
Figure 1.
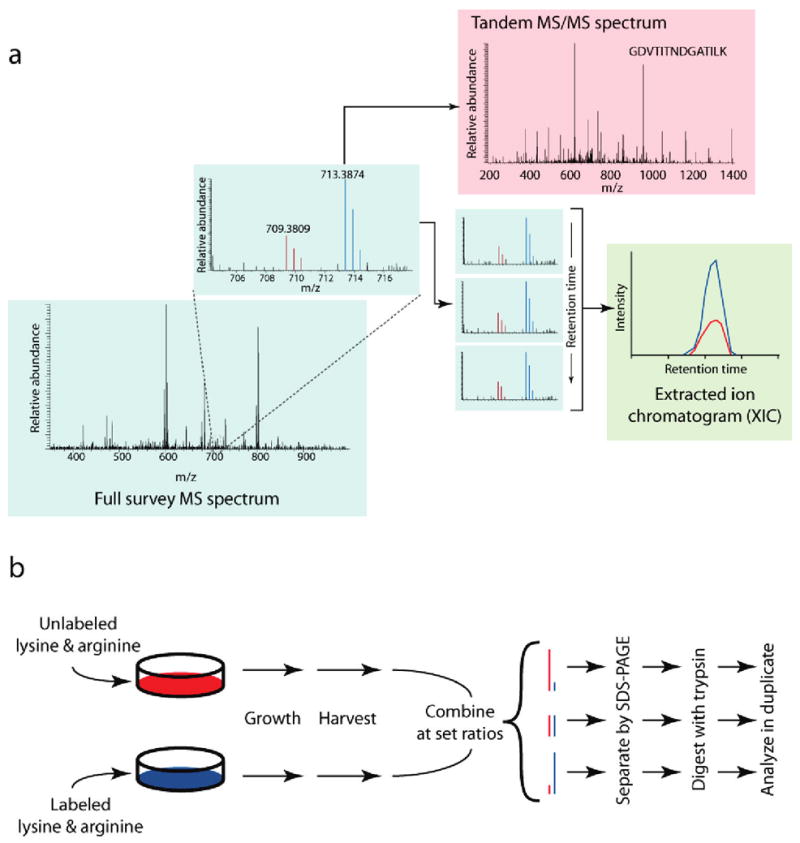
Experimental design. (a) Typical workflow for quantitative proteomic methods using stable isotopes. Stable isotope labeling produces two chemically identical peptide pools which differ only in their masses. This difference in mass is easily resolved within the full (MS) survey spectrum (blue box) into separate isotopic envelopes composed of all spectral peaks for the labeled and unlabeled species (red and blue peaks; see inset). To identify the eluting peptide, the mass spectrometer isolates and fragments the peptide ion of either the labeled (blue) or unlabeled (red) species to produce a tandem MS/MS spectrum (red box). The relative spectral peak intensities of both species are culled from successive MS spectra into an extracted ion chromatogram (green box). These chromatographic peaks, when compared across time, correlate with the change in abundance between the two peptide species. (b) Stable-isotope-labeled protein mixtures for testing signal-to-noise and quantitative accuracy. Jurkat lymphoblastic T-cell lines were grown using the SILAC method in two separate cultures differing in their growth media: one culture contained 13C615N2-lysine and 13C615N4-arginine, while the other contained natural forms of these amino acids. Cells were harvested, lysed, and combined at set protein concentration ratios of 5:1, 2.5:1, 1:1, 1:2.5, and 1:5. Sample mixtures were then gel-separated, trypsin-digested and analyzed in duplicate by LC-MS/MS techniques.