

Abstract
EphA2, a member of the receptor tyrosine kinase (RTK) family, is commonly expressed by a broad range of cancer types, where its level of (over)expression correlates with poor clinical outcome. Since tumor cell expressed EphA2 is a non-mutated “self” protein, specific CD8+ T cells are subject to self-tolerance mechanisms and typically exhibit only moderate-to-low functional avidity, rendering them marginally competent to recognize EphA2+ tumor cells in vitro or in vivo. We have recently reported that the ability of specific CD8+ T cells to recognize EphA2+ tumor cells can be augmented after the cancer cells are pretreated with EphA2 agonists that promote proteasomal degradation and upregulated expression of EphA2/class I complexes on the tumor cell membrane (Wesa et al., J. Immunol. 2008;181:7721-7). In the current study we show that treatment of EphA2+ tumor cells with the irreversible HSP90 inhibitor, 17-DMAG, similarly enhances their recognition by EphA2-specific CD8+ T cell lines and clones in vitro via a mechanism that is dependent on proteasome and TAP function, as well as, the retrotranslocation of EphA2 into the tumor cytoplasm. When 17-DMAG and agonist anti-EphA2 mAb are co-applied, T cell recognition of tumor cells is further increased over that observed for either agent alone. These studies suggest that EphA2 represents a novel HSP90 client protein and that the treatment of cancer patients with 17-DMAG-based “pulse” therapy may improve the anti-tumor efficacy of CD8+ T effector cells reactive against EphA2-derived epitopes.
Keywords: EphA2, HSP90 inhibitor, 17-DMAG, CD8+ T cells, Proteasome
Introduction
EphA2, a member of the RTK family of molecules, is a 130kDa (Type I) glycoprotein that mediates intercellular interactions via binding to its ligands Ephrin-A1, -A3, -A4 and -A5 expressed on an opposing cell surface (1). This RTK is expressed at low levels on a broad range of epithelial tissues in normal adults, including lung, spleen, kidney and liver (2), where it is primarily localized to sites of cell-to-cell contact and plays a role in contact inhibition of cell growth/migration that is critical for the organization and formation of epithelial layers in EphA2+ tissues (3). In addition to epithelial cells, activated endothelial cells also express EphA2 in association with tissue neovascularization in adults (4). In contrast to non-transformed cells, EphA2 is commonly overexpressed in a range of cancer types, including melanoma and many carcinomas (5-12), where it serves as an oncoprotein and a facilitator of metastasis (3, 13). Clinical observations suggest that the level of EphA2 overexpression by tumor cells is an indicator of poor prognosis, since it has been linked to reduced time to disease recurrence, and enhanced disease progression and metastatic spread (7, 9, 14, 15).
As a consequence, EphA2 represents an attractive target for therapeutic intervention in the majority of patients with solid tumors, with several treatment strategies considered for translation into the clinic. One strategy involves the implementation of agents (agonist mAb or recombinant ligands) that promote the proteasome-mediated degradation of tumor EphA2 protein, thereby limiting its oncogenic function (16, 17). We have recently determined that such reagents also promote a corollary enhancement in tumor cell presentation of EphA2 peptides in tumor cell MHC class I complexes, thereby facilitating tumor cell recognition and eradication by low-to-modest avidity CD8+ T cells (18). Since EphA2-specific CD8+ T cells have been detected in the peripheral blood of patients with renal cell carcinoma (RCC, ref. 8), prostate carcinoma (19) or glioma (20), and the frequencies of these protective T cells would be anticipated to be augmented as a consequence of active vaccination (2, 21), combinational therapies that sensitize EphA2+ tumors for specific CD8+ T cell eradication may yield enhanced clinical benefits in the cancer setting (22).
Interestingly, many RTK serve as client proteins for the molecular chaperone HSP90 (see http://www.picard.ch/downloads/Hsp90interactors.pdf), a protein designed to stabilize and refold denatured proteins into their native conformations to preserve their function and utility in normal and stressed cells (23). HSP90 is commonly overexpressed in tumor cells, where it is believed to protect client oncogenic/survival proteins that support tumor progression and metastasis, in part, by preventing their proteasome-dependent destruction (24). Our current report suggests that EphA2 represents a previously unknown HSP90 client protein. Furthermore, treatment of tumor cells with 17-DMAG, a well-tolerated clinical inhibitor of HSP90 (24, 25), results in the proteasome-dependent degradation of tumor EphA2 and in augmented tumor cell recognition by anti-EphA2 CD8+ T cells in vitro.
Materials and Methods
Cell lines and Media
SLR20 (EphA2+, HLA-A2neg; ref. 8), SLR22 (EphA2+, HLA-A2+; ref. 8) and SKOV3 (EphA2+, HLA-A2neg; kindly provided by Dr. Nora Disis, University of Washington), as well as, the SLR20.A2 (EphA2+, HLA-A2+) and SKOV3.A2 (EphA2+, HLA-A2+) cell lines (established via transduction of the corresponding parental cell lines with a recombinant retrovirus encoding HLA-A2.1 provided by Dr. Peter Cresswell, Yale University; ref. 18) were free of mycoplasma contaimination and maintained as previously reported (8).
HSP90 inhibitor and Peptides
HSP90 inhibitor 17-(Dimethylaminoethylamino)-17-demethoxygeldanamycin (17-DMAG, NSC 707545) was obtained by National Cancer Institute (Bestheda, Maryland). HLA-A2 presented EphA2 peptides, EphA258-66 (IMNDMPIYM; ref. 19) and EphA2883-891 (TLADFDPRV; ref. 8) were synthesized (at > 96% purity) as previously described (18). The ICP471-35 and ICP4735-1 synthetic peptides (26) were kindly provided by Dr. Peter Cresswell.
Western blot
RCC cell lines at 80-90% confluency were incubated with 17-DMAG (10-1000 nM) in 2% human serum supplemented RPMI-1640 media for 24-48h, as indicated in text. To assess the impact of proteasome function, TAP function, endosomal acidification and retrotranslocation in EphA2 protein degradation promoted by the HSP90 inhibitor, MG-132 (5-10 μM; Sigma-Aldrich, St. Louis, MO), ICP471-35 peptide (10 μg/ml), chloroquine (30-100 μM; Sigma-Aldrich) and Pseudomonas aeruginosa Exotoxin A (10-50 μg/ml; Sigma-Aldrich), respectively, were added at the initiation of 24h tumor cell cultures, as indicated in individual experiments. As a negative control for the ICP471-35 peptide in these studies, the “reverse”, scrambled ICP4735-1 peptide (26) was used at a concentration of 10 μg/ml. Harvested cells were lysed and western blotting performed as previously reported (18). Polyclonal anti-EphA2 Ab and horseradish peroxidase (HRP)-conjugated goat anti-rabbit antibody (both from Santa Cruz Biotechnology, Santa Cruz, CA) were used to detect EphA2. Monoclonal antibodies against TAP-1 and TAP-2 (NOB-1 and NOB-2, respectively, were kindly provided by Dr. Soldano Ferrone, University of Pittsburgh), with HRP-conjugated goat anti-mouse IgG (Santa Cruz) was used to probe blots.
Flow Cytometry
Control or treated tumor cells were phenotyped using anti-EphA2 mAb (B2D6, Upstate Biologicals, Inc., Lake Placid, NY) or anti-pan class I mAb (W6/32; Serotec Inc., Raleigh, NC) by flow cytometry as previously described (18).
Proteasome function analysis
SLR20 cells were transfected with the proteasome sensor vector (PSV; BD Biosciences) using lipofectamine 2000 (Invitrogen) and selected in cultures containing G418 (Invitrogen), thus generating SLR20.PSV cells. PSV expresses a fluorescent substrate for the proteasome (27), which accumulates in the cytoplasm of cells if proteasome function is inhibited. SLR20.PSV cells were grown to 80-90% confluency, before being cultured in the absence or presence of 17-DMAG or the proteasome inhibitors MG-132 (Sigma-Aldrich) or PS-341 (Bortezomib; kindly provided by Dr. Ram Ganapathi, Cleveland Clinic Foundation) at the indicated concentrations for 24h at 37°C and 5% CO2 tension. Fluorescence was detected in the FITC bandwidth (i.e. 488nm) by flow cytometry.
T cell lines and clones
Bulk CD8+ T cell lines and clones specific for EphA258-66 or EphA2883-891 were generated as previously described (18).
Tumor recognition assays
Tumor recognition by anti-EphA2 T cells was evaluated by IFN-γ ELISPOT assays as described before (8, 18) or using a commercial hIFN-γ ELISA (BD-Biosciences). For both the ELISPOT and ELISA protocols, tumor cells were treated with 100-500 nM 17-DMAG and/or 10 μg/ml anti-EphA2 mAb208 (18) for 24-48h, prior to their harvest using Trypsin-EDTA (Invitrogen). After washing with PBS (Invitrogen), tumor cells were co-cultured with anti-EphA2 T cell lines/clones at an effector:target cell ratio of 1:1 for 24h at 37°C and 5% CO2 tension. In some assays, where indicated, the class I-restricted nature of CD8+ T cell recognition of tumor cells was assessed by inclusion of 10 μg/well W6/32 (pan HLA-class I) mAb. To assess the impact of proteasome function, TAP function, endosomal acidification and retrotranslocation on 17-DMAG-treated tumor cells by anti-EphA2 CD8+ T cells, MG-132 (10 μM), ICP471-35 peptide (10 μg/ml), chloroquine (100 μM) or Pseudomonas aeruginosa Exotoxin A (10-50 μg/ml), respectively, were added to tumor cells during the 24h treatment period. After harvest, tumor cells were washed twice with PBS, prior to using these cells as targets for T cell recognition.
Statistical Analyses
Two-tailed Student's t tests were used to evaluate the difference between groups, with p values < 0.05 considered significant.
Results
The HSP90 inhibitor 17-DMAG induces EphA2 degradation that may be blocked by inhibitors of proteasome function, but not endosomal acidification
The EphA2 (over)expressing RCC cell line SLR20 was incubated in the absence or presence of 17-DMAG (0-1000 nM) for 24-48h. The resultant cells were then analyzed for EphA2 protein levels by Western Blotting (i.e. total protein; Fig. 1A) and flow cytometry (i.e. cell surface protein; Fig. 1B). In both cases, tumor EphA2 levels were reduced at both 24h and 48h post-treatment with 17-DMAG treatment (IC50 approximately 250 nM), although the pool of EphA2 protein most sensitive to 17-DMAG effects may be intracellular given the somewhat greater degree of reduction noted in the Western Blotting- vs. flow cytometry-based assays. Inclusion of the proteasome inhibitor MG-132 blocked the ability of 17-DMAG to promote EphA2 protein loss (Fig. 1C), suggesting HSP90 effects on EphA2 are at least partially proteasome-dependent. In contrast, inclusion of chloroquine, which disrupts endosomal acidification (28), failed to impact 17-DMAG-induced degradation of EphA2 protein (Fig. 1C). Single and combinational (17-DMAG + inhibitors) drug treatments did not lead to tumor cell death based on retention of control β-actin signal in the Western Blot experiments and appropriate forward/side scatter gating profiles in the flow cytometry assays (Fig. 1 and data not shown).
Figure 1. HSP90 inhibitor 17-DMAG promotes the loss of tumor EphA2 protein (via degradation) in a dose-, time- and proteasome-dependent manner.
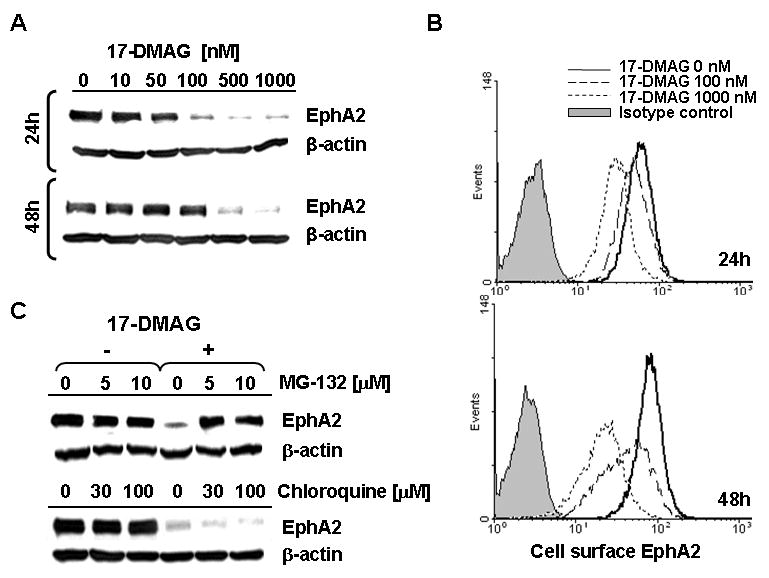
A. The EphA2+ SLR20 RCC line was incubated in the absence or presence of 17-DMAG (10-1000 nM) for 24h or 48h at 37°C, before generation of cell lysates and Western Blot analysis to determine levels of EphA2 protein expression. β-actin was monitored as an internal control protein. B. SLR20 cells were treated as above, with cell surface expression of EphA2 protein monitored by flow cytometry. Differences in tumor cell MFI expression of EphA2 were significant for 17-DMAG-treated vs. control, untreated tumor cells evaluated in flow-based assays (i.e. p = 0.008 at 24h for 500 nM 17-DMAG treated (MFI = 28 +/- 13) vs. untreated (MFI = 60 +/- 12). C. SLR20 cells were treated with 500 nM 17-DMAG in the absence or presence of MG-132 (10 μM) or chloroquine (100 μM) prior to Western Blot analysis in order to analyze the dependency of EphA2 (vs. control β-actin) protein loss on proteasome function or endosomal acidification, respectively. Data in each panel are representative of 4 independent experiments performed.
Treatment of tumor cells with HSP90 inhibitor does not significantly alter major components of the tumor MHC class I antigen processing machinery (APM)
We next assessed the impact of drug treatment on the expression and/or function of components of tumor cell MHC class I APM. Proteasome activity was analyzed using a model employing SLR20 cells transfected with a proteasome sensor vector (i.e. SLR20.PSV cells), in which intracellular fluorescent protein accumulates when proteasome function is inhibited. As depicted in Supplemental Fig. 1A, the proteasome inhibitors MG-132 and PS-341 both increased the fluorescence of SLR20.PSV cells, while 17-DMAG had minimum effect even after 48h at concentrations in excess of that required to promote EphA2 degradation (as shown in Fig. 1). Western Blot analysis revealed no changes in tumor cell expression of the TAP-1 or TAP2 proteins (Supplemental Fig. 1B), and flow cytometric analyses documented no appreciable alterations in tumor cell class I molecule expression (Supplemental Fig. 1C) after 17-DMAG treatment.
17-DMAG treatment of tumor cells transiently enhances tumor recognition by bulk CD8+ T cells specific for EphA2
Bulk CD8+ T cell lines and clones reactive against EphA2 peptides were generated from normal HLA-A2+ donors via in vitro stimulation. As shown in Fig. 2, bulk peptide-primed CD8+ T cells recognized EphA2+, HLA-A2+ RCC lines (SLR22 (Fig. 2A) and SLR20.A2 (Fig. 2B, Supplemental Fig. 2)) in a manner that was class I-restricted (i.e. inhibited by W6/32 mAb; Fig. 2A). These same T cell lines reacted poorly against the EphA2+, but HLA-A2neg SLR20 cell line (Fig. 2B). Pretreatment of SLR20.A2 with 17-DMAG for 24h enhanced tumor cell recognition by anti-EphA2 T cells (p < 0.05; Fig. 2B; Supplemental Fig. 2B). However, this enhanced immune recognition was transient in nature, since tumor cells pre-treated for 48h with 17-DMAG were recognized to a degree that was comparable with untreated tumor cells (Fig. 2B).
Figure 2. Recognition of EphA2+ SLR20.A2 tumor cells by bulk anti-EphA2 CD8+ T cells is enhanced after treatment with 17-DMAG.
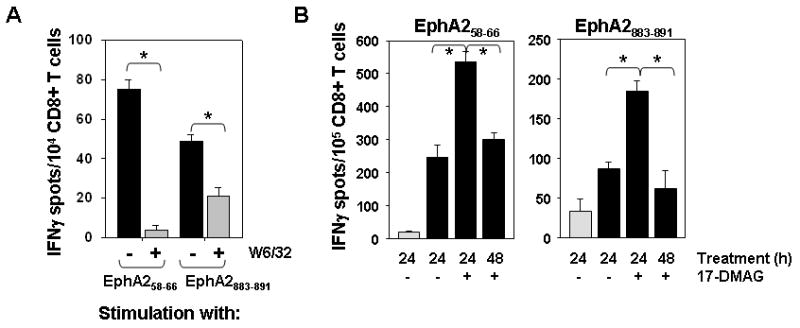
A. HLA-A2-restricted CD8+ T cells were generated in vitro by repeated stimulation with autologous DC pulsed with the EphA258-66 or EphA2883-891 peptide epitopes. These bulk CD8+ T cells were then co-cultured with the EphA2+, HLA-A2+ SLR22 RCC line alone (black bars) or in the presence of blocking anti-class I mAb W6/32 (gray bars) for 24h at 37°C in IFN-γ ELISPOT assays. B. SLR20 (EphA2+, HLA-A2neg; light gray bars) or SLR20.A2 (EphA2+, HLA-A2+; black bars) cells were cultured in the absence or presence of 500 nM 17-DMAG for 24h or 48h, prior to analysis of these target cells in IFN-γ ELISPOT assays. Bulk anti-EphA258-66 or anti-EphA2883-891 CD8+ T cell lines were used as responder cells. *p < 0.05 for SLR20.A2 treated with 17-DMAG vs. untreated SLR20.A2 at 24h. *p < 0.05 vs. 17-DMAG only treatment. In all panels, data are reported as the mean +/- SD of triplicate determinations from a single representative IFN-γ ELISPOT assay, with 3 independent assays performed in each case.
To determine whether 17-DMAG could sensitize tumor cells of an alternate lineage to anti-EphA2 CD8+ T cell recognition, we treated SKOV3.A2 ovarian carcinoma cells (EphA2+, HLA-A2+; Fig. 3A) with 17-DMAG for 24h in vitro. Like SLR20.A2 cells, 17-DMAG (500 nM)-treated SKOV3.A2 cells displayed reduced EphA2 protein expression (vs. control untreated tumor cells; Fig. 3B and Fig. 3C). Furthermore, 17-DMAG treatment of SKOV3.A2 (but not SKOV3 parental) tumor cells enhanced their recognition by anti-EphA258-66 bulk CD8+ T cells in an MHC class I-restricted manner (Fig. 3D). As shown in Fig. 3E, enhanced T cell recognition of 17-DMAG-treated SKOV3.A2 cells was effectively ablated by the proteasome inhibitor, but not by the lysosomotropic drug (chloroquine). Notably, MG-132 also reduced basal recognition of SKOV3.A2 cells by anti-EphA2 T cells (Fig. 3E), supporting the intrinsic loading of tumor cell class I complexes with EphA2 peptides via a proteasome-dependent pathway.
Figure 3. 17-DMAG promotes enhanced recognition of the EphA2+, HLA-A2+ ovarian carcinoma cell line SKOV3.A2 in vitro.
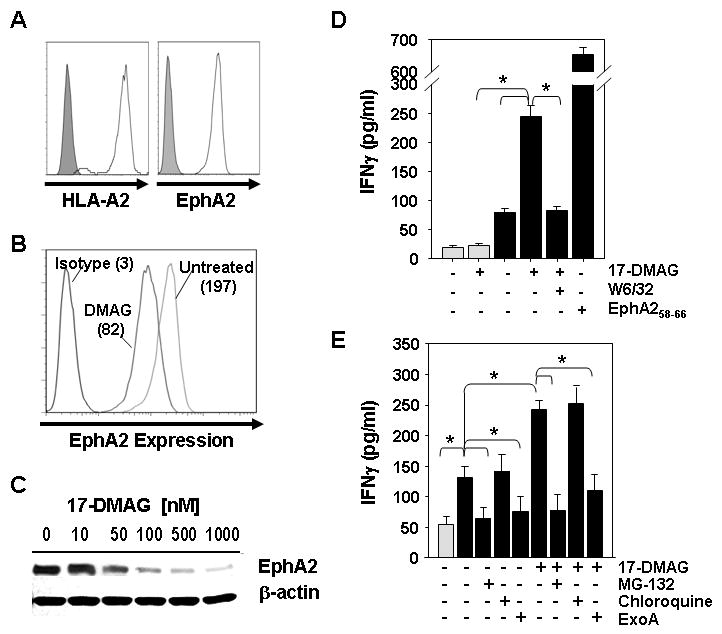
A. SKOV3.A2 tumor cells were analyzed for expression of cell surface HLA-A2 and EphA2 proteins by flow cytometry using specific mAbs (open profiles) vs. isotype control mAbs (filled profiles). To assess the impact of HSP90 inhibition, SKOV3.A2 tumor cells were untreated or treated with 17-DMAG (10-1000 nM) for 24h and analyzed for EphA2 expression by flow cytometry (B.) and Western Blotting (C.), as well as, for their ability to be recognized by bulk anti-EphA258-66 T cells (D.). In the flow cytometry analyses; p = 0.001 for 500 nM 17-DMAG treated (MFI = 74 +/- 16) vs. untreated (MFI = 190 +/- 18)). In T cell assays, cell-free supernatants were harvested after a 24h T cell (105)-tumor cell (104; +/- 500 nM 17-DMAG pretreatment) co-culture period and analyzed for IFN-γ content by specific ELISA. E. SKOV3 or SKOV3.A2 tumor cells were untreated or treated with 500 nM 17-DMAG +/- MG-132 (10 μM) or chloroquine (100 μM) or ExoA (50 μg/ml) for 24h, before being used as targets for bulk anti-EphA258-66 CD8+ T cells (106) responses monitored using IFN-γ ELISA. In panels D. and E., SKOV3 histograms are indicated by light gray bars, while SKOV3.A2 histograms are denoted by black bars. All ELISA data is reported as the mean +/- SD of triplicate assay determinations; *p < 0.05 for all indicated comparisons. Data in all panels are from 1 representative experiment of 3 performed.
17-DMAG treatment of tumor cells enhances tumor recognition by anti-EphA2 CD8+ T cells via a retrotranslocation- and TAP-dependent mechanism
Since 17-DMAG promotes enhanced recognition by CD8+ T cells capable of recognizing peptides derived from both the extracellular (i.e. EphA258-66) and the intracellular (i.e. EphA2883-891) domains of this RTK, this suggests that both domains of this transmembrane protein must become accessible for proteasomal processing into peptides that are loaded into nascent HLA-A2 complexes in tumor cells. Current paradigms (29-31) suggest that this may be accomplished through a retrotranslocation process involving sec61-dependent “ratcheting” of the target protein into the cytoplasm, making it a substrate for the proteasome. To test this hypothesis, we added the sec61 inhibitor ExoA (26) to tumor cells during culture with 17-DMAG, before their analysis in western blotting and T cell assays. As shown in Fig. 4, ExoA inhibits 17-DMAG-induced degradation of tumor cell EphA2 protein (Fig. 4A) and prevents enhanced recognition of 17-DMAG-treated SLR20.A2 tumor cells by anti-EphA2 (clone 15/9) CD8+ T cells (Fig. 4B, 4C). Similar results were obtained using a bulk anti-EphA258-66 CD8+ T cell line and the SKOV3.A2 tumor cell line (Fig. 3E).
Figure 4. 17-DMAG treatment improves recognition of the SLR20.A2 RCC cell line by a low avidity, anti-EphA258-66 CD8+ T cell clone in a TAP- and retrotranslocation-dependent manner.
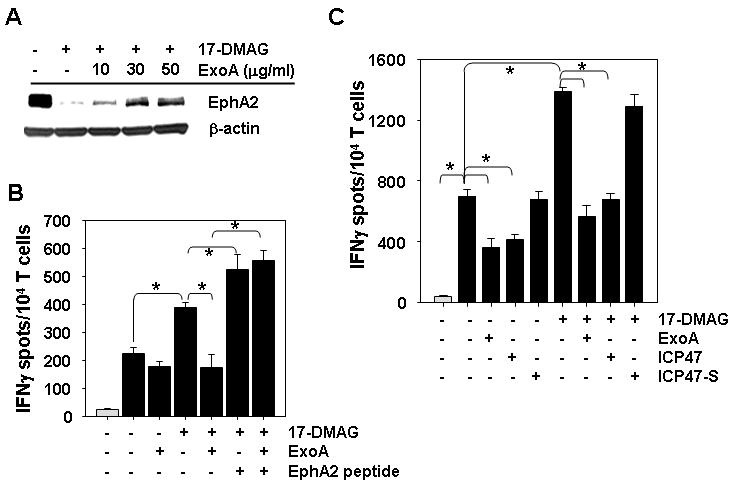
A. SLR20.A2 tumor cells were untreated or treated for 24h with 500 nM 17-DMAG +/- 10, 30 or 50 μg/ml ExoA, then analyzed for expression of EphA2 vs. control β-actin protein by western blotting. B. Untreated SLR20 cells (light gray bars) or SL20.A2 cells (black bars) pretreated as in panel A using 10 μg/ml ExoA were used as targets for recognition by anti-EphA258-66 CD8+ T cell clone 15/9 in IFN-γ ELISPOT assays. Drug-treated tumor cells pulsed with exogenous EphA258-66 peptide (10 μg/ml) to clone 15/9 was included as a positive control. C. SLR (light gray bars) and SLR20.A2 (black bars) tumor cells were untreated or treated with 500 nM 17-DMAG +/- 50 μg/ml ExoA, 10 μg/ml ICP471-35 peptide or 10 μg/ml ICP4735-1 scrambled peptide for 24h, prior to use as target cells for clone 15/9 T cell recognition in IFN-γ ELISPOT assays. All ELISPOT data are reported as the mean +/- SD of triplicate determinations. In all cases, data are representative of that obtained in 3 independent assays performed. * p < 0.05 for all indicated comparisons.
The TAP dependency of the sensitizing effects of 17-DMAG on tumor cell recognition by specific T cells was next addressed. We observed that co-treatment of tumor cells with 17-DMAG and an N-terminal fragment of the ICP47 protein (ICP471-35), a (Herpes Simplex) viral inhibitor of TAP (26), ablated enhanced T cell recognition when compared to tumor cells treated with 17-DMAG alone (p < 0.05). This effect was specific, as inclusion of a scrambled peptide ICP35-1 (bearing the reverse AA sequence of ICP471-35) exhibited no such inhibitory effect (Fig. 4C).
Combined treatment of EphA2+, HLA-A2+ tumor cells with both 17-DMAG and agonist (anti-EphA2) mAb208 results in superior recognition by a low avidity CD8+ T cells specific for EphA2
As we have previously reported that EphA2 agonist mAb208 promotes the proteasomal destruction of tumor EphA2 protein and enhances specific CD8+ T cell recognition of treated tumor cells (18), we next investigated whether the combined use of both 17-DMAG and mAb208 would result in an even greater degree of tumor cell recognition by specific T cells when compared to either treatment modality alone. As depicted in Figs. 5A and 5B, while both 17-DMAG and mAb208 resulted in reduced EphA2 expression on SLR20.A2 cells, combined treatment with both reagents yielded an even greater degree of EphA2 protein loss. T cell assays similarly support the greatest degree of tumor cell recognition by CD8+ T cells (targeting either of the EphA2 epitopes) after SLR20.A2 cells are pretreated with both 17-DMAG and mAb208 vs. either reagent alone (p < 0.05; Figs. 5C and 5D).
Figure 5. Superior in vitro recognition of the SLR20.A2 tumor cells by anti-EphA2 CD8+ T cells after combined treatment with 17-DMAG and agonistic anti-EphA2 mAb208.
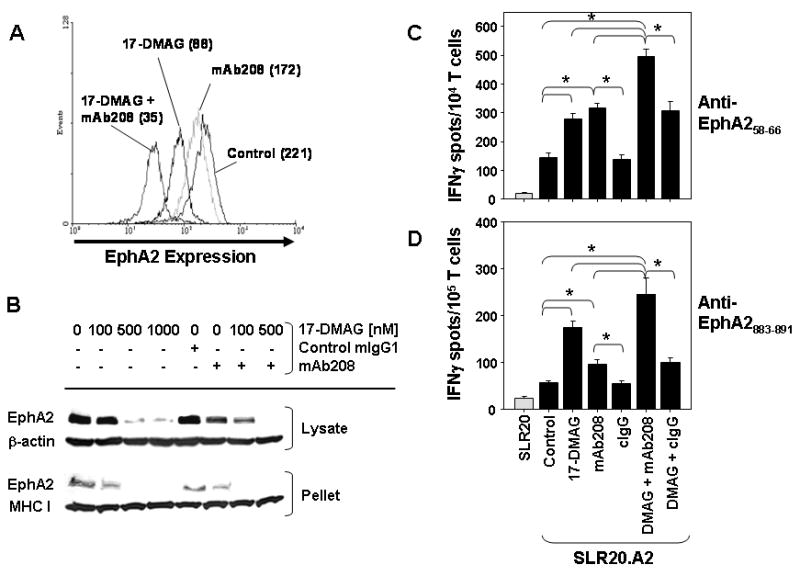
A., SLR20.A2 cells were untreated or treated for 24h with 17-DMAG (100 nM) +/- 10 μg/ml of mAb208 or control IgG (cIgG), after which cells were washed and analyzed by flow cytometry for expression of cell surface EphA2 molecules. Data in parentheses represents the mean fluorescence intensity value for each cell population analyzed and are representative of 4 independent assays performed. p < 0.05 for differences in the MFI between all cohorts in pair-wise comparisons. In B., SLR20.A2 cells were untreated or treated with 17-DMAG (100-1000 nM) +/- control IgG (10 μg/ml) or agonist anti-EphA2 mAb208 (10 μg/ml) for 24h. After harvest, cells were lysed in TritonX-100-containing buffer. After centrifugation, the soluble lysate and pellet were recovered and analyzed via Western Blotting for comparative levels of EphA2. β-actin or MHC class I heavy chain proteins served as control proteins, as indicated. In panels C. and D., SLR20 control cells (light gray bars) or SLR20.A2 (black bars) tumor cells pre-treated for 24h (as indicated) were used as targets for recognition by anti-EphA258-66 clone 15/9 or bulk anti-EphA2883-891 T cells, respectively, in IFN-γ ELISPOT assays. *p < 0.05 for all indicated comparisons. All data are representative of that obtained in 3 independent experiments performed.
Discussion
Immunotherapies (including cancer vaccines) designed to stimulate specific T cell-mediated immunity have thus far yielded rather modest objective clinical response rates, despite their ability to enhance circulating frequencies of tumor-specific T cells in many treated patients (32). This may reflect the host availability of an only low-to-moderate avidity repertoire that has survived negative selection (33, 34), and while such T cells may be activated by specific vaccination, they frequently fail to recognize tumor cells that naturally present low stochastic frequencies of relevant MHC/tumor peptide complexes (35). Poor immune reactivity of tumor cells may be further exacerbated by defects in the tumor APM (36, 37).
If tolerance selection restricts the clinical utility of the anti-tumor CD8+ T cell repertoire due to functional avidity constraints, we hypothesized that tumor cells might instead be manipulated in order to exceed the cognate Ag threshold requirements for effective immune surveillance. In particular, we believe that by conditionally enhancing the proteasomal processing of tumor antigens, such as EphA2, the level of class I/EphA2 peptide complexes might be increased on the tumor cell surface, allowing for improved immune recognition (22). Indeed, we have now determined that; 1) EphA2 is a novel client protein of HSP90 that is susceptible to 17-DMAG-induced degradation; 2) 17-DMAG treatment of EphA2+ tumor (RCC and ovarian carcinoma) cells improves recognition by low avidity anti-EphA2 CD8+ T cells; 3) enhanced T cell recognition of 17-DMAG-treated tumor cells is MHC-dependent and appears unrelated to tumor cell expression of co-stimulatory/co-inhibitory molecules which remains unchanged upon HSP90 antagonism (Supplemental Fig. 3), and 4) EphA2+ tumor cell recognition by specific CD8+ T cells may be further enhanced by combined treatment with 17-DMAG and EphA2 agonists (vs. treatment with either single modality).
Notably, tumor recognition by T cells reactive against peptides found in the extracellular (i.e. EphA258-66) as well as intracellular (i.e. EphA2883-891) domains of the target protein was improved by treatment with 17-DMAG. Mechanistically, we observed that enhanced T cell recognition of 17-DMAG-conditioned tumor cells was ablated upon inclusion of proteasome (MG-132), TAP (ICP471-35) or sec61 (Pseudomonas aeruginosa Exotoxin A) inhibitors. In contrast, there was minimal impact associated with the inclusion of the lysosomotropic agent chloroquine, which interferes with endosomal acidification and lysosomal processing of protein antigens. These data suggest that the major pool of EphA2 protein undergoing (constitutive as well as) enhanced proteasomal processing as a consequence of 17-DMAG inhibition likely enters the tumor cytosol via a retrotranslocation event (26, 29-31). At present, we cannot distinguish whether this pool of EphA2 protein derives from an early endosomal compartment (i.e. internalized after interaction with the EphA2 ligands co-expressed by adjacent tumor cells that is unaffected by chloroquine) and/or from newly-synthesized, mis-folded EphA2 proteins within the exocytic pathway. However, given the observed quantitative variance in 17-DMAG-induced EphA2 degradation as imaged using flow cytometry vs. Western Blotting analyses, and the apparent synergy of agonist mAb208 and 17-DMAG in promoting EphA2 protein loss and enhanced T cell recognition, it could be suggested that mAb208 primarily affects the membrane pool of EphA2 protein, while 17-DMAG primarily affects the intracellular pool of this protein. In either case, derivative EphA2 peptides would then appear to be integrated into “empty” class I complexes after TAP-dependent transfer into the MHC class I loading compartment.
Our data suggest that 17-DMAG enhances tumor cell recognition by EphA2-specific CD8+ T cells in a transient manner (i.e. at 24h but not 48h) after drug treatment, despite the comparable level of drug impact on EphA2 protein levels at both time points. One possible explanation for this dichotomy may involve differential rates at which the diverse array of HSP90 client proteins undergo proteasomal destruction upon application of 17-DMAG, making the competitor substrates for proteasomal processing variable over time after drug treatment. If this hypothesis is correct, EphA2 may be more efficiently processed and/or be less effectively competed for loading into MHC class I complexes during the first 24h of HSP90 inhibitor administration. In extended studies, we noted that the ability of 17-DMAG treated tumor cells to be loaded by exogenous peptide was not differential at the 24h vs. 48h time points post-DMAG application (Supplemental Fig. 4A) and that there was no change in the prevalence of “empty” HLA-A2 complexes on the tumor cell surface as a consequence of drug treatment (Supplemental Fig. 4B). These findings mitigate concerns that the class I APM is substantially altered in tumor cells after extended treatment (i.e. 48h vs. 24h) with the HSP inhibitor.
Based on our current and recently published studies (18), both EphA2 agonists (mAb, recombinant EphrinA1-Fc) and 17-DMAG (alone or in combination) are capable of enhancing anti-EphA2 T cell recognition of (EphA2+, HLA-A2+) tumor cell lines. In the clinic, EphA2 agonists would be expected to be superior with regard to specificity in targeting the EphA2 antigen vs. multiple client proteins in the case of HSP90 inhibitors. However, the promiscuous and coordinate effects of HSP90 inhibitors on multiple oncoprotein clients would argue for the use of these drugs in combinational immunotherapies targeting multiple antigens (such as EphA2, EGFR and Her2/neu, etc.). HSP90 inhibitors may be preferred to agonists based on their selective accumulation in tumor vs. normal EphA2+ tissue sites. For example, 17-AAG binds to tumor cell-derived HSP90 up to 2-logs more tightly than it does to normal cell-derived HSP90 (38). Furthermore, in mouse models, 17-DMAG persists within tumor lesions far longer than in normal tissues, with detectable levels selectively observed in tumors 48h after a single i.v. administration of drug (39). This suggests that drug dosing far below the MTD may be capable of modulating T cell recognition of cancer cells within the tumor microenvironment in vivo. However, it is also possible that the application of 17-DMAG and/or EphA2 agonists may promote increased immune recognition of normal EphA2+ tissues that express a fully functional class I APM. This raises the specter of autoimmune pathology that would need to be closely monitored in prospective clinical trials applying these agents, particularly should they be combined. In this regard, no untoward, immune-mediated effects on patient EphA2+ organs (i.e. lungs, kidney) have been reported in clinical trials employing HSP90 inhibitors to date (40), and EphA2 agonist therapies have yet to be investigated in phase I trials.
In conclusion, our results suggest that an effective combinational immunotherapy for clinical translation (22) may be defined by “pulse” 17-DMAG administration to improve EphA2+ tumor cell recognition by CD8+ T cells. In patients, anti-EphA2 T cells may be elicited in response to specific vaccination or they could be provided via adoptive transfer. The efficacy of such combinational approaches might be further improved by inclusion of EphA2 agonists or Type-I or -II interferon co-administration to further improve tumor cell APM function and the MHC class I (and II) presentation of targeted epitopes by tumor cells in vivo (41, 42).
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants R01 CA114071 and P50 CA121973 (to WJS). The authors also wish to thank Drs. Robert Binder, Jeff Brodsky, Merrill Egorin, Paul Robbins, Russell Salter, Alessandra Giodini and Peter Cresswell for their helpful discussions, careful review and critical comments provided during the preparation of this manuscript.
References Cited
- 1.Surawska H, Ma PC, Salgia R. The role of ephrins and Eph receptors in cancer. Cytokine Growth Factor Rev. 2004;15:419–33. doi: 10.1016/j.cytogfr.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 2.Hatano M, Kuwashima N, Tatsumi T, et al. Vaccination with EphA2-derived T cell-epitopes promotes immunity against both EphA2-expressing and EphA2-negative tumors. J Transl Med. 2004;2:40. doi: 10.1186/1479-5876-2-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zantek ND, Azimi M, Fedor-Chaiken M, Wang B, Brackenbury R, Kinch MS. E-cadherin regulates the function of the EphA2 receptor tyrosine kinase. Cell Growth Differ. 1999;10:629–38. [PubMed] [Google Scholar]
- 4.Cheng N, Brantley DM, Chen J. The ephrins and Eph receptors in angiogenesis. Cytokine Growth Factor Rev. 2002;13:75–85. doi: 10.1016/s1359-6101(01)00031-4. [DOI] [PubMed] [Google Scholar]
- 5.Walker-Daniels J, Hess AR, Hendrix MJ, Kinch MS. Differential regulation of EphA2 in normal and malignant cells. Am J Pathol. 2003;162:1037–42. doi: 10.1016/S0002-9440(10)63899-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abraham S, Knapp DW, Cheng L, et al. Expression of EphA2 and Ephrin A-1 in carcinoma of the urinary bladder. Clin Cancer Res. 2006;12:353–60. doi: 10.1158/1078-0432.CCR-05-1505. [DOI] [PubMed] [Google Scholar]
- 7.Miyazaki T, Kato H, Fukuchi M, Nakajima M, Kuwano H. EphA2 overexpression correlates with poor prognosis in esophageal squamous cell carcinoma. Int J Cancer. 2003;103:657–63. doi: 10.1002/ijc.10860. [DOI] [PubMed] [Google Scholar]
- 8.Tatsumi T, Herrem CJ, Olson WC, et al. Disease stage variation in CD4+ and CD8+ T-cell reactivity to the receptor tyrosine kinase EphA2 in patients with renal cell carcinoma. Cancer Res. 2003;63:4481–9. [PubMed] [Google Scholar]
- 9.Herrem CJ, Tatsumi T, Olson KS, et al. Expression of EphA2 is prognostic of disease-free interval and overall survival in surgically treated patients with renal cell carcinoma. Clin Cancer Res. 2005;11:226–31. [PubMed] [Google Scholar]
- 10.Nasreen N, Mohammed KA, Antony VB. Silencing the receptor EphA2 suppresses the growth and haptotaxis of malignant mesothelioma cells. Cancer. 2006;107:2425–35. doi: 10.1002/cncr.22254. [DOI] [PubMed] [Google Scholar]
- 11.Thaker PH, Deavers M, Celestino J, et al. EphA2 expression is associated with aggressive features in ovarian carcinoma. Clin Cancer Res. 2004;10(15):5145–50. doi: 10.1158/1078-0432.CCR-03-0589. [DOI] [PubMed] [Google Scholar]
- 12.Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. EphA2: a determinant of malignant cellular behavior and a potential therapeutic target in pancreatic adenocarcinoma. Oncogene. 2004;23:1448–56. doi: 10.1038/sj.onc.1207247. [DOI] [PubMed] [Google Scholar]
- 13.Zelinski DP, Zantek ND, Stewart JC, Irizarry AR, Kinch MS. EphA2 overexpression causes tumorigenesis of mammary epithelial cells. Cancer Res. 2001;61:2301–6. [PubMed] [Google Scholar]
- 14.Kinch MS, Moore MB, Harpole DH., Jr Predictive value of the EphA2 receptor tyrosine kinase in lung cancer recurrence and survival. Clin Cancer Res. 2003;9:613–8. [PubMed] [Google Scholar]
- 15.Kinch MS, Carles-Kinch K. Overexpression and functional alterations of the EphA2 tyrosine kinase in cancer. Clin Exp Metastasis. 2003;20:59–68. doi: 10.1023/a:1022546620495. [DOI] [PubMed] [Google Scholar]
- 16.Coffman KT, Hu M, Carles-Kinch K, et al. Differential EphA2 epitope display on normal versus malignant cells. Cancer Res. 2003;63:7907–12. [PubMed] [Google Scholar]
- 17.Noblitt LW, Bangari DS, Shukla S, et al. Decreased tumorigenic potential of EphA2-overexpressing breast cancer cells following treatment with adenoviral vectors that express EphrinA1. Cancer Gene Ther. 2004;11:757–66. doi: 10.1038/sj.cgt.7700761. [DOI] [PubMed] [Google Scholar]
- 18.Wesa AK, Herrem CJ, Mandic M, et al. Conditional Enhancement in Specific CD8+ T cell Recognition of EphA2+ Tumors In Vitro and In Vivo After Treatment with Ligand Agonists. J Immunol. 2008;181:7721–7. doi: 10.4049/jimmunol.181.11.7721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alves PM, Faure O, Graff-Dubois S, et al. EphA2 as target of anticancer immunotherapy: identification of HLA-A*0201-restricted epitopes. Cancer Res. 2003;63:8476–80. [PubMed] [Google Scholar]
- 20.Hatano M, Eguchi J, Tatsumi T, et al. EphA2 as a glioma-associated antigen: a novel target for glioma vaccines. Neoplasia. 2005;7:717–22. doi: 10.1593/neo.05277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamaguchi S, Tatsumi T, Takehara T, et al. Immunotherapy of murine colon cancer using receptor tyrosine kinase EphA2-derived peptide-pulsed dendritic cell vaccines. Cancer. 2007;110:1469–77. doi: 10.1002/cncr.22958. [DOI] [PubMed] [Google Scholar]
- 22.Storkus WJ, Herrem C, Kawabe M, et al. Improving immunotherapy by conditionally enhancing MHC class I presentation of tumor antigen-derived Peptide epitopes. Crit Rev Immunol. 2007;27:485–93. doi: 10.1615/critrevimmunol.v27.i5.60. [DOI] [PubMed] [Google Scholar]
- 23.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–72. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 24.Neckers L. Heat shock protein 90: the cancer chaperone. J Biosci. 2007;32:517–30. doi: 10.1007/s12038-007-0051-y. [DOI] [PubMed] [Google Scholar]
- 25.Glaze ER, Lambert AL, Smith AC, et al. Preclinical toxicity of a geldanamycin analog, 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin (17-DMAG), in rats and dogs: potential clinical relevance. Cancer Chemother Pharmacol. 2005;56:637–47. doi: 10.1007/s00280-005-1000-9. [DOI] [PubMed] [Google Scholar]
- 26.Ackerman AL, Giodini A, Cresswell P. A role for the endoplasmic reticulum protein retrotranslocation machinery during crosspresentation by dendritic cells. Immunity. 2006;25:607–17. doi: 10.1016/j.immuni.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 27.Li X, Zhao X, Fang Y, et al. Generation of destabilized green fluorescent protein as a transcription reporter. J Biol Chem. 1998;273:34970–5. doi: 10.1074/jbc.273.52.34970. [DOI] [PubMed] [Google Scholar]
- 28.Maxfield FR. Weak bases and ionophores rapidly and reversibly raise the pH of endocytic vesicles in cultured mouse fibroblasts. J Cell Biol. 1982;95:676–81. doi: 10.1083/jcb.95.2.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kamhi-Nesher S, Shenkman M, Tolchinsky S, Fromm SV, Ehrlich R, Lederkremer GZ. novel quality control compartment derived from the endoplasmic reticulum. Mol Biol Cell. 2001;12:1711–23. doi: 10.1091/mbc.12.6.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakatsukasa K, Huyer G, Michaelis S, Brodsky JL. Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell. 2008;132:101–12. doi: 10.1016/j.cell.2007.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scott DC, Schekman R. Role of Sec61p in the ER-associated degradation of short-lived transmembrane proteins. J Cell Biol. 2008 Jun 30;181(7):1095–105. doi: 10.1083/jcb.200804053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–15. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lyman MA, Nugent CT, Marquardt KL, Biggs JA, Pamer EG, Sherman LA. The fate of low affinity tumor-specific CD8+ T cells in tumor-bearing mice. J Immunol. 2005;174:2563–72. doi: 10.4049/jimmunol.174.5.2563. [DOI] [PubMed] [Google Scholar]
- 34.Lustgarten J, Dominguez AL, Cuadros C. The CD8+ T cell repertoire against Her-2/neu antigens in neu transgenic mice is of low avidity with antitumor activity. Eur J Immunol. 2004;34:752–61. doi: 10.1002/eji.200324427. [DOI] [PubMed] [Google Scholar]
- 35.Dutoit V, Taub RN, Papadopoulos KP, Talbot S, Keohan ML, Brehm M, Gnjatic S, Harris PE, Bisikirska B, Guillaume P, Cerottini JC, Hesdorffer CS, Old LJ, Valmori D. Multiepitope CD8+ T cell response to a NY-ESO-1 peptide vaccine results in imprecise tumor targeting. J Clin Invest. 2002;110:1813–22. doi: 10.1172/JCI200216428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Slingluff CL, Jr, Speiser DE. Progress and controversies in developing cancer vaccines. J Transl Med. 2005;3:18. doi: 10.1186/1479-5876-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seliger B, Maeurer MJ, Ferrone S. Antigen-processing machinery breakdown and tumor growth. Immunol Today. 2000;21:455–64. doi: 10.1016/s0167-5699(00)01692-3. [DOI] [PubMed] [Google Scholar]
- 38.Kamal A, Thao L, Sensintaffar J, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–10. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 39.Eiseman JL, Lan J, Lagattuta TF, et al. Pharmacokinetics and pharmacodynamics of 17-demethoxy17-[[(2-dimethylamino)ethyl]amino] geldanamycin (17DMAG, NSC 707545) in C.B-17 SCID mice bearing MDA-MB-231 human breast cancer xenografts. Cancer Chemother Pharmacol. 2005;55:21–32. doi: 10.1007/s00280-004-0865-3. [DOI] [PubMed] [Google Scholar]
- 40.Lattouf JB, Srinivasan R, Pinto PA, Linehan WM, Neckers L. Mechanisms of disease: the role of heat-shock protein 90 in genitourinary malignancy. Nat Clin Pract Urol. 2006;3:590–601. doi: 10.1038/ncpuro0604. [DOI] [PubMed] [Google Scholar]
- 41.Matsui M, Machida S, Itani-Yohda T, Akatsuka T. Downregulation of the proteasome subunits, transporter, and antigen presentation in hepatocellular carcinoma, and their restoration by interferon-γ. J Gastroenterol Hepatol. 2002;17:897–907. doi: 10.1046/j.1440-1746.2002.02837.x. [DOI] [PubMed] [Google Scholar]
- 42.Seliger B, Dunn T, Schwenzer A, Casper J, Huber C, Schmoll HJ. Analysis of the MHC class I antigen presentation machinery in human embryonal carcinomas: evidence for deficiencies in TAP, LMP and MHC class I expression and their upregulation by IFN-γ. Scand J Immunol. 1997;466:625–32. doi: 10.1046/j.1365-3083.1997.d01-176.x. [DOI] [PubMed] [Google Scholar]
- 43.Zeh HJ, 3rd, Leder GH, Lotze MT, et al. Flow-cytometric determination of peptide-class I complex formation. Identification of p53 peptides that bind to HLA-A2. Hum Immunol. 1994;39:79–86. doi: 10.1016/0198-8859(94)90105-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.