase. A subgroup of genes of specific interest, such as genes for HSPs and proteins involved in oxidative stress, was analyzed further by quantitative real-time PCR (Table 3, marked with an asterisk), and no significant differences were found between retinas of AAV-LEDGF– and PBS-injected eyes.
Table 3.
RNA Array Analysis
| Gene Symbol |
x-Fold Difference (AAV-Ledgf/Control) |
P (t-Test) |
|---|---|---|
| Apoptosis | ||
| Bcl2a1* | 2.07 | 0.109 |
| Il10 | 2.02 | 0.435 |
| Prok2* | 1.89 | 0.009 |
| Tnfrsf1b | 2.12 | 0.011 |
| Stress and toxicity | ||
| Bcl2l1* | 1.53 | 0.028 |
| Casp8 | 1.46 | 0.002 |
| Ccnd1 | 1.50 | 0.041 |
| Cdkn1a | 1.25 | 0.024 |
| Ddit3 | 1.33 | 0.036 |
| Ercc2 | 1.64 | 0.028 |
| Fmo1* | 2.12 | 0.067 |
| Gadd45a | 1.43 | 0.028 |
| Gpx1* | 1.33 | 0.043 |
| Gpx2* | 1.61 | 0.017 |
| Gsr* | 1.33 | 0.022 |
| Gstm3 | 1.43 | 0.0002 |
| Hmox1* | 2.12 | 0.071 |
| Hsph1* | 1.64 | 0.004 |
| Hspa1a* | 2.12 | 0.064 |
| Hspa4* | 1.53 | 0.030 |
| Hspa5* | 1.27 | 0.045 |
| Hspd1* | 1.64 | 0.015 |
| Nfkb1 | 1.27 | 0.010 |
| Pcna | 1.33 | 0.028 |
| Rad50 | 1.57 | 0.005 |
| RGD1564823_predicted | 1.33 | 0.023 |
| Serpine1* | 2.27 | 0.103 |
| Tp53 | 1.43 | 0.048 |
| Tradd | 1.50 | 0.004 |
| Ugt1a6 | 1.37 | 0.037 |
| Ung | 1.25 | 0.027 |
| Rpl13a* | 0.90 | 0.016 |
| Ldha* | 1.33 | 0.019 |
| Actb | 1.11 | 0.016 |
Probabilities in bold show significantly higher (P < 0.05, n = 3 where each is an average of three repeats per eye) expression in AAV-Ledgf–injected eyes versus PBS control eyes.
Genes were further analyzed by real-time PCR.
Our previous study17 indicated higher levels of small HSPs in retinas from RCS rat eyes treated with a bolus of GST-LEDGF protein versus control eyes, and intravitreous injection of HSP25 protein protected photoreceptors in RCS rats and light exposed Sprague-Dawley rats from degeneration.26 To test the possibility directly that upregulation of small heatshock proteins could produce functional protection, eight RCS rats were treated at 3 weeks of age with a subretinal injection of Ad-HSP27, and the contralateral eye was injected with an E1-defective empty adenovirus as the control. ERGs were recorded at 10 weeks of age and revealed functional protection in Ad-HSP27-treated eyes (Fig. 5), with the mean improvement in scotopic b-wave amplitude in six treated eyes equivalent to 3 log units of stimulus intensity. Response amplitudes of a seventh animal were higher in the Ad-HSP27-treated eye, but they did not reach our criterion for protection of 100% increase minimum. Only one of the eight animals gave no evidence of functional protection in the Ad-HSP27-treated eye. Eyes treated with empty adenovirus showed amplitudes similar to those in PBS-treated eyes, suggesting that the virus itself had no protection effect.
Figure 5.

Functional protection induced by Ad-HSP27. (A) ERG dark- and light-adapted responses after treatment of the eye with Ad-HSP27 versus the responses of contralateral control eye receiving empty adenovirus vector. (B) Intensity curves of dark- and light-adapted responses of RCS rats treated with subretinal injection of Ad-HSP27 in one eye and empty adenovirus in the control eye. Animals were treated at age 3 weeks and examined at 10 weeks. Average and SE bars are shown. *Significant amplitude differences (paired Student's t-test, n = 6, P ≤ 0.05).
Discussion
This study showed that transduction of retinal cells with AAV-Ledgf protects photoreceptors and helps maintain their function in an animal model of inherited retinal degeneration. The current results remove the ambiguity inherent in our previous findings with intravitreous injection of recombinant LEDGF protein in which the protection may be attributed to the GST fusion protein rather than LEDGF itself.17 Although upregulation of small HSPs was reported as a possible mechanism for the LEDGF protection pathway,11,17 the present study does not provide direct evidence that LEDGF upregulated small HSPs. Our previous study delivered a relatively massive bolus of LEDGF protein and evaluated HSP25 30 hours later in 6-week-old RCS rats. By comparison, the AAV-Ledgf construct resulted in Ledgf message being upregulated to a level only about two times the endogenous level and an increase in LEDGF protein that was just detectable at the experimental endpoint. It is possible that a small increase in LEDGF is sufficient to induce protection, as has previously been shown for regulatory proteins.27 The production of LEDGF is likely to be tightly regulated, and thus may be downregulated in the presence of sustained viral mediated expression. In addition, we do not know the level of LEDGF protein expression over the period of treatment, and it is possible that the functional and morphologic states of the retina at the endpoint are actually a result of prior higher LEDGF expression. The low levels of HSPs in this study compared to our previous study, in which they were measured 30 hours after LEDGF administration, could be due to the 11-week age of the animals, a time when both LEDGF expression and the number of photoreceptors remaining to express HSP are low. Thus, we cannot rule out that there were higher levels of expression of HSP at an earlier stage that contributed to photoreceptor protection.
The Tat sequence in LEDGF allows it to cross the plasma membrane efficiently28 and allows LEDGF to move into and be taken up from the extracellular space and facilitates survival of cells that were not directly transduced by the AAV. Indeed, cell count in the nasal side was not lower than in the temporal side, where the AAV-Ledgf was injected. A comparable effect was described for spread of virus-modified toxins from cell to cell through gap junctions, termed the bystander effect.29 Furthermore, LEDGF can penetrate blood–brain barriers.11 These features make LEDGF a good candidate for use as an in vivo retinal cell survival factor by local administration into the eye. Preliminary results showed that systemic intraperitoneal and intramuscular administration of AAV-Ledgf are effective routes for transferring LEDGF to the retina in mice.30
Correlation of the functional and morphologic states of the retina showed that the b-wave declined linearly with cell loss in the RCS rat. This result agrees with those in a previous study on photoreceptor degeneration from light damage.31 It seems that the magnitude of functional differences between treated and untreated eyes was greater than the morphologic differences, which were at a single row of cells in some places. It is possible that some of the photoreceptor cells, though apparent in the tissue, no longer maintain normal function. The electronegative component of the RCS ERG did not change with early photoreceptor loss, suggesting that the remaining photoreceptor function was sufficient for its saturation. As degeneration progressed and additional photoreceptors were lost, the electronegative potential was no longer saturated and gradually declined, indicating that it may serve as an indirect measure of the state of the photoreceptors beyond the stage of b-wave loss. The amplitude of the electronegative potential conceivably also benefits from rewiring in proximal retinal layers during outer retinal degeneration.32
As was noted in the RCS retina by LaVail and Battelle,33 we found slower cell loss in the superior hemisphere in untreated RCS retinas. Photoreceptor protection by LEDGF was greater in this superior region. It appears therefore that AAV-Ledgf protection was more effective for a slower rate of degeneration.
The suitability of a rescue agent to work across multiple genetic etiologies of degeneration is an important consideration in seeking a universal solution to inherited photoreceptor neurodegenerations. We repeated the AAV-Ledgf protection strategy in a group of nine P23H rhodopsin transgenic line 1 rats but we found no evidence of protection, even though the rate of functional degeneration in P23H line 1 rats is slower than in the RCS rats, as the b-wave persists, even at age 30 weeks.34 These results corroborate our previous results with a lack of protective effect of bolus LEDGF protein administered without a viral vector to these animals.17 On the other hand, LEDGF protein protected retinas from light damage.17 One fundamental difference between the models is that, in the RCS and in light-damaged retinas, apoptosis is inflicted on otherwise healthy photoreceptor cells,35,36 and protection would restore photoreceptor cell health. This is in contrast to mutation-induced apoptosis in the P23H rhodopsin retina in which abnormal rhodopsin protein is synthesized,37 and LEDGF may not be able to ameliorate the damage from this inherent metabolic disorder. In other words, based on these in vivo studies, we suggest that LEDGF deters photoreceptor degeneration by supporting otherwise healthy photoreceptor cells, but when the cause of degeneration is innate to the photoreceptor cells themselves, LEDGF has no protection effect.
Our results of protection by LEDGF in cases of degeneration factors external to the photoreceptors are consistent with previous studies showing that LEDGF protects against cell death induced by outside stressors, such as serum starvation and heat shock11 and suggests that both light damage and RCS degeneration are stress-related. In the case of RCS rats, the stress may be caused by the restriction of free exchange of metabolites between photoreceptors and the RPE and choroid by the debris build up in the subretinal space.19 In the case of P23H rhodopsin rats, the photoreceptor specific mutation provokes a metabolic stress in the rod cells.38 Similar to our findings, Ranchon et al.39 found that phenyl-N-tert-butylnitrone (PBN) protects against light damage but not against the inherited degeneration induced by P23H rhodopsin mutation. Together, these findings suggest that even though apoptosis is the ultimate cause of cell death common to these photoreceptor degenerations, methods for intervening with cell death may have to be specific. Furthermore, it is possible that the apoptotic pathways themselves differ.39,40 Therefore, search for a protective strategy against photoreceptor degeneration should consider these distinctions, and therapy design must be tested on a broad spectrum of disorders.
The mechanism of LEDGF protection remains unclear. We looked for but did not find a decrease in mRNA of caspases or other apoptosis-related genes in the AAV-Ledgf–treated eye. We found the mRNA of many genes related to stress and toxicity significantly upregulated in the AAV-Ledgf–treated group relative to the control. However, generally a cutoff of twofold up- or downregulation is used to define differential expression.41 By this criterion, several genes were borderline increased in the AAV-Ledgf–treated eyes relative to the contralateral control eyes. Further examination in a follow-up qRT-PCR analysis did not corroborate the significant differences. It is possible that the extraretinal tissue that was in the sample in addition to the photoreceptor cells masked greater elevations. Although we found that a small Hsp (HSP27) administered subretinally with a viral vector preserved retinal function in the RCS rat, this does not mean that LEDGF-induced protection is necessarily associated with HSPs. Lack of association between LEDGF, HSPs, and protection was also found after intravitreous injection of LEDGF protein in P23H rhodopsin rats. Hsp25 was upregulated in these retinas with no evidence of functional or morphologic protection.17 A further instance of a lack of coupling of HSPs from LEDGF is in embryonic chick retina. Small HSPs and αβ-crystallin levels were not upregulated in embryonic chick retinal cells, even though they acquired resistance to starvation and heat stress in the presence of LEDGF.42
Another question that interested us was the protection capacity of AAV-Ledgf in the RCS rats. Comparison of cell count in the superior retina, where the majority of photoreceptor protection occurs, referenced to the normal rate of degeneration, as well as functional differences suggest a delay in degeneration of approximately 2 weeks (Fig. 3C). It is possible that, as the photoreceptors in the RCS rat die subsequent to failure of the RPE cells to phagocytose shed outer segments,19 a combination therapy of AAV-Ledgf plus RPE cell transplantation43,44 may result in greater retinal protection in this model.
Unlike vectors based on adenovirus or herpes simplex virus, AAV is not associated with significant immune response in the eye.45 Proof of concept for restoring retinal structure and function using AAV was shown a decade ago for the P23H rat and for the Prph2Rd2/Rd2 mouse model of retinitis pigmentosa.46,47 Since then, several neurotrophic factors including CNTF, BDNF, and GDNF have been investigated for their potential to delay photoreceptor degeneration when delivered by AAV vectors.48 To use LEDGF as a general photoreceptor rescue agent, one would want to know the time window of protection after AAV-Ledgf treatment. This question would require a longitudinal study that is beyond the scope of the current work. However, preliminary follow-up on a single RCS animal injected at 3 weeks of age showed ERG functional protection at 11 and at 15 weeks, but the protection effect did not persist at 19 and 23 weeks (data not shown). It was reported that, in the rat eye, maximum expression of a protein delivered by AAV via subretinal injection occurs 8 weeks after injection, as our present results also suggest, and that expression gradually decreases thereafter,49 with need for reapplication. In humans however, wild-type AAV integrates stably into a site-specific chromosomal locus50 and long-term protein expression is expected. Achieving efficient long-term expression of a gene after a single intraocular delivery is key to translating the use of AAV into gene therapy treatment for human life-long retinal diseases.
Table 1.
Functional Effect of AAV-Ledgf
| Animals (n) |
Age at Injection (wk) |
Elapsed Time* (wk) |
Cases Showing Protection† |
|---|---|---|---|
| 9 | 5–7 | 4.5–5.5 | 6 |
| 6 | 5–7 | 8–9 | 6 |
| 19 | 2.5–3 | 8–9 | 12 |
| 2 | 14 | 3 | 0 |
Time from injection to ERG recording.
Protection threshold is a 100% increase in ERG amplitude at maximum stimulus intensity.
Acknowledgments
The authors thank Jinbo Li for assistance with histology, Maria Santos for husbandry help, and Juanita Marner for help with manuscript preparation.
Supported by the Intramural Research Program of the National Institutes of Health, National Institute on Deafness and Other Communication Disorders, and the National Eye Institute.
Footnotes
Disclosure: D. Raz-Prag, None; Y. Zeng, None; P.A. Sieving, None; R.A. Bush, None
References
- 1.RetNet. RetNet: Retinal Information Network. The University of Texas Health Science Center; Houston, TX: [June 2, 2009]. Available at http://www.sph.uth.tmc.edu/RetNet. [Google Scholar]
- 2.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 3.Chang GQ, Hao Y, Wong F. Apoptosis: final common pathway of photoreceptor death in rd, rds, and rhodopsin mutant mice. Neuron. 1993;11:595–605. doi: 10.1016/0896-6273(93)90072-y. [DOI] [PubMed] [Google Scholar]
- 4.Li ZM, Milam AH. Apoptosis in retinitis pigmentosa. In: Anderson R, LaVail MM, Hollyfield JG, editors. Degenerative Diseases of the Retina. New York: Plenum; 1995. pp. 1–8. [Google Scholar]
- 5.Davidson FF, Steller H. Blocking apoptosis prevents blindness in Drosophila retinal degeneration mutants. Nature. 1998;391:587–591. doi: 10.1038/35385. [DOI] [PubMed] [Google Scholar]
- 6.Delyfer MN, Leveillard T, Mohand-Said S, Hicks D, Picaud S, Sahel JA. Inherited retinal degenerations: therapeutic prospects. Biol Cell. 2004;96:261–269. doi: 10.1016/j.biolcel.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 7.Nakamura H, Izumoto Y, Kambe H, et al. Molecular cloning of complementary DNA for a novel human hepatoma-derived growth factor: its homology with high mobility group-1 protein. J Biol Chem. 1994;269:25143–25149. [PubMed] [Google Scholar]
- 8.Izumoto Y, Kuroda T, Harada H, Kishimoto T, Nakamura H. Hepatoma-derived growth factor belongs to a gene family in mice showing significant homology in the amino terminus. Biochem Biophys Res Commun. 1997;238:26–32. doi: 10.1006/bbrc.1997.7233. [DOI] [PubMed] [Google Scholar]
- 9.Ganapathy V, Daniels T, Casiano CA. LEDGF/p75: a novel nuclear autoantigen at the crossroads of cell survival and apoptosis. Autoimmun Rev. 2003;2:290–297. doi: 10.1016/s1568-9972(03)00063-6. [DOI] [PubMed] [Google Scholar]
- 10.Stec I, Nagl SB, van Ommen GJ, den Dunnen JT. The PWWP domain: a potential protein-protein interaction domain in nuclear proteins influencing differentiation? FEBS Lett. 2000;473:1–5. doi: 10.1016/s0014-5793(00)01449-6. [DOI] [PubMed] [Google Scholar]
- 11.Shinohara T, Singh DP, Fatma N. LEDGF, a survival factor, activates stress-related genes. Prog Retin Eye Res. 2002;21:341–358. doi: 10.1016/s1350-9462(02)00007-1. [DOI] [PubMed] [Google Scholar]
- 12.Amin V, Cumming DV, Coffin RS, Latchman DS. The degree of protection provided to neuronal cells by a pre-conditioning stress correlates with the amount of heat shock protein 70 it induces and not with the similarity of the subsequent stress. Neurosci Lett. 1995;200:85–88. doi: 10.1016/0304-3940(95)12074-e. [DOI] [PubMed] [Google Scholar]
- 13.Lowenstein DH, Chan PH, Miles MF. The stress protein response in cultured neurons: characterization and evidence for a protective role in excitotoxicity. Neuron. 1991;7:1053–1060. doi: 10.1016/0896-6273(91)90349-5. [DOI] [PubMed] [Google Scholar]
- 14.Rordorf G, Koroshetz WJ, Bonventre JV. Heat shock protects cultured neurons from glutamate toxicity. Neuron. 1991;7:1043–1051. doi: 10.1016/0896-6273(91)90348-4. [DOI] [PubMed] [Google Scholar]
- 15.Bruey JM, Ducasse C, Bonniaud P, et al. Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat Cell Biol. 2000;2:645–652. doi: 10.1038/35023595. [DOI] [PubMed] [Google Scholar]
- 16.Kamradt MC, Chen F, Cryns VL. The small heat shock protein alpha B-crystallin negatively regulates cytochrome c- and caspase-8-dependent activation of caspase-3 by inhibiting its autoproteolytic maturation. J Biol Chem. 2001;276:16059–16063. doi: 10.1074/jbc.C100107200. [DOI] [PubMed] [Google Scholar]
- 17.Machida S, Chaudhry P, Shinohara T, et al. Lens epithelium-derived growth factor promotes photoreceptor survival in light-damaged and RCS rats. Invest Ophthalmol Vis Sci. 2001;42:1087–1095. [PubMed] [Google Scholar]
- 18.Dowling JE, Sidman RL. Inherited retinal dystrophy in the rat. J Cell Biol. 1962;14:73–109. doi: 10.1083/jcb.14.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.D'Cruz PM, Yasumura D, Weir J, et al. Mutation of the receptor tyrosine kinase gene Mertk in the retinal dystrophic RCS rat. Hum Mol Genet. 2000;9:645–651. doi: 10.1093/hmg/9.4.645. [DOI] [PubMed] [Google Scholar]
- 20.Auricchio A, Hildinger M, O'Connor E, Gao GP, Wilson JM. Isolation of highly infectious and pure adeno-associated virus type 2 vectors with a single-step gravity-flow column. Hum Gene Ther. 2001;12:71–76. doi: 10.1089/104303401450988. [DOI] [PubMed] [Google Scholar]
- 21.Hildinger M, Auricchio A, Gao G, Wang L, Chirmule N, Wilson JM. Hybrid vectors based on adeno-associated virus serotypes 2 and 5 for muscle-directed gene transfer. J Virol. 2001;75:6199–6203. doi: 10.1128/JVI.75.13.6199-6203.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flannery JG, Zolotukhin S, Vaquero MI, LaVail MM, Muzyczka N, Hauswirth WW. Efficient photoreceptor-targeted gene expression in vivo by recombinant adeno-associated virus. Proc Natl Acad Sci U S A. 1997;94:6916–6921. doi: 10.1073/pnas.94.13.6916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bennett J, Duan D, Engelhardt JF, Maguire AM. Real-time, noninvasive in vivo assessment of adeno-associated virus-mediated retinal transduction. Invest Ophthalmol Vis Sci. 1997;38:2857–2863. [PubMed] [Google Scholar]
- 24.Shin JH, Piao CS, Lim CM, Lee JK. LEDGF binding to stress response element increases alphaB-crystallin expression in astrocytes with oxidative stress. Neurosci Lett. 2008;435:131–136. doi: 10.1016/j.neulet.2008.02.029. [DOI] [PubMed] [Google Scholar]
- 25.Singh DP, Fatma N, Kimura A, Chylack LT, Jr, Shinohara T. LEDGF binds to heat shock and stress-related element to activate the expression of stress-related genes. Biochem Biophys Res Commun. 2001;283:943–955. doi: 10.1006/bbrc.2001.4887. [DOI] [PubMed] [Google Scholar]
- 26.Bush RA, Machida S, Cahaudhry P, et al. Photoreceptor protection by small heat shock proteins in animal models. 6th Annual Vision Research conference; 2002. Abstract O4.1. [Google Scholar]
- 27.Muller-Hill B, Crapo L, Gilbert W. Mutants that make more lac repressor. Proc Natl Acad Sci U S A. 1968;59:1259–1264. doi: 10.1073/pnas.59.4.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frankel AD, Bredt DS, Pabo CO. Tat protein from human immunodeficiency virus forms a metal-linked dimer. Science. 1988;240:70–73. doi: 10.1126/science.2832944. [DOI] [PubMed] [Google Scholar]
- 29.Ishii-Morita H, Agbaria R, Mullen CA, et al. Mechanism of ‘bystander effect’ killing in the herpes simplex thymidine kinase gene therapy model of cancer treatment. Gene Ther. 1997;4:244–251. doi: 10.1038/sj.gt.3300379. [DOI] [PubMed] [Google Scholar]
- 30.Glushakova LG, Gorbatuyk M, Lu Y, et al. Transfer of LEDGF to the mouse retina via systemic AAV vector administration. Mol Ther. 2006;13:S192. [Google Scholar]
- 31.Sugawara T, Sieving PA, Bush RA. Quantitative relationship of the scotopic and photopic ERG to photoreceptor cell loss in light damaged rats. Exp Eye Res. 2000;70:693–705. doi: 10.1006/exer.2000.0842. [DOI] [PubMed] [Google Scholar]
- 32.Pu M, Xu L, Zhang H. Visual response properties of retinal ganglion cells in the royal college of surgeons dystrophic rat. Invest Ophthalmol Vis Sci. 2006;47:3579–3585. doi: 10.1167/iovs.05-1450. [DOI] [PubMed] [Google Scholar]
- 33.LaVail MM, Battelle BA. Influence of eye pigmentation and light deprivation on inherited retinal dystrophy in the rat. Exp Eye Res. 1975;21:167–192. doi: 10.1016/0014-4835(75)90080-9. [DOI] [PubMed] [Google Scholar]
- 34.Machida S, Raz-Prag D, Fariss RN, Sieving PA, Bush RA. Photopic ERG negative response from amacrine cell signaling in RCS rat retinal degeneration. Invest Ophthalmol Vis Sci. 2008;49:442–452. doi: 10.1167/iovs.07-0291. [DOI] [PubMed] [Google Scholar]
- 35.Edwards RB, Szamier RB. Defective phagocytosis of isolated rod outer segments by RCS rat retinal pigment epithelium in culture. Science. 1977;197:1001–1003. doi: 10.1126/science.560718. [DOI] [PubMed] [Google Scholar]
- 36.Reme CE. The dark side of light: rhodopsin and the silent death of vision. The Proctor lecture. Invest Ophthalmol Vis Sci. 2005;46:2672–2682. doi: 10.1167/iovs.04-1095. [DOI] [PubMed] [Google Scholar]
- 37.Naash MI, Hollyfield JG, al-Ubaidi MR, Baehr W. Simulation of human autosomal dominant retinitis pigmentosa in transgenic mice expressing a mutated murine opsin gene. Proc Natl Acad Sci U S A. 1993;90:5499–5503. doi: 10.1073/pnas.90.12.5499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anderson RE, Maude MB, McClellan M, Matthes MT, Yasumura D, LaVail MM. Low docosahexaenoic acid levels in rod outer segments of rats with P23H and S334ter rhodopsin mutations. Mol Vis. 2002;8:351–358. [PubMed] [Google Scholar]
- 39.Ranchon I, LaVail MM, Kotake Y, Anderson RE. Free radical trap phenyl-N-tert-butylnitrone protects against light damage but does not rescue P23H and S334ter rhodopsin transgenic rats from inherited retinal degeneration. J Neurosci. 2003;23:6050–6057. doi: 10.1523/JNEUROSCI.23-14-06050.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hao W, Wenzel A, Obin MS, et al. Evidence for two apoptotic pathways in light-induced retinal degeneration. Nat Genet. 2002;32:254–260. doi: 10.1038/ng984. [DOI] [PubMed] [Google Scholar]
- 41.Hegde P, Qi R, Abernathy K, et al. A concise guide to cDNA microarray analysis. Biotechniques. 2000;29:548–550. doi: 10.2144/00293bi01. 552-544, 556 passim. [DOI] [PubMed] [Google Scholar]
- 42.Nakamura M, Singh DP, Kubo E, Chylack LT, Jr, Shinohara T. LEDGF: survival of embryonic chick retinal photoreceptor cells. Invest Ophthalmol Vis Sci. 2000;41:1168–1175. [PubMed] [Google Scholar]
- 43.Sheedlo HJ, Li L, Turner JE. Photoreceptor cell rescue at early and late RPE-cell transplantation periods during retinal disease in RCS dystrophic rats. J Neural Transplant Plast. 1991;2:55–63. doi: 10.1155/NP.1991.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang S, Lu B, Girman S, Holmes T, Bischoff N, Lund RD. Morphological and functional rescue in RCS rats after RPE cell line transplantation at a later stage of degeneration. Invest Ophthalmol Vis Sci. 2008;49:416–421. doi: 10.1167/iovs.07-0992. [DOI] [PubMed] [Google Scholar]
- 45.Anand V, Duffy B, Yang Z, Dejneka NS, Maguire AM, Bennett J. A deviant immune response to viral proteins and transgene product is generated on subretinal administration of adenovirus and adeno-associated virus. Mol Ther. 2002;5:125–132. doi: 10.1006/mthe.2002.0525. [DOI] [PubMed] [Google Scholar]
- 46.Lewin AS, Drenser KA, Hauswirth WW, et al. Ribozyme rescue of photoreceptor cells in a transgenic rat model of autosomal dominant retinitis pigmentosa. Nat Med. 1998;4:967–971. doi: 10.1038/nm0898-967. [DOI] [PubMed] [Google Scholar]
- 47.Ali RR, Sarra GM, Stephens C, et al. Restoration of photoreceptor ultrastructure and function in retinal degeneration slow mice by gene therapy. Nat Genet. 2000;25:306–310. doi: 10.1038/77068. [DOI] [PubMed] [Google Scholar]
- 48.Buch PK, Bainbridge JW, Ali RR. AAV-mediated gene therapy for retinal disorders: from mouse to man. Gene Ther. 2008;15(11):849–857. doi: 10.1038/gt.2008.66. [DOI] [PubMed] [Google Scholar]
- 49.Rolling F, Shen WY, Barnett NL, et al. Long-term real-time monitoring of adeno-associated virus-mediated gene expression in the rat retina. Clin Exp Ophthalmol. 2000;28:382–386. doi: 10.1046/j.1442-9071.2000.00341.x. [DOI] [PubMed] [Google Scholar]
- 50.Kotin RM, Siniscalco M, Samulski RJ, et al. Site-specific integration by adeno-associated virus. Proc Natl Acad Sci U S A. 1990;87:2211–2215. doi: 10.1073/pnas.87.6.2211. [DOI] [PMC free article] [PubMed] [Google Scholar]