

Abstract
Purpose
To evaluate the effect of bevacizumab on the pharmacokinetics (PK) of irinotecan and its active metabolite. Exploratory analyses of the impact of variability in uridine diphosphate glucuronosyltransferase 1A (UGT1A) genes on irinotecan metabolism and toxicity were conducted.
Methods
This was an open-labeled, fixed-sequence study of bevacizumab with FOLFIRI (irinotecan, leucovorin, and infusional 5-fluorouracil). Pharmacokinetic assessments were conducted in cycles 1 and 3.
Results
Forty-five subjects were enrolled. No difference in dose-normalized AUC0-last for irinotecan and SN-38 between irinotecan administered alone or in combination with bevacizumab was identified. Leukopenia was associated with higher exposure to both irinotecan and SN-38. UGT1A1 polymorphisms were associated with variability in irinotecan PK. Gastrointestinal toxicity was associated with UGT1A6 genotype. No other associations between UGT1A genotypes and toxicity were detected.
Conclusion
Bevacizumab does not affect irinotecan PK when administered concurrently. A variety of pharmacogenetic relationships may influence the pharmacokinetics of irinotecan and its toxicity.
Background
Bevacizumab (rhuMab VEGF, Avastin®, Genentech, Inc., South San Francisco) is a humanized antibody against vascular endothelial growth factor (VEGF) that blocks the binding of VEGF to its cell surface receptor, resulting in disruption of the angiogenic signaling cascade. Bevacizumab was first approved by the United States Food and Drug Administration for the treatment of patients with advanced colorectal cancer based on a phase III study which compared bevacizumab in combination with IFL (irinotecan, bolus fluorouracil, and leucovorin) to IFL alone. In this clinical trial, increases in the incidence of grade 3 or 4 diarrhea and leukopenia were observed in the bevacizumab-containing arm [11]. A limited sampling pharmacokinetic substudy performed on 123 patients enrolled in this study suggested that the addition of bevacizumab to IFL was associated with a 33% increase in the AUC0-5 of SN-38 (the most active metabolite of irinotecan), and that this may have corresponded to higher levels of toxicity in patients receiving bevacizumab [8]. However, the PK substudy was not definitive because of the short sampling time for irinotecan and the large inter-subject variability. To formally address the issue of a potential pharmacokinetic interaction between bevacizumab and irinotecan, a controlled trial was undertaken. An exploratory pharmacogenetic study was also conducted, since the disposition of irinotecan is known to vary in a fashion partially dependent upon genetic variation in its metabolic pathways.
Irinotecan is a prodrug that is metabolized to its active form, SN-38, by carboxyesterases. SN-38 is subsequently inactivated via a glucuronidation process to SN-38 glucuronide (SN-38G). Inactivation of SN-38 is catalyzed by members of the uridine diphosphate glucuronosyltransferase 1A (UGT1A) and CYP3A4 systems [18]. Although the most commonly studied enzyme involved in the glucuronidation of SN-38 is UGT1A1, data have emerged for the roles of UGT1A7, UGT1A6, and UGT1A9 isoforms in the glucuronidation process [7, 15, 20, 34].
Irinotecan pharmacokinetics show significant interpatient variability. Recent data have been inconsistent regarding the role of UGT1A gene polymorphisms in mediating irinotecan toxicity [2, 3, 10, 12, 19, 21, 29]. This inconsistency is likely a function of the redundant affinity of several UGT1A isoforms for SN-38 as well as the complex genetics of the UGT1A loci [3, 7, 18]. Several studies have indicated an association between low activity UGT1A1 alleles and increased neutropenia in patients treated with irinotecan [10, 12, 14, 19]. These findings led to an FDA recommendation that irinotecan dosing be lowered in patients homozygous for the low activity UGT1A1*28 allele [22]. However, few studies have evaluated the role of UGT1A polymorphisms in toxicity associated with the most commonly used irinotecan regimens in patients with colorectal cancer, i.e. irinotecan in combination with infusional 5-FU [30].
The primary objective of the current clinical trial was to formally investigate whether bevacizumab impacts the PK of irinotecan and SN-38 in a controlled, fully powered clinical trial. The commonly used FOLFIRI regimen (irinotecan, leucovorin, and bolus 5-FU followed by continuous infusion 5-FU over 46 hours) [31] was selected as the platform for this study. Furthermore, we also explored the association of pharmacogenetic parameters of the UGT1A gene on the pharmacokinetic and toxicity profiles of irinotecan in this regimen.
Methods
Study Design and Patient Eligibility
This was a phase I, open-label, fixed sequence clinical trial conducted at three study centers utilizing the combination of 5-fluorouracil, leucovorin, irinotecan, and bevacizumab. This clinical trial was approved by the institutional review board at each participating institution. All patients provided written informed consent prior to entering both the study and the substudy.
Eligible patients had histologically-confirmed advanced solid tumors for which treatment with FOLFIRI plus bevacizumab was medically reasonable. Additional selection criteria included age >18 years; ECOG performance status 0 or 1; adequate organ function including absolute neutrophil count ≥ 1500 /uL, platelets ≥ 100,000 /uL, total bilirubin ≤ 1.5 mg/dL, AST < 3× upper limit of normal or < 5× upper limit of normal if liver metastases, creatinine ≤ 2.0 mg/dL, hemoglobin ≥ 9 g/dL, and International Normalized Ratio (INR) ≤ 1.5 unless receiving warfarin sodium.
Patients were excluded if they had received prior irinotecan or bevacizumab therapy; prior monoclonal antibody therapy; major surgical procedure or chemotherapy within 28 days; or history of serious systemic disease including myocardial infarction or stroke within 6 months prior to Day 0, unstable angina, clinically significant peripheral vascular disease, blood pressure > 150/100 mmHg, or New York Heart Association grade II or greater congestive heart failure; CNS or brain metastases; lung carcinoma; urine protein/creatinine ratio ≥ 1.0 at screening; evidence of bleeding diatheses or coagulopathy; non-healing wound, ulcer, or bone fracture; or history of abdominal fistula, gastrointestinal perforation, or intra-abdominal abscess within 26 days of Day 0. The use of concomitant drugs including St. John's Wart, phenytoin, valproic acid, phenobarbital, cyclosporine, indinavir, nelfinavir, ritonavir, saquinovir, fluconazole, itraconazole, or ketoconazole within 30 days prior to Day 0 was prohibited.
Treatment
Patients were treated with the FOLFIRI regimen as described by Tournigand et al [31] (irinotecan 180 mg/m2 IV administered over 90 minutes plus racemic leucovorin 400 mg/m2 IV administered over 2 hours, followed by 5-fluorouracil 400 mg/m2 IV bolus followed by 2400 mg/m2 continuous IV infusion over 46 hours every two weeks). Bevacizumab 5 mg/kg IV was administered over 30 minutes every two weeks, with the initial two doses given over 90 and 60 minutes respectively. During cycle 1, patients received FOLFIRI alone on Day 0 and bevacizumab was administered on Day 2 after the last irinotecan PK sample was drawn. In subsequent cycles, bevacizumab was administered prior to FOLFIRI on the same day. Dose adjustments were made based on interval toxicities. The primary study period during which PK sampling was obtained was Cycles 1 through 3. Subjects deriving benefit from treatment with FOLFIRI plus bevacizumab could continue treatment every two weeks for a period of up to two years. Treatment discontinuation was permitted for disease progression, adverse events, discretion of treating physician, or subject withdrawal of consent.
Clinical Assessments
Toxicity was graded according the National Cancer Institute Common Terminology Criteria for Adverse Events version 3 [1] during cycles 1-3. All subjects who received any study treatment were included in the safety analysis population. In order to assess the risk of toxicity after the first cycle and its relationship to UGT1A polymorphism, the highest grade of toxicity during cycle 1 and over cycles 1-3 was evaluated with respect to genotype.
Pharmacokinetic Assessments
For irinotecan and SN-38 PK, plasma samples were collected at the following time points on the first day of Cycles 1 and 3: prior to the start of the irinotecan infusion, 45 and 90 minutes after the initiation of irinotecan infusion, and post-infusion at 5, 10, 15, and 30 minutes and 1, 2, 4, 6, 8, 10, 24, 30, and 48 hours. For bevacizumab peak or trough concentrations, serum samples were collected prior to and ten minutes after the completion of the bevacizumab infusion during Cycles 1, 2, and 3.
Plasma samples were analyzed for irinotecan, SN-38, and 7-ethyl-10-[4-N-(5-aminopentanoic acid)-1-piperidino]carbonyloxycamptothecin (APC) by mass spectrometry with minimum quantifiable concentrations of 5.0 ng/ml, 1.0 ng/ml, and 2.0 ng/ml, respectively (Cedra Corp., Austin). Serum bevacizumab concentrations were measured by enzyme-linked immunosorbent assay (Genentech, Inc., South San Francisco) with a minimum quantifiable concentration of 78 ng/mL.
Non-compartmental analysis methods were used to calculate PK parameters using concentrations at or above the limit of quantification. PK parameters were determined for irinotecan, SN-38, SN-38G, and APC, and included AUC0-last and AUC0-infinity, clearance, maximum concentration (Cmax), time to maximum concentration (Tmax), mean residence time, terminal half-life, and steady-state volume of distribution. Calculations were performed using WinNonlin, Version 4.1 (Pharsight Corporation, Mountain View, California).
UGT1A Genotyping
Before cycle 1, an optional blood sample was collected from patients for germline analysis of genes relevant to irinotecan metabolism. Blood was collected in Vacutainer tubes and stored at -70°C until processing. Buffy coat from whole blood was used to isolate genomic DNA via automation on the Gentra AutoPure LS using the PureGene chemistry. Candidate genes were selected based on known involvement in the metabolic pathway of irinotecan and functionally significant genetic polymorphisms.
For UGT1A1 genotyping, a 119 bp region of the promoter was amplified by PCR using Jumpstart™ REDTaq® ReadyMix™ PCR Reaction Mix (Sigma) in a 25 μL reaction with 20-30 ng genomic DNA as the template. PCR and pyrosequencing were carried out using primers and thermal cycling conditions according to the methods described by Saeki et al [27]. This method allows for discrimination of the n=5, 6, 7 and 8 TATA repeat that defines the common UGT1A1 alleles. UGT1A1*28 is defined as n=7 TATA repeats and is associated with lower UGT1A1 enzyme activity than the n=6 TATA repeat UGT1A1*1 allele. For UGT1A6 T19G (S7A, rs6759893), A541G (T181A, rs2070959), A552C (R184S, rs1105879) polymorphisms, a 238bp fragment containing the codon 7 SNP and a 215bp fragment containing the codon 181 and 184 SNPs were amplified by PCR using Jumpstart™ REDTaq® ReadyMix™ PCR Reaction Mix (Sigma) in a 25 μL reaction with 20-30 ng genomic DNA as the template. PCR and pyrosequencing were carried out using primers and thermal cycling conditions according to the methods described by Carlini et al [3]. UGT1A7 and UGT1A9 SNPs were genotyped by BigDye® Terminator cycle sequencing on an ABI PRISM® 3100 Genetic Analyzer (Applied Biosystems) according to the methods of Carlini et al [3]. PCR reactions contained Jumpstart™ REDTaq® ReadyMix™ PCR Reaction Mix (Sigma) and 20-30 ng genomic DNA template in a 25 μL volume.
UGT1A isoforms were binned in cases where enzymatic activity could be predicted. UGT1A6 and UGT1A7 genotypes were binned according to predicted enzyme activity [3, 20, 34]. For UGT1A6, genotype bins were categorized as high, moderate, low, and unknown enzyme activity. UGT1A7 genotype bins were categorized as high or low enzyme activity. Genotype bins for UGT1A6 and UGT1A7 were used for the pharmacogenetic substudy analyses. Genotypes for UGT1A1 and UGT1A9 were utilized for the pharmacogenetic substudy analyses, as there is little data to support the prediction of UGT1A9 activity from genotype. (Table 1)
Table 1. UGT1A Genotype Frequency and Genotype Bins (Total n=37).
| UGT1A1 Genotype Frequency (n) | ||||
| *1/*1 (14) | ||||
| *1/*28 (16) | ||||
| *28/*28 (7) | ||||
| UGT1A9 Genotype Frequency (n) | ||||
| 9/9 (14) | ||||
| 9/10 (18) | ||||
| 10/10 (5) | ||||
| UGT1A Genotype Bin | High Enzyme Activity (n) | Moderate Enzyme Activity (n) | Low Enzyme Activity (n) | Unknown Activity (n) |
| UGT1A6 | *2/*2 (6) | *1/*1 (12) | *1/*2 (13) | *2/*3 (2) |
| *1/*3 (2) | *1/*4 (2) | |||
| UGT1A7 | *2/*2 (3) | |||
| *1/*1 (5) | *2/*3 (3) | |||
| *1/*2 (6) | *3/*3 (6) | |||
| *1/*3 (12) | *3/*8 (1) | |||
| *8/*8 (1) | ||||
Toxicity categories assessed were diarrhea; leukopenia; gastrointestinal toxicity (including mucositis, nausea, vomiting, diarrhea, and dehydration); and other toxicities including fever, asthenia, cardiovascular, respiratory, nervous system, skin, constipation, anorexia, and abdominal pain. Toxicity data were available during cycles 1-3, while pharmacokinetic data were available for cycles 1 and 3 of FOLFIRI plus bevacizumab treatment.
Statistical Methods
Statistical analyses to determine drug-drug interaction were based on the FDA recommendations for in vivo drug metabolism/drug interaction studies [32] and pre-specified prior to study initiation and assaying of serum samples. Analyses utilized AUC0-last, which was normalized to a dose of 1 mg/m2 of administered irinotecan. The primary statistical analysis was the estimation of the geometric mean ratio (GMR) of the AUC0-last in the presence of bevacizumab (Cycle 3) to the AUC0-last in the absence of bevacizumab (Cycle 1). Confidence intervals (CI) were calculated for GMRs by calculating the difference between the log-transformed AUC0-last using the t-distribution and then back-transformed to the ratio scale. The 90% CI for the GMR of irinotecan was compared with pre-specified bounds (0.8, 1.25) while the 90% CI for the GMR of SN-38 was compared with pre-specified bounds (0.7, 1.43) due to a larger intra-subject variability [32]. The equivalence hypotheses compare the pharmacokinetics of irinotecan with and without bevacizumab. If the 90% CI of the GMR for irinotecan was completely contained within pre-specified (0.8, 1.25), and the 90% CI of the GMR for SN-38 was completely contained within pre-specified (0.7, 1.43), and the point estimate of the GMR for SN-38 was within pre-specified (0.8, 1.25), then it would be concluded that bevacizumab had no significant effect on the pharmacokinetics of irinotecan.
Pharmacogenomic analyses with regard to UGT1A were exploratory. The method of Guo and Thompson [9] was used to test for Hardy-Weinberg equilibrium. One-sided Jonckheere-Terpstra tests were used to assess the association of discrete toxicity categories and pharmacokinetic measures. The Kruskal-Wallis [28] test was used to assess the relation of genotype (UGT1A1 and UGT1A9), or genotype bins (UGT1A6 and UGT1A7), to pharmacokinetic measures of Cmax and AUC of irinotecan and SN-38. Permutation tests [28] were used to assess the significance of associations in sparse contingency table chi-squared tables of toxicity versus genotype or enzyme activity bins.
Results
Demographics and Disease Characteristics
Forty-five subjects were enrolled between January 4, 2005 and October 26, 2005 at three institutions. Patient characteristics included a median age 55.0 years (range 30-74), 64% male, 92% Caucasian, and ECOG performance status 0 (53%) and 1 (47%). The most common primary tumor types included esophagus (12), ovary (9), and pancreas (5). The most frequent sites of metastases included the liver (44%) and lymph nodes (42%).
Pharmacokinetic and Drug-Drug Interaction Analysis
The drug-drug interaction PK analysis population included 36 subjects. This population excluded the 6 subjects who did not complete all three cycles of treatment, 2 patients who did not have adequate PK sample to support AUC calculation, and 1 patient who did not receive bevacizumab in Cycle 3. Twenty-two of the 36 patients (61%) received prior systemic therapy with four having two or more prior regimens. Twenty-seven of 36 patients experienced a dose delay and/or reduction of irinotecan due to diarrhea and/or leukopenia.
Dose-normalized concentration–time profiles for irinotecan and SN-38 concentrations are displayed in Figures 1 and 2, respectively. The Cmax, Tmax, elimination half lives, clearance, steady state volumes of distribution, and pharmacokinetic properties of irinotecan and its metabolites for cycles 1 and 3 are listed in Table 2. These parameters are consistent with previous reports [5, 6, 23]. The GMR of the AUC0–last in the presence of bevacizumab (at Cycle 3) to the AUC0–last in the absence of bevacizumab (at Cycle 1) was calculated for both irinotecan and SN-38 and is presented in Table 2 together with the 90% confidence interval (CI). The GMR point estimates and 90% CIs are within pre-specified boundaries, which indicate that the presence of bevacizumab does not alter the pharmacokinetics of irinotecan.
Fig. 1.
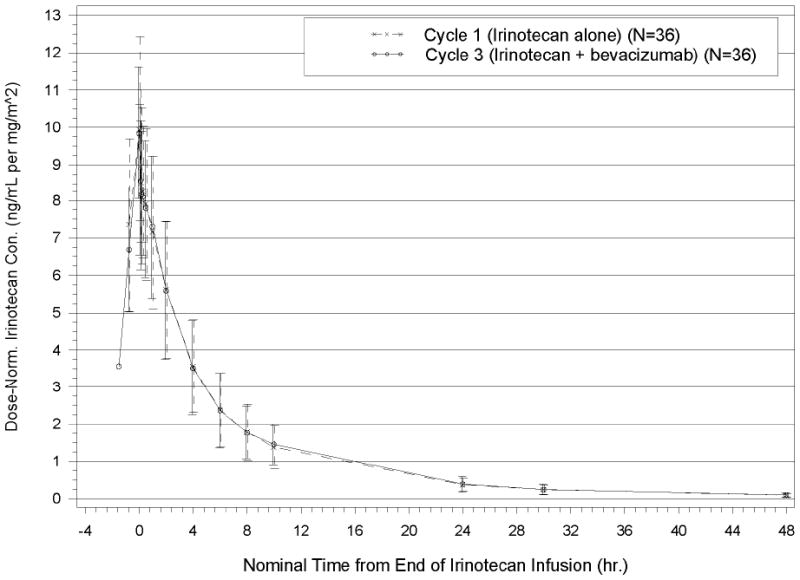
Dose-Normalized Plasma Irinotecan Concentration–Time Profiles for Cycle 1 and Cycle 3 (Mean±SD): Primary PK Analysis Population
Fig. 2.
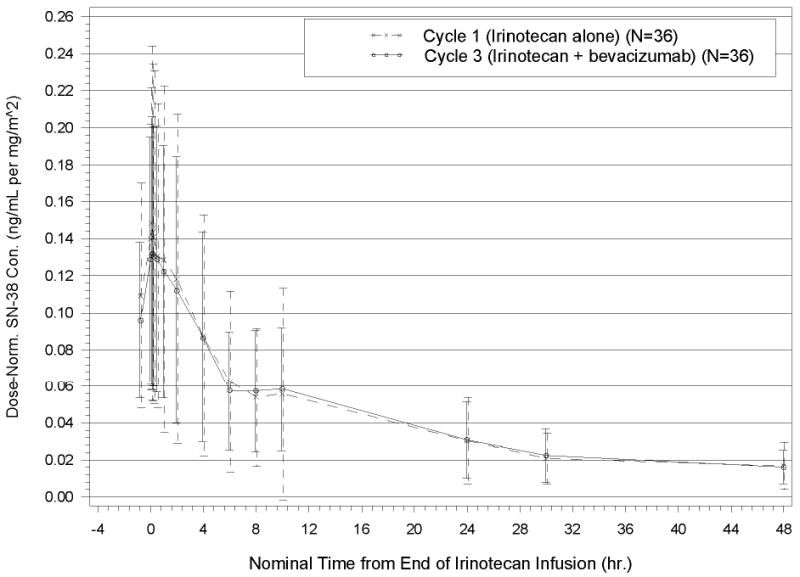
Dose-Normalized Plasma SN-38 Concentration–Time Profiles for Cycle 1 and Cycle 3 (Mean±SD): Primary PK Analysis Population
Table 2.
Irinotecan, SN-38, APC, and SN-38G Plasma Pharmacokinetic Parameters
| Mean (SD) | |||||
|---|---|---|---|---|---|
| Cycle | Parameter | Irinotecan | SN-38 | APC | SN-38G |
| 1 (prior to bevacizumab administration) | Cmax (ng/mL) | 1,870 (425) | 29.6 (18.6) | 235 (159) | 81.1 (39.0) |
| Tmax (hr) | 1.57 (0.316) | 2.18 (1.02) | 3.49 (1.89) | 2.26 (0.74) | |
| Elimination half-life (hr) | 11.9 (2.26) | 21.1 (8.34) | 10.5 (2.11) | 17.5 (5.40) | |
| Clearance (mL/hr/m2) | 17,500 (6050) | 0.542 (0.274) | 0.089 (0.056) | 0.166 (0.079) | |
| Volume of distribution (mL/m2) | 158,000 (51500) | 14.9 (9.09) | 1.20 (0.744) | 3.80 (1.74) | |
| Geometric Mean | 58.6 | 1.68 | 13.3 | 5.71 | |
| 3 (after bevacizumab administration) | Cmax (ng/mL) | 1,610 (276) | 23.7 (11.3) | 147 (115) | 59.8 (25.6) |
| Tmax (hr) | 1.58 (0.209) | 2.04 (1.03) | 3.74 (1.31) | 3.07 (1.45) | |
| Elimination half-life (hr) | 12.1 (2.22) | 18.9 (6.47) | 9.84 (2.68) | 16.4 (7.66) | |
| Clearance (mL/hr/m2) | 17,500 (5750) | 0.561 (0.304) | 0.120 (0.084) | 0.186 (0.090) | |
| Volume of distribution (mL/m2) | 161,000 (49,100) | 13.3 (7.05) | 1.67 (1.23) | 3.99 (1.97) | |
| Geometric Mean | 58.3 | 1.69 | 9.76 | 5.08 | |
| Geometric Mean Ratio (Cycle 3/Cycle 1 (90% CI) | 0.99 (0.93, 1.07) | 1.01 (0.92, 1.11) | 0.74 (0.64, 0.85) | 0.89 (0.80, 0.99) | |
Volume of distribution: for irinotecan, it is the steady-state volume of distribution (mL/m2); for SN-38, APC, and SN-38G, it is WinNonlin output that is a composite of metabolite volume of distribution, clearance, and fraction metabolized.
For each irinotecan metabolite (SN-38, APC, and SN-38G), clearance is apparent clearance, which is CL/fm, where fm is the fraction of the parent drug that is metabolized to that metabolite.
Serum samples were collected pre- and post-infusion at Cycles 1 and 3 to confirm bevacizumab exposure. Table 3 summarizes bevacizumab peak and trough concentrations for the analysis population. Bevacizumab peak concentrations in this trial were lower than those observed in previous studies, perhaps due to differences in sampling times and inherent difficulty in reliably measuring peak concentrations, while the trough concentrations were consistent with previous experience [17].
Table 3.
Serum Bevacizumab Peak and Trough Concentrations (ng/mL)
| Cycle | Concentration | No. of samples | Mean | SD | %CV | Median | Minimum | Maximum |
|---|---|---|---|---|---|---|---|---|
| 1 | Peak | 35 | 64,900 | 31,300 | 48 | 57,200 | 20,200 | 134,000 |
| Trough | 31 | 23,100 | 13,700 | 59 | 20,800 | 12,500 | 90,200 | |
| 2 | Peak | 32 | 85,400 | 42,300 | 50 | 80,200 | 22,900 | 185,000 |
| Trough | 36 | 26,100 | 7,490 | 29 | 25,600 | 16,000 | 40,700 | |
| 3 | Peak | 36 | 94,600 | 35,400 | 37 | 99,800 | 38,100 | 183,000 |
Adverse Events
Thirty-nine patients received all scheduled FOLFIRI treatments at full or reduced doses, while 38 patients received FOLFIRI and bevacizumab at full or reduced doses. Forty-three patients had at least one adverse event during cycles 1-3, with 4 patients having adverse events leading to study discontinuation. The most common toxicities overall were nausea (60%), vomiting (20%), diarrhea (51%), leukopenia (51%), sweating (24%), and dehydration (16%). The most common grade 3 or 4 adverse events were leukopenia (38%), dehydration (9%), asthenia (4%), nausea (4%), and diarrhea (4%). Twenty-two patients had a grade 3 or 4 adverse event, with 17/45 (38%) patients having grade 3 or 4 leukopenia.
Pharmacogenetic and Pharmacodynamic Substudy
Thirty-nine patients consented to participation in the pharmacogenetic substudy. Genotyping was unsuccessful in two patients, one patient had incomplete adverse event reporting, and eight patients were not evaluable for pharmacokinetics. Therefore, 37 patients had genotyping, 29 had genotyping and PK, 30 had PK and toxicity data, and 36 had genotyping and toxicity data. Of the patients who consented to the substudy, 30 were included in the drug-drug interaction PK analysis population. All genotypes appeared to follow Hardy-Weinberg equilibrium, including UGT1A1*28.
Within the substudy population, leukopenia during cycle 1 was associated with the Cmax of SN-38 during cycle 1 (p=0.049), as well as the AUCs of irinotecan (p=0.007) and its active metabolite SN-38 (p=0.016). These relationships were confirmed when highest grade of leukopenia across cycles 1-3 was considered. In contrast, there was no association between diarrhea and the pharmacokinetic parameters of irinotecan (p=0.21 for Cmax and p=0.26 for AUC0-last) or its active metabolite (p=0.87 for Cmax SN38 and p=0.71 for AUC0-last SN38). Other toxicities such as mucositis, nausea, vomiting, and dehydration were also not associated with Cmax or AUC0-last of irinotecan, SN-38, or SN-38G. This finding was consistent when considering cycle 1 alone and combined cycles 1-3.
We conducted exploratory analyses of the association between genotype and PK parameters. As shown in Table 4, there was an association between UGT1A1 genotype and mean Cmax and AUC0-last of irinotecan and SN-38. UGT1A7 genotype bin trended toward an association with mean Cmax of SN-38 and mean AUC0-last of SN-38. However, this may be largely attributed to the impact of higher values seen in those patients with the UGT1A7 3/3 genotype compared with the other genotypes in this genotype bin (Table 4) or influenced by genetic linkage among UGT1A alleles. There was no association between UGT1A6 or UGT1A9 genotype and PK parameters.
Table 4.
UGT 1A1 and UGT1A7 Genotype vs. SN-38 Pharmacokinetics during Cycle 1
| Genotype (n) | Mean Cmax SN-38 ng/mL (Standard Deviation) | P value | Mean AUC0-last SN-38 ng/mL • hr/(mg/m2) (Standard Deviation) | P value |
|---|---|---|---|---|
| UGT 1A1 1/1 (n=9) | 23.2 (11.3) | p=0.03 | 1.65(1.50) | p=0.01 |
| UGT1A1 1/28 (n=15) | 27.3 (19.9) | 1.73 (1.22) | ||
| UGT1A1 28/28 (n=5) | 48.8 (18.5) | 3.45 (1.05) | ||
| UGT1A7 1/1 (n=3) | 21.9 (7.46) | p=0.09 | 1.48 (0.21) | p=0.12 |
| UGT1A7 1/2 (n=5) | 29.7 (18.5) | 2.25 (2.09) | ||
| UGT1A7 1/3 (n=10) | 27.1 (23.9) | 1.761 (1.52) | ||
| UGT1A7 2/2 (n=3) | 28.9 (9.584) | 1.344 (0.46) | ||
| UGT1A7 2/3 (n=2) | 18.5 (1.20) | 1.46 (0.09) | ||
| UGT1A7 3/3 (n=5) | 42.9 (22.0) | 2.78 (1.37) |
No associations were detected between UGT1A genotypes or genotype bins and diarrhea, leukopenia, or combined GI toxicities during Cycle 1. No association between leukopenia across Cycles 1-3 and UGT1A genotypes or genotype bins was detected. However, an association between UGT1A6 genotype bin and overall gastrointestinal toxicity was observed across Cycles 1-3 (p=0.028), with greater toxicity seen in patients with low and moderate activity genotypes. UGT1A7 genotype bin was not associated with toxicity.
Discussion
The data we present demonstrate that the pharmacokinetics of irinotecan was not altered by the presence of bevacizumab. This study was undertaken as follow-up of an exploratory PK analysis conducted within the bevacizumab colorectal cancer licensing trial that suggested elevated SN-38 exposure in the presence of bevacizumab [8]. We employed a single-arm design in which each patient served as his/her own control, and pharmacokinetic analysis covered 4 to 5 irinotecan half-lives. These features allowed for adequate capture of the concentration-time curves to estimate all parameters necessary to definitively address the potential interaction between bevacizumab and irinotecan.
In order to control for irinotecan dose reduction between cycles, dose normalization of the PK parameters were utilized to avoid an imbalanced comparison between Cycle 1 and Cycle 3 irinotecan and SN-38 PK. Irinotecan and SN-38 pharmacokinetics are linear in the doses administered in this study [5, 26]. Bevacizumab trough concentrations were consistent with previous observations, indicating that patients in this study received clinically relevant exposure to bevacizumab.
We used this formal PK study as an opportunity to explore the interaction between irinotecan metabolizing enzyme polymorphisms, pharmacokinetics, and toxicity. Consistent with other reports, irinotecan and SN-38 exposure were associated with leukopenia in our study [5, 14, 23, 24, 33]. Notably, GI toxicities such as diarrhea had no association with PK parameters. This lack of association between plasma PK and diarrhea suggests that plasma levels of irinotecan and SN-38 may not be the only driving force behind the development of diarrhea. The contribution of local intestinal exposure to SN-38 via hydrolysis of SN-38G by glucuronidases in the microflora may also play a role in the variable severity of irinotecan-induced diarrhea [16, 25]. However, this lack of association is complicated by the low incidence of severe diarrhea observed in this small patient population.
Recent data have raised the question of whether UGT1A genotype should be used to help guide irinotecan dosing [29, 30]. In particular, the UGT1A1*28 allele has been associated with the development of neutropenia following irinotecan therapy, and these data lead to an FDA recommendation for a reduced starting dose in patients homozygous for this allele [12-14]. In a recent study of 250 patients with metastatic colorectal cancer receiving FOLFIRI as first-line treatment, the UGT1A1 *28 allele was associated with a higher risk of grade 3 or 4 neutropenia during the first cycle, but this association was not observed when the entire treatment period was considered [30]. We sought to establish a mechanism for this observation by assessing genetics, PK, and toxicity in the same patients. Although we were able to confirm a relationship between UGT1A1 polymorphisms and SN-38 exposure, we did not find an association between genotype and toxicity, particularly leukopenia. We also previously reported a lack of association between UGT1A1 genotype and toxicity in patients treated with irinotecan plus capecitabine [3]. In that previous study, UGT1A7 and UGT1A9 low activity genotypes were associated with reduced gastrointestinal toxicity. Consistent with our findings, other studies also did not support an association of UGT1A1 genotype with diarrhea [14, 19, 29, 30].
A significant limitation of our analysis of UGT1A pharmacogenomics is the small sample size of this study, and the potential bias in enrolling patients with the UGT1A1*28 allele associated with restricting the eligibility criteria to a total bilirubin ≤ 1.5 mg/dL. Although we did observe Hardy-Weinberg equilibrium among enrolled subjects, this criterion may have biased the study against enrolling those most vulnerable to SN-38-related toxicities. Our results must therefore be viewed as exploratory.
We observed an association between UGT1A6 enzymatic activity level and gastrointestinal toxicities across all three cycles. In a previous study, we did not find an association between UGT1A6 genotype and toxicity in patients receiving capecitabine plus irinotecan [4]. This discordance between studies may relate to differences in the chemotherapy regimens investigated, patient populations, or small sample sizes of the studies conducted. Clearly, further work in this area is necessary to dissect the interplay between various UGT1A alleles, haplotypes, and toxicities with particular chemotherapy combinations and schedules.
In conclusion, bevacizumab does not impact the pharmacokinetics of irinotecan administered in the FOLFIRI regimen. Our exploratory pharmacogenetic analyses confirmed the relationship between UGT1A1 low activity alleles and increased SN-38 exposure, but not toxicity. Controversy remains regarding UGT1A1 genotyping for patients initiating irinotecan, and our exploratory pharmacogenomic substudy results suggest the need for larger trials to explore the contribution of other UGT1A isoforms on pharmacokinetics and toxicity of irinotecan.
Acknowledgments
Jacques Gaudreault and Dan Spyker for their input and insights into the study design.
This study was supported by Genentech and Cancer Center Support Grant NCI-P30 CA 006927 to Fox Chase Cancer Center.
References
- 1.Common Terminology Criteria for Adverse Events version 3.0, US Department of Health and Human Services National Institutes of Health.
- 2.Ando Y, Saka H, Ando M, Sawa T, Ueoka H, Yokoyama A, Saitoh S, Shimokata K, Hasegawa Y. Polymorphisms of UDP-glucuronosyltransferase gene and irinotecan toxicity: a pharmacogenetic analysis. Can Res. 2000;60:6921–6926. [PubMed] [Google Scholar]
- 3.Carlini LE, Meropol NJ, Bever J, Andria ML, Hill T, Gold P, Rogatko A, Wang H, Blanchard RL. UGT1A7 and UGT1A9 polymorphisms predict response and toxicity in colorectal cancer patients treated with capecitabine/irinotecan. Clin Can Res. 2005;11:1226–1236. [PubMed] [Google Scholar]
- 4.Carlini LE, Meropol NJ, Chen YM, McGarry C, Hill T, Gold P, Blanchard RL. Pharmacogenetic analysis of UGT1A1, UGT1A6, UGT1A7 and thymidylate synthase in a phase II study of capecitabine plus irinotecan in patients with metastatic colorectal cancer. J Clin Oncol. 2004;22:3623. Meeting Abstracts. [Google Scholar]
- 5.Chabot GG, Abigerges D, Catimel G, Culine S, de Forni M, Extra JM, Mahjoubi M, Herait P, Armand JP, Bugat R, Clavel M, Marty ME. Population pharmacokinetics and pharmacodynamics of irinotecan (CPT-11) and active metabolite SN-38 during phase I trials. Ann Oncol. 1995;6:141–51. doi: 10.1093/oxfordjournals.annonc.a059109. [DOI] [PubMed] [Google Scholar]
- 6.de Jonge MJ, Verweij J, de Bruijn P, Brouwer E, Mathijssen RH, van Alphen RJ, de Boer-Dennert MM, Vernillet L, Jacques C, Sparreboom A. Pharmacokinetic, metabolic, and pharmacodynamic profiles in a dose escalating study of irinotecan and cisplatin. J Clin Oncol. 2000;18:195–203. doi: 10.1200/JCO.2000.18.1.195. [DOI] [PubMed] [Google Scholar]
- 7.Gagne J, Montminy V, Belanger P, Journault K, Gaucher G, Guillemette C. Common human UGT1A polymorphisms and the altered metabolism of irinotecan active metabolite 7-ethyl-10-hydroxycamptothecin (SN-38) Mol Pharmacol. 2002;62:608–617. doi: 10.1124/mol.62.3.608. [DOI] [PubMed] [Google Scholar]
- 8.Genentech. Avastin Package Insert 2007 [Google Scholar]
- 9.Guo SW, Thompson EA. Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics. 1992;48:361–372. [PubMed] [Google Scholar]
- 10.Han J, Lim H, Sin ES, Yoo Y, Park YH, Lee J, Jang I, Lee DO, Lee JS. Comprehensive analysis of UGT1A polymorphisms predictive for pharmacokinetics and treatment outcome in patients with non-small cell lung cancer treated with irinotecan and cisplatin. J Clin Oncol. 2006;24:2237–2244. doi: 10.1200/JCO.2005.03.0239. [DOI] [PubMed] [Google Scholar]
- 11.Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. New Engl J Med. 2004;350:2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 12.Innocenti F, Undevia SD, Iyer L, Chen PX, Das S, Kocherginsky M, Karrison T, Janisch L, Ramirez J, Rudin CM, Vokes EE, Ratain MJ. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J Clin Oncol. 2004;22:1382–1388. doi: 10.1200/JCO.2004.07.173. [DOI] [PubMed] [Google Scholar]
- 13.Innocenti F, Vokes EE, Ratain MJ. Irinogenetics: what is the right star? J Clin Oncol. 2006;24:2221–2224. doi: 10.1200/JCO.2005.05.2464. [DOI] [PubMed] [Google Scholar]
- 14.Iyer L, Das S, Janisch L, Wen M, Ramirez J, Karrison T, Fleming GF, Vokes EE, Schilsky RL, Ratain MJ. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenetics J. 2002;2:43–47. doi: 10.1038/sj.tpj.6500072. [DOI] [PubMed] [Google Scholar]
- 15.Iyer L, King CD, Whitington PF, Green MD, Roy SK, Thephly TR, Coffman BL, Ratain MJ. Genetic predisposition to the metabolism of irinotecan (CPT-11): role of uridine diphosphate glucuronosyltransferase isoform 1A1 in the glucuronidation of its active metabolite (SN38) in human liver microsomes. J Clin Invest. 1998;101:847–854. doi: 10.1172/JCI915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kehrer DFS, Sparreboom A, Verweij J, de Bruijn P, Nierop CA, van de Schraaf J, Ruijgrok EJ, de Jonge MJA. Modulation of Irinotecan-induced Diarrhea by Cotreatment with Neomycin in Cancer Patients. Clin Cancer Res. 2001;7:1136–1141. [PubMed] [Google Scholar]
- 17.Lu JF, Gaudreault J, Novotny W, Lum BL, Bruno R. A population pharmacokinetic (PK) model for bevacizumab (Avastin). ASCPT meeting; Miami, Florida. 2005. [Google Scholar]
- 18.Mathijssen RH, van Alphen RJ, Verweij J, Loos WJ, Nooter K, Stoter G, Sparreboom A. Clinical pharmacokinetics and metabolism of irinotecan (CPT-11) Clin Can Res. 2001;7:2182–2194. [PubMed] [Google Scholar]
- 19.McLeod HL, Parodi L, Sargent DJ, Marsh S, Green E, Abreu P, Cisar LA, Goldberg RM. UGT1A1*28, toxicity and outcome in advanced colorectal cancer: Results from Trial N9741. J Clin Oncol. 2006;24:3520. Meeting Abstracts. [Google Scholar]
- 20.Nagar S, Zalatoris JJ, Blanchard RL. Human UGT1A6 pharmacogenetics: identification of a novel SNP, characterization of allele frequencies and functional analysis of recombinant allozymes in human liver tissue and in cultured cells. Pharmacogenetics. 2004;14:487–499. doi: 10.1097/01.fpc.0000114771.78957.cb. [DOI] [PubMed] [Google Scholar]
- 21.O'Dwyer PJ, Catalano RB. Uridine Diphosphate Glucuronosyltransferase (UGT) 1A1 and Irinotecan: Practical Pharmacogenomics Arrives in Cancer Therapy. J Clin Oncol. 2006;24:4534–4538. doi: 10.1200/JCO.2006.07.3031. [DOI] [PubMed] [Google Scholar]
- 22.Pfizer. Camptosar Package Insert 2007 [Google Scholar]
- 23.Pitot HC, Goldberg RM, Reid JM, Sloan JA, Skaff PA, Erlichman C, Rubin J, Burch PA, Adjei AA, Alberts SA, Schaaf LJ, Elfring G, Miller LL. Phase I dose-finding and pharmacokinetic trial of irinotecan hydrochloride (CPT-11) using a once-every-three-week dosing schedule for patients with advanced solid tumor malignancy. Clin Cancer Res. 2000;6:2236–44. [PubMed] [Google Scholar]
- 24.Poujol S, Bressolle F, Duffour J, Abderrahim AG, Astre C, Yehou M, Pinguet F. Pharmacokinetics and pharmacodynamics of irinotecan and its metabolites from plasma and saliva data in patients with metastatic digestive cancer receiving Folfiri regimen. Cancer Chemother Pharmacol. 2006;58:292–305. doi: 10.1007/s00280-005-0166-5. [DOI] [PubMed] [Google Scholar]
- 25.Ratain MJ. Irinotecan Dosing: Does the CPT in CPT-11 Stand for “Can't Predict Toxicity”? J Clin Oncol. 2002;20:7–8. doi: 10.1200/JCO.2002.20.1.7. [DOI] [PubMed] [Google Scholar]
- 26.Rothenberg ML, Kuhn JG, Schaaf LJ, Rodriguez GI, Eckhardt SG, Villalona-Calero MA, Rinaldi DA, Hammond LA, Hodges S, Sharma A, Elfring GL, Petit RG, Locker PK, Miller LL, von Hoff DD. Phase I dose-finding and pharmacokinetic trial of irinotecan (CPT-11) administered every two weeks. Ann Oncol. 2001;12:1631–1641. doi: 10.1023/a:1013157727506. [DOI] [PubMed] [Google Scholar]
- 27.Saeki M, Saito Y, Jinno H, Tohkin M, Kurose K, Kaniwa N, Komamura K, Ueno K, Kamakura S, Kitakaze M, Ozawa S, Sawada J. Comprehensive UGT1A1 genotyping in a Japonese population by pyrosequencing. Clin Chem. 2003;49:1182–1185. doi: 10.1373/49.7.1182. [DOI] [PubMed] [Google Scholar]
- 28.Siegel S, Castellan NJ. Non-parametric statistics for the behavior sciences. McGraw-Hill; 1988. [Google Scholar]
- 29.Stewart CF, Panetta JC, O'Shaughnessy MA, Throm SL, Fraga CH, Owens T, Lui T, Billups C, Rodriguez-Galindo C, Gajjar A, Furman WL, McGregor LM. UGT1A1 promoter genotype correlates with SN-38 pharmacokinetics, but no severe toxicity in patients receiving low-dose irinotecan. J Clin Oncol. 2007;25:2594–2600. doi: 10.1200/JCO.2006.10.2301. [DOI] [PubMed] [Google Scholar]
- 30.Toffoli G, Cecchin E, Corona G, Russo A, Buonadonna A, D'Andrea M, Pasetto LM, Pessa S, Errante D, De Pangher V, Giusto M, Medici M, Gaion F, Sandri P, Galligioni E, Bonura S, Boccalon M, Biason P, Frustaci S. The role of UGT1A1*28 polymorphism in the pharmacodynamics and pharmacokinetics of irinotecan in patients with metastatic colorectal cancer. J Clin Oncol. 2006;24:3061–3068. doi: 10.1200/JCO.2005.05.5400. [DOI] [PubMed] [Google Scholar]
- 31.Tournigand C, Andre T, Achille E, Lledo G, Flesh M, Mery-Mignard D, Quinaux E, Couteau C, Buyse M, Ganem G, Landi B, Colin P, Louvet C, de Gramont A. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol. 2004;22:229–237. doi: 10.1200/JCO.2004.05.113. [DOI] [PubMed] [Google Scholar]
- 32.U.S. Department of Health and Human Services FaDA, CDER, CBER. Guidance for Industry. Rockville, MD: 1999. In vivo drug metabolism/drug interaction studies—study design, data analysis, and recommendations for dosing and labeling. [Google Scholar]
- 33.van der Bol JM, Mathijssen RH, Loos WJ, Friberg LE, van Schaik RHN, de Jonge MJ, Planting AS, Verweij J, Sparreboom A, de jonge FA. Cigarette smoking and irinotecan treatment: pharmacokinetic interaction and effects on neutropenia. J Clin Oncol. 2007;25:1–8. doi: 10.1200/JCO.2006.09.6115. [DOI] [PubMed] [Google Scholar]
- 34.Villeneuve L, Girard H, Fortier L, Gagne J, Guillemette C. Novel functional polymorphisms in the UGT1A7 and UGT1A9 glucuronidating enzymes in Caucasian and African-american subjects and their impact on the metabolism of 7-ethyl-10-hydroxycamptothecin and flavopiridol anticancer drugs. JPET. 2003;307:117–128. doi: 10.1124/jpet.103.054072. [DOI] [PubMed] [Google Scholar]