

Abstract
One of the long-term goals in developing advanced biomaterials is to generate protein-like nanostructures and functions from a completely nonnatural polymer. Toward that end, we introduced a high-affinity zinc-binding function into a peptoid (N-substituted glycine polymer) two-helix bundle. Borrowing from well-understood zinc-binding motifs in proteins, thiol and imidazole moieties were positioned within the peptoid such that both helices must align in close proximity to form a binding site. We used fluorescence resonance energy transfer (FRET) reporter groups to measure the change of the distance between the two helical segments and to probe the binding of zinc. We systematically varied the position and number of zinc-binding residues, as well as the sequence and size of the loop that connects the two helical segments. We found that certain peptoid two-helix bundles bind zinc with nanomolar affinities and high selectivity compared to other divalent metal ions. Our work is a significant step toward generating biomimetic nanostructures with enzyme-like functions.
Introduction
Sequence-specific heteropolymers are growing in importance as useful tools in chemical biology, drug discovery, delivery, and materials science.1–4 Recent advances in synthetic chemistry have made it possible to generate many different types of nonnatural sequence-specific heteropolymers, providing a test of folding principles as well as potential therapeutic and diagnostic materials. Ultimately, we aim to create stable nanostructures with protein-like functions from nonnatural polymers.5–7 But many challenges remain before we can design a sequence that can fold into a defined tertiary structure. Despite decades of study, the rules that govern the kinetics and thermodynamics of folding polymer chains into stable tertiary structures are still not fully understood.
Among sequence-specific heteropolymers, N-substituted glycine oligomers, or peptoids, are of particular interest.2,3 The high efficiency of the solid-phase submonomer method allows the synthesis of peptoid chains up to 48 monomers in length.8,9 The length can be further increased by conjugating individual chains together to provide materials in the size range of proteins.6,10–13 The diversity of peptoid side chains is much greater than that found in proteins, since hundreds of primary amines can be directly incorporated.
Peptoids have been shown to have a wide variety of biological activities and are stable to proteolysis.14 Numerous short oligomers (<8-mers) have been found that bind to therapeutically relevant proteins, acting as antagonists, inhibitors, or activators.15–22 Peptoid substitutions into peptides (to make peptide/peptoid hybrids) have been shown to significantly enhance binding affinity.20–22 Longer bioactive peptoid oligomers have also been discovered that inhibit protein–protein interactions or mimic polypeptide function, such as magainin or lung surfactant.23,24 Finally, peptoids have utility as materials for nucleic acid and drug delivery.9,25,26
However, most of the above activities are from relatively unstructured oligomers. If we want to mimic the sophisticated functions of proteins, we need to be able to form defined peptoid tertiary structure folds and introduce functional side chains at defined locations.
Peptoid oligomers can already be folded into helical secondary structures. They can be readily generated by incorporating bulky chiral side chains into the oligomer.27–31 Such helical secondary structures are extremely stable to chemical denaturants and temperature.32 The unusual stability of the helical structure may be a consequence of the steric hindrance of backbone φ angle by the bulky chiral side chains.27,31
Recently, we synthesized compact, multihelical peptoid structures by linking helical units together.6 The tertiary structure was probed by fluorescence resonance energy transfer (FRET) and circular dichroism (CD). However, in order to generate well-defined tertiary structures with biological functions, we feel it is necessary to screen directly for function. In this paper, we introduced a structure-dependent function into our helical bundle motifs. We chose to screen for high-affinity and selective zinc binding, since zinc-binding motifs are well understood in biology. Zinc typically stabilizes native protein structures or acts as a cofactor for enzyme catalysis.33,34 Zinc also binds to cellular cysteine-rich metallothioneins solely for storage and distribution.35 The binding of zinc is typically mediated by cysteines and histidines.33,34 In order to create a zinc-binding site, we incorporated thiol and imidazole side chains into a peptoid two-helix bundle.
We positioned the thiols and imidazoles so that they would bind zinc only if the two helices folded into a two-helix bundle. We varied the locations and numbers of thiols and imidazoles, as well as the loop size and sequence, in order to investigate sequence–structure–function relationships of zinc binding and selectivity.
Results and Discussion
Minimalist Design of Zinc-Binding Peptoid Two-Helix Bundles
Our aim was a minimalist design of a peptoid two-helix bundle as a platform for zinc binding. Hence our monomer “alphabet” included only three types of residues: a chiral hydrophobic monomer [(S)-N-(1-phenylethyl)glycine (Nspe)], a chiral anionic monomer [(S)-N-(1-carboxylethyl)glycine (Nsce)], and a cationic monomer [N-(2-aminoethyl)glycine (Nae)] (Figure 1). Each helix was forced to contain a chiral monomer in two-thirds of the monomer positions since these side chains are known to enforce helicity. The Nspe and Nsce side chains impose steric hindrance into the backbone, generating a polyproline type I-like helix with three residues per turn.31 Our designs are thus based on a repeating three-residue motif. The hydrophobic phenyl rings of Nspe are incorporated at every third position, the repeat period of peptoids, to drive the chain to form an amphipathic helix. For the loop region, we used Gly-Pro-Gly-Gly (GPGG), which has a propensity to form type II β-turns in proteins.36 As controls, we also included several other loop sequences (see below). Our aim was an amphiphilic two-helix bundle scaffold that would fold into a compact structure in aqueous solution, driven by interhelical hydrophobic interactions. The minimalist design approach using amphipathic helices has previously been used for generating helix-bundle proteins.37,38
Figure 1.
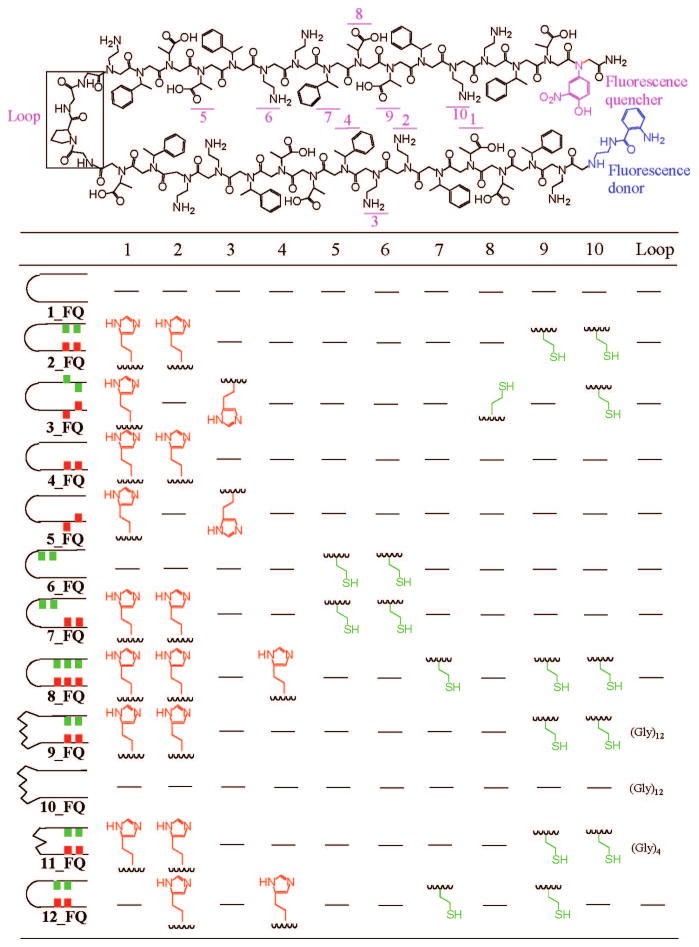
Chemical structures and sequences of peptoids used in our study. Ten positions and one loop were chosen for substitutions with other groups.
In order to construct a zinc-binding site, we adopted a well-known zinc-binding motif, Cys2His2.34,39 The thiols and imidazoles were incorporated into the two-helix bundle peptoid as shown in Figure 1. Since the thiols and imidazoles are put into both helical segments of the two-helix bundle, it is expected that the zinc will stabilize the folded state of the two-helix bundles by holding the two helical segments in close proximity.
Synthesis and Purification of Peptoids
All peptoid two-helix bundles were synthesized in solid phase by an automated robot synthesizer and were purified to more than 95% purity by reverse-phase HPLC (see Experimental Section). We determined the molecular weights of the peptoids by mass spectrometry (Supporting Information Table S1).
FRET Assay for Folding and Self-Association
In order to measure the proximity of two helical segments, we put a fluorescence donor (anthranilamide) at one end of the peptoid and a quencher (nitrophenol) at the other for intramolecular FRET (Figures 1 and 2). These FRET reporters have been used previously to probe the folded state of peptoids.6 As a control, to test whether separate peptoid molecules associate with each other, we put the donor on one molecule and the quencher on the other, for intermolecular FRET. As a reference for measuring the FRET efficiency, we synthesized peptoids having identical sequences but with only a fluorescence donor and no quencher. The intra- and intermolecular FRET efficiencies were obtained from the equation shown in Figure 2.
Figure 2.
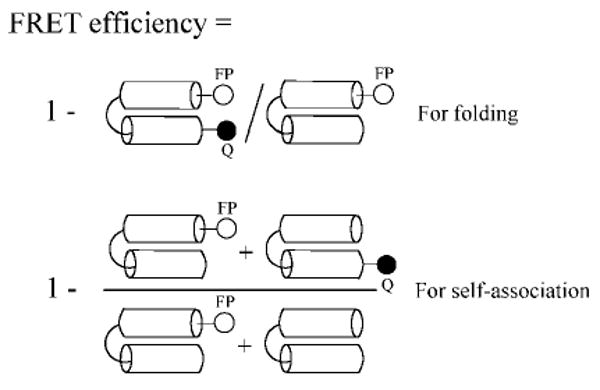
Schematic diagram for intra- and intermolecular FRET efficiency. Intramolecular FRET indicates the folding of a two-helix bundle; intermolecular FRET indicates self-association between peptoids. Fluorescence intensities from both numerator and denominator were measured to calculate the FRET efficiency.
We assigned each peptoid two-helix bundle a number (Figure 1 and Supporting Information Table S1) and indicated at the end of the number where the FRET reporters are incorporated; FQ for both fluorescence donor and quencher, F for only fluorescence donor only, and Q for quencher only. A number without letters was used for peptoids without FRET reporters.
Folding Stability of Peptoid Two-Helix Bundles
We performed equilibrium denaturant titration with acetonitrile, a hydrophobic solvent, as a probe of a cooperatively folded core (Figure 3a). The two-helix bundle peptoid 1_FQ that does not contain the thiols and imidazoles showed a cooperative unfolding transition with acetonitrile. This is evidence for a hydrophobic folding core. We fitted the unfolding transition data to a two-state model in order to obtain thermodynamic parameters ΔGu(H2O) and m value (see Experimental Section).
Figure 3.
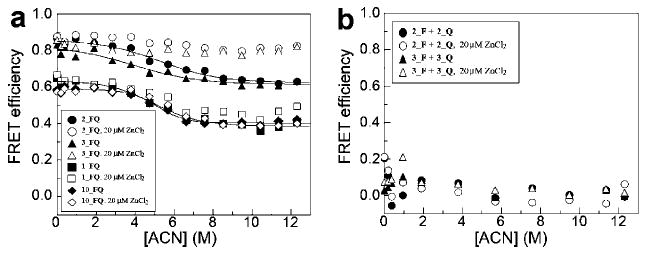
Equilibrium acetonitrile titration for (a) intramolecular FRET and (b) intermolecular FRET. (a) 2_FQ, 3_FQ, 1_FQ, and 10_FQ were titrated with acetonitrile in the absence and presence of 10 mol equiv of ZnCl2. The fluorescence intensities from reference molecules (2_F, 3_F, and 1_F) were also measured in order to obtain the intramolecular FRET efficiency as described in Figure 2. (b) 2_F + 2_Q and 3_F + 3_Q were titrated with acetonitrile in the absence and presence of 10 mol equiv of ZnCl2. The fluorescence intensities from reference molecules (2_F + 2 and 3_F + 3) were also measured in order to obtain the intermolecular FRET efficiency as described in Figure 2. Tris-HCl (5 mM, pH 7.5) was used for the buffer. The concentrations of each individual peptoid for intra- and intermolecular FRET are 2 μM and 1 μM, respectively.
To see the effect of a long and flexible loop on the folding of the two-helix bundle, we constructed another two-helix bundle, 10_FQ, that has (Gly)12 in the loop region. In proteins, long loop insertions have been studied previously.40–43 The effect of a long loop in an unstructured region of proteins was either negligible or had a low free energy cost. Similarly, the effect of the long loop on the folding of our peptoid two-helix bundle slightly induced a more compact and stable structure. 10_FQ showed a cooperative unfolding transition with a similar range of FRET efficiency as 1_FQ (Figure 3a). The ΔGu(H2O) and m values increase by 0.9 kcal mol−1 and 0.17 kcal mol−1 M−1, respectively (Table 1).
Table 1.
Thermodynamic Parameters for Folding of Peptoids Measured by Equilibrium Acetonitrile Titration
| peptoid | ΔGu(H2O), kcal mol−1 | m, kcal mol−1 M−1 |
|---|---|---|
| 1_FQ | 2.5 ± 0.5 | 0.50 ± 0.10 |
| 10_FQ | 3.4 ± 0.5 | 0.67 ± 0.10 |
| 2_FQ | 2.1 ± 0.5 | 0.39 ± 0.09 |
| 3_FQ | 1.4 ± 0.7 | 0.35 ± 0.13 |
The peptoids 2_FQ and 3_FQ, which contain two thiols and two imidazoles, were also titrated with acetonitrile to see how the hydrophobic folding core is affected by these side chains (Figure 3a). The two imidazoles and the two thiols in each helical segment were separated by either one (2_FQ) or two residues (3_FQ). Both peptoids showed a cooperative unfolding transition (Figure 3a). As compared to 1_FQ, the FRET efficiencies over the range of acetonitrile concentrations increased by ∼0.2 (Figure 3a). However, the m values of 2_FQ and 3_FQ decreased, as shown in Table 1. The m value typically correlates with the change of solvent-accessible area between folded and unfolded states.44 Some of the buried surface area was not exposed in the presence of acetonitrile in these peptoids. We then added 10 mol equiv of zinc (20 μM) in order to see if there is any change in folding thermodynamics. In both 2_FQ and 3_FQ, zinc increased the FRET efficiency in the presence of acetonitrile (Figure 3a). There is no indication of unfolding by acetonitrile in the presence of zinc. This means that zinc stabilizes the two-helix bundle by holding the two helical segments together. In control peptoids (1_FQ and 10_FQ), 10 mol equiv of zinc slightly increased the FRET efficiency in the presence of acetonitrile in 1_FQ or had no effect on the acetonitrile-dependent FRET efficiency in 10_FQ (Figure 3a).
To confirm that the zinc induced intramolecular folding, rather than intermolecular peptoid–peptoid association, we measured intermolecular FRET by constructing the same sequences of peptoids as 2_FQ and 3_FQ but with fluorescence donor only in one peptoid and quencher only in the other (Figure 2). We carried out the equilibrium acetonitrile titration using mixtures of these peptoids (Figure 3b). We found that the intermolecular FRET efficiencies were not changed in the presence of 10 mol equiv of zinc in mixtures of 2_F + 2_Q and 3_F + 3_Q. To see if there is a concentration-dependent self-association, we measured the intermolecular FRET efficiencies at various peptoid concentrations from 2 to 32 μM (Supporting Information Figure S1). There was no indication of self-association below 5 μM peptoid, although in a couple of cases (2_F + 2_Q and 9_F + 9_Q) self-association was detected above 8 μM in the presence of 5 mol equiv of zinc. In the absence of zinc, peptoids that we tested were monomeric, which was also confirmed by analytical gel filtration (Supporting Information Figure S2). We conclude that zinc binds to the peptoid and stabilizes the structure of the two-helix bundles without inducing self-association at 2 μM peptoid.
We measured circular dichroic spectra in peptoids 2 and 3 to see if there is a change in secondary structure when the peptoids bind zinc. As shown in Figure 4, there was no significant change in CD signal in the far-UV region in the presence of 10 mol equiv of zinc. This means that the secondary structure of the peptoids was not changed. Therefore, zinc-induced folding of the peptoid two-helix bundles occurs by the docking of two helical segments without changing the secondary structure. There was no significant change of CD signal at different concentrations of peptoid or acetonitrile in either the absence or presence of 5 mol equiv of zinc (Supporting Information Figures S3–S5), which is in line with our observation that the helical secondary structure of these peptoids are stable.
Figure 4.
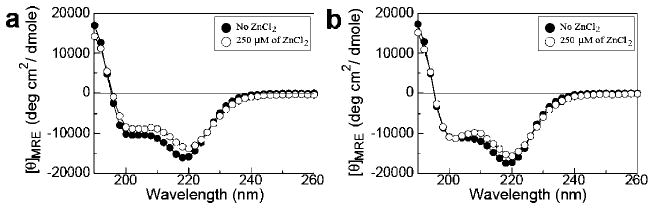
Effect of zinc on the secondary structure of peptoids. CD spectra were measured at far-UV region in peptoids (a) 2 and (b) 3, in the absence (●) and presence (○) of 10 mol equiv of ZnCl2.
Using guanidine hydrochloride (GdnHCl), a denaturant commonly used in protein unfolding, we also performed an equilibrium titration in peptoids 1_FQ, 2_FQ, and 8_FQ. GdnHCl did not appear to unfold these peptoids cooperatively (Supporting Information Figure S6). The detailed mechanism of how proteins are unfolded cooperatively by GdnHCl is not fully understood yet. But we believe that guanidine is less effective in denaturing peptoids than proteins because of the lack of backbone hydrogen bonds in the peptoids. The cooperative and noncooperative responses to acetonitrile and GdnHCl, respectively, are in line with our previous results from other peptoid two- and three-helix bundles.6 We found that 10 mol equiv of zinc was not effective in folding peptoids 2_FQ and 8_FQ in the presence of GdnHCl (Supporting Information Figure S6).
Zinc-Binding Peptoid Two-Helix Bundles
We systematically varied the locations and numbers of thiols and imidazoles, in addition to the monomer sequences and sizes of the loops, to see how those factors affect the binding of zinc (Figure 1).
Using the change of the intermolecular FRET, zinc-binding affinities were measured in the presence of acetonitrile (Figure 5 and Table 2). In the absence of acetonitrile, the change of FRET efficiency was too small to measure the affinity precisely (Figure 3a and Supporting Information Figure S7). Thus, we added acetonitrile in order to obtain a significant change of FRET efficiency with better signal-to-noise ratio. The affinity was estimated by fitting the data with a single zinc-binding model (see Experimental Section). Interestingly, acetonitrile did not affect the zinc-binding affinity in peptoids 2_FQ and 3_FQ: zinc-binding affinities were similar at 30%, 40%, and 50% (v/v) acetonitrile in water (Supporting Information Figure S8). When a molar excess of ethylenediaminetetraacetic acid (EDTA) was added, the FRET efficiency decreased back to the value where zinc was absent (Supporting Information Figure S8). The peptoids returned to the unfolded state because the bound zinc was sequestered by EDTA.
Figure 5.
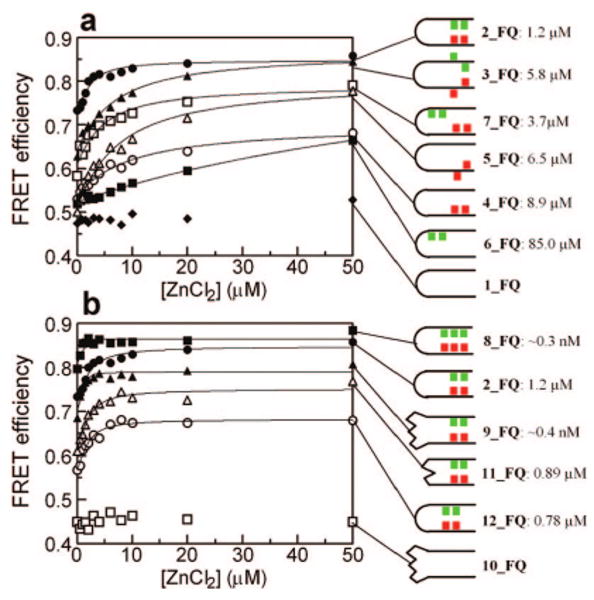
Measuring zinc-binding affinities for various peptoid two-helix bundles. Each peptoid (2 μM) was titrated with ZnCl2 in the presence of 30% acetonitrile in water.
Table 2.
Apparent Zinc-Binding Dissociation Constants for Peptoids and FRET Efficiencies for the Zinc-Bound State of Peptoidsa
| peptoid | apparent kd (M) | ΔΔGzn (kcal/mol) | FRET efficiency |
|---|---|---|---|
| 2_FQ | (1.2 ± 0.5) × 10−6 | 0 | 0.86 ± 0.01 |
| 3_FQ | (5.8 ± 1.0) × 10−6 | 0.9 | 0.85 ± 0.01 |
| 4_FQ | (8.9 ± 2.2) × 10−6 | 1.0 | 0.68 ± 0.01 |
| 5_FQ | (6.5 ± 1.4) × 10−6 | 1.2 | 0.78 ± 0.01 |
| 6_FQ | (8.5 ± 3.0) × 10−5 | 2.5 | 0.66 ± 0.02 |
| 7_FQ | (3.7 ± 1.5) × 10−6 | 0.7 | 0.79 ± 0.01 |
| 8_FQ | ∼0.3 × 10−9 b | −4.9 | 0.88 ± 0.01 |
| 9_FQ | ∼0.4 × 10−9 b | −4.7 | 0.81 ± 0.02 |
| 11_FQ | (8.9 ± 3.4) × 10−7 | −0.2 | 0.77 ± 0.01 |
| 12_FQ | (7.8 ± 2.8) × 10−7 | −0.3 | 0.68 ± 0.01 |
The free energy difference of the zinc-bound state (ΔΔG zn) was calculated from ΔΔG zn = −RT ln (kd_ref/kd_sample). R is the gas constant and T is temperature. We took the kd of 2_FQ as the reference kd.
Zinc-binding dissociation constants were obtained from the competition assay with EGTA (Figure 6).
In the control peptoids 1_FQ and 10_FQ, where the thiols and imidazoles are absent, zinc was not effective for folding of these peptoids, as shown in Figure 5. Each helical unit contains five carboxyl groups, so that one-third of our helices are negatively charged. However, the zinc-binding ability of these carboxyl groups alone was not sufficient to stabilize helix bundle formation, since the FRET efficiency was relatively low and showed no dependence on zinc ion concentration. In compounds where half a binding site was introduced, for example, when two thiols (6_FQ) or two imidazoles (4_FQ and 5_FQ) were installed into one helical segment, we began to see evidence of helix bundle formation, although the zinc binding affinities were weak (Figure 5). It is likely that the thiols or imidazoles are able to coordinate zinc in concert with some of the carboxyl groups on the opposite helix (in peptoids 6_FQ, 4_FQ, and 5_FQ).
Comparing 4_FQ (kd = 8.9 μM) with 6_FQ (kd = 85 μM), we found that imidazoles were 10-fold more efficient than thiols for zinc binding. In a peptoid that has two thiols at the ninth and tenth positions (13_FQ), we found that there was no significant change in FRET efficiency upon addition of zinc (Supporting Information Figure S9). Combined together, imidazoles are more efficient than thiols for zinc-induced folding in the peptoid two-helical bundles.
Two imidazoles were incorporated into peptoids, separated by one (4_FQ) or two (5_FQ) residues in order to see the effect of the relative locations of imidazoles along the sequence. The binding affinity slightly increased, from kd = 8.9 μM (4_FQ) to kd = 6.5 μM (5_FQ), and the FRET efficiency of the zinc-bound state increased by 0.1 (Figure 5 and Table 2) when the two imidazoles were separated further. Since the two imidazoles in 5_FQ are more efficient for the zinc binding than those in 4_FQ, it suggests that the carboxyl groups located at the eighth and ninth positions in Figure 1 contribute a tighter zinc binding in 5_FQ than in 4_FQ.
However, in the presence of two thiols and imidazoles, 2_FQ, which has those groups separated by one residue, is more efficient for the binding of zinc than 3_FQ, which has those groups separated by two residues (Figure 5 and Table 2). The binding affinity increased 5-fold, from kd = 5.8 μM (3_FQ) to kd = 1.2 μM (2_FQ), suggesting that the two thiols and imidazoles are geometrically more optimized for zinc binding in 2_FQ than in 3_FQ.
The peptoids should fold into antiparallel two-helix bundles. Thus, we expect that the binding of zinc to 7_FQ should be weaker than to 2_FQ. In comparison of peptoid 2_FQ to 7_FQ (kd = 3.7 μM), the binding affinity decreased 3-fold and the FRET efficiency of the zinc-bound state decreased by 0.1 (Figure 5 and Table 2). This result indicates that the two thiols and imidazoles face each other for tighter zinc binding in 2_FQ than in peptoid 7_FQ, where the two thiols are located further from the two imidazoles in the tertiary structure, even though they are closer in the primary sequence.
We synthesized 12_FQ to see the effect of the relative location of the zinc-binding site. The zinc-binding site is moved by two residues as compared to 2_FQ (Figure 1). When 12_FQ is compared to 2_FQ, we found that the binding affinity slightly improved (12_FQ, kd = 0.78 μM), but the FRET efficiency of the zinc bound state decreased by ∼0.2 (Figure 5 and Table 2). In this peptoid (12_FQ), one of the thiols and imidazoles replaces the hydrophobic group. The strength of the interhelical hydrophobic interactions in peptoid 12_FQ should be weaker than that in 2_FQ. Thus, peptoid 12_FQ was less compact, decreasing the FRET efficiency over the entire range of acetonitrile concentrations.
In order to see the effect of a more flexible loop on the binding of zinc, we synthesized a two-helix bundle (11_FQ), which has the sequence GGGG in the loop. When 11_FQ is compared with 2_FQ, which has GPGG in the loop, we found that the binding affinity slightly improved (11_FQ, kd = 0.89 μM), but the FRET efficiency of the zinc-bound state decreased by 0.1 (Figure 5 and Table 2). The GPGG loop helped bring the ends of two helical segments together.
High-Affinity and Selective Zinc-Binding Peptoids
We observed tight zinc binding in peptoids 8_FQ and 9_FQ. In 8_FQ, three thiols and imidazoles were incorporated into each helical segment. In 9_FQ, two helical segments were connected by a long and flexible loop of 12 glycines. In both peptoids, the FRET efficiency already reached a plateau in the zinc titration when 1:1 equivalents of both peptoid (2 μM) and zinc (2 μM) were mixed (Figure 5b). This implies that the zinc-binding dissociation constant is lower than the concentration we used for the zinc titration. To obtain the binding affinities more precisely, we carried out a competition assay using ethylene glycol bis(2-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) (Figure 6). EGTA has a zinc-binding affinity of 4.5 × 10−10 M (Experimental Section). In our competition assays, a 1:1 mixture of peptoid (2 μM) and zinc (2 μM) was titrated with EGTA. EGTA itself decreased the FRET efficiency in the presence of 30% acetonitrile (Figure 6), unfolding the peptoid further. The data for EGTA titration were fit by eq 3 to obtain K, the equilibrium constant for the exchange reaction. By use of eq 4, the zinc binding affinities of 8_FQ and 9_FQ were estimated to be ∼0.3 nM for 8_FQ and ∼0.4 nM for 9_FQ. The zinc-binding affinities of these two peptoids are similar to those found in zinc-binding proteins.45–47 Another competition assay using the zinc-binding fluorescent dye FluoZin-1 and FluoZin-3 confirmed the tight zinc binding for 8_FQ and 9_FQ (Supporting Information Figure S10). We measured the intermolecular FRET in order to see if there is zinc-induced self-association in these peptoids. We found that zinc did not induce a self-association in 2_F + 2_Q, 8_F + 8_Q, and 9_F + 9_Q (Supporting Information Figure S11).
Figure 6.
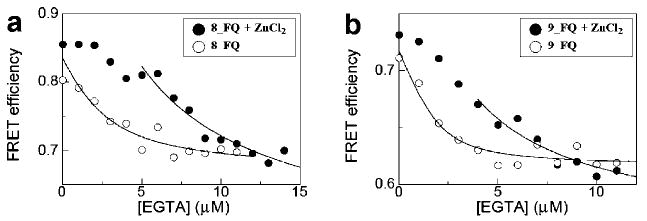
Competition assay between peptoid and EGTA for zinc. (●) A 1:1 mixture of peptoid (2 μM) and zinc (2 μM) was titrated with EGTA for (a) 8_FQ and (b) 9_FQ. (○) Titration of peptoid with EGTA in the absence of zinc. Solid line indicates data points used for data fitting.
Given the high-affinity binding to zinc, we next investigated binding selectivity relative to other divalent metal ions. The peptoid 9_FQ (2 μM) was titrated with other divalent metal ions Mg2+, Mn2+, Ca2+, Ni2+, Co2+, Cu2+, and Cd2+ in the range 0–50 μM (Figure 7). Among various divalent metal ions, zinc made the most significant change in the FRET efficiency with a binding affinity an order of magnitude higher than other metal ions (Figure 7). This kind of selectivity is also found in biological systems. For example, in the transcription factor TFIIIA, the zinc-binding affinity is 3 orders of magnitude higher than that for cobalt.47
Figure 7.
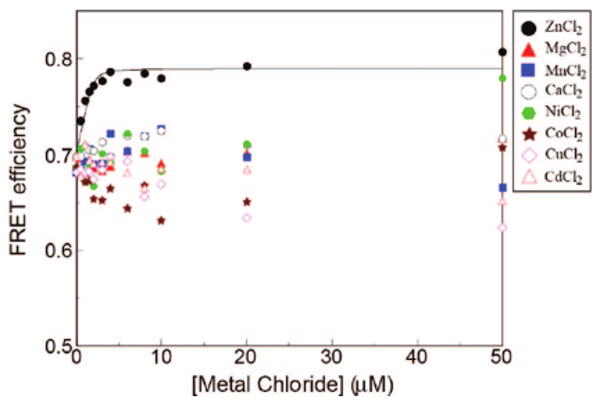
Effect of various divalent metal ions on the folding of 9_FQ in the presence of 30% acetonitrile. The peptoid concentration was 2 μM and the metal counterion was chloride in all cases.
In 8_FQ, the zinc-binding site contains three thiols and three imidazoles, overlapping with the sites from 2_FQ and 12_FQ. However, 2_FQ and 12_FQ had only micromolar zinc-binding affinities. We believe the overlapping multivalency in 8_FQ is responsible for the tight binding, as has also been found for protein–protein and ligand–protein interactions.48,49 It is likely that the zinc ion occupies one of two binding sites when the concentration of zinc is less than the concentration of peptoid (Figure 8). However, we cannot exclude the possibility that the tight zinc-binding affinity of 8_FQ originates from the tight packing of this peptoid, based on the weak correlation between the folded state and zinc-binding dissociation constants (Supporting Information Figure S12). This correlation suggests that peptoids with tighter packing pay less cost for high-affinity zinc binding.
Figure 8.
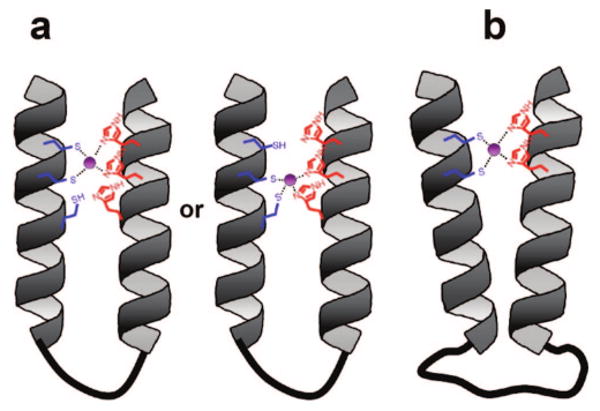
Schematic diagrams of peptoid two-helix bundles that showed nanomolar dissociation constants for zinc. (a) Schematic model of 8_FQ. One of the overlapping binding sites is likely to be occupied by zinc in the presence of a 1:1 mixture of peptoid and zinc. (b) Schematic model of 9_FQ. The long flexible linker of (Gly)12 allows two helical segments to search and find a conformation for tight zinc binding. The zinc is colored purple.
Interestingly, the length of the flexible loop linking the two helices had a dramatic effect on zinc-binding affinity. When the length of the loop increased from (Gly)4 to (Gly)12, zinc-binding affinity increased by 3 orders of magnitude (11_FQ and 9_FQ). To see if the higher zinc-binding affinity was due to the long loop region, we constructed a control two-helix bundle (10_FQ) that had the same loop size but did not contain the thiols and imidazoles. The zinc had no effect on the folding of 10_FQ (Figures 3a and 5b). Thus, the tight zinc binding in peptoid 9_FQ is a consequence of the two thiols and two imidazoles, and not other structural elements. We believe that the longer linker in 9_FQ may simply help release some overconstraints that may occur in our peptoids having smaller loops. It suggests that longer flexible linkers in the loop regions accommodate optimal zinc-coordination geometry (Figure 8).
Number of Zinc-Binding Sites and Coordination Geometry
The number of zinc-binding sites for our peptoids was estimated by Hill and Job plots. A slope of 1 was obtained from a Hill plot for the zinc-binding data presented in Figure 5 (Supporting Information Figure S13). The Job plot for peptoids 2_FQ, 3_FQ, 8_FQ, and 9_FQ showed a kink around 0.5 (Supporting Information Figure S14). Both results indicate that there was one zinc-binding site for our peptoids. The competition assay using FluoZin-1 and FluoZin-3 was also in line with this argument because the fluorescence signal was saturated in the presence of 1 mol equiv of zinc (Supporting Information Figure S10). In order to gain insight into the coordination structure for Zn(II)-bound peptoids, we titrated the peptoids 2, 3, 8, and 9 with Co(II). Co(II) has been used as a surrogate to probe the coordination geometry for Zn(II)-bound proteins.45,50 There was strong absorbance for ligand-to-metal charge transfer, but no pronounced d–d orbital transition at 500∼800 nm, with extinction coefficients lower than 100 M−1 cm−1 for these peptoids (Supporting Information Figures S15 and S16). Thus, we believe that the coordination number is not 4 but either 5 or 6. In the coordination sphere for Zn(II)-bound peptoids, water likely occupies one or two coordination sites.
Conclusions
One of the ultimate goals in the area of bioinspired heteropolymers is to create precisely folded nanostructures with protein-like functions. Using peptoid chain molecules, we created zinc-binding nanostructures that folded up into two-helix bundles. The formation of the tertiary structure in these peptoids is governed by the docking of preorganized peptoid helices as shown in this and our previous studies.6 This peptoid folding is relatively simple compared to protein folding, where many levels of structural transitions are involved such as helix-coil transition, formation of various secondary and tertiary structures, etc. This simple folding behavior in peptoids allows us to design and build useful self-assembled nanostructures. We believe that this approach can be coupled with structure-based design and combinatorial screening to discover robust nano-structured materials capable of precise molecular recognition and catalysis.
Experimental Section
Peptoid Synthesis
Peptoid oligomers were synthesized on an automated robot synthesizer using a solid-phase submonomer cycle as described previously.8 Amine submonomers used for our peptoid synthesis are listed in Supporting Information. Rink amide resin (0.57 mmol/g, Novabiochem, San Diego, CA) was used to generate C-terminal amide peptoids. After histamine was added into the peptoid, chloroacetic acid was used for all subsequent steps of acylation in order to reduce side product formation as described previously.51
In the loop region of peptoids, amino acids such as glycine and proline were incorporated into the peptoid. Fmoc-Gly-OH (Novabiochem) or Fmoc-Pro-OH (Novabiochem) in N,N-dimethylformamide (0.8 mmol in 2 mL of DMF) was added to the resin-bound amine with 0.4 M hydroxybenzotriazole in DMF and 137 μL of N,N′-diisopropylcarbodiimide (DIC) (0.92 mmol). The reaction mixture was incubated at 35 °C for 60 min. When the Fmoc-Gly-OH was added next to a peptoid backbone, the reaction mixture was added twice for efficient coupling. Fmoc group was then deprotected with 20% piperidine in DMF for further synthesis.
The crude peptoid products (50 μmol of resin) were cleaved from the resins with 95:5 Trifluoroacetic acid (TFA)/water (v/v) for 20 min at room temperature. For thiol-containing peptoids, the trityl groups were removed in the presence of triethylsilane. The mixture of 92.5:5:2.5 TFA/water/triethylsilane (v/v/v) was added, and the sample was incubated for 20 min at room temperature to cleave the peptoids from the resins. The cleavage solution was filtered and evaporated under a stream of nitrogen to remove the TFA. The crude peptoid product was then dissolved in a mixture of water and acetonitrile and subjected to further purification by reverse-phase HPLC with a Vydac C4 column (10 μm, 22 mm × 250 mm). For thiol-containing peptoids, the peptoids were treated with 100 mM dithiothreitol (DTT) prior to HPLC in order to ensure that all thiols are reduced. All final products were analyzed by analytical reverse-phase HPLC (5–95% gradient at 0.8 mL/min over 30 min at 60 °C with a C4 Duragel G C4, 5 μm, 50 × 2 mm column) and electrospray mass spectrometry (Hewlett-Packard Series 1100). Final peptoid products were lyophilized, dissolved either in water or buffer (100 mM tris-HCl, pH 7.0), and stored at −70 °C. Approximate peptoid concentrations were determined by use of the extinction coefficients of anthranilic acid (ε = 2000 M−1 cm−1 at 315 nm) and 2-nitrophenol (ε = 3500 M−1 cm−1 at 424 nm)52 when they are incorporated into peptoids; otherwise, peptoids were quantified by weight after lyophilization.
Fluorescence, Absorbance, and Circular Dichroic Spectroscopy
FluoZin-1 and FluoZin-3 were obtained from Invitrogen (Carlsbad, CA). The water used to prepare metal-free buffer solutions was purified with the Super-Q Plus water purification system (Millipore Corp.). All buffer solutions were prepared from Trizma base (SigmaUltra grade) obtained from Sigma–Aldrich. Zinc ion solutions were prepared from zinc chloride (99.999% purity) from Aldrich. Fluorescence from the anthranilamide side chain (Naae) of the peptoids was measured either with a fluorescence 96-well plate reader (Fluo Star, BMG LabTechnologies, Inc., or SpectraMAX Gemini EM, Molecular Devices Corp.) or with a Fluoromax-4 spectrofluorometer from Horiba Jobin Yvon Inc. The titrations of peptoids with ZnCl2 or acetonitrile were performed in the presence of 5 mM Tris-HCl (pH 7.5) to monitor the change in fluorescence. The fluorescent probe inside peptoids was excited at 320 nm, and emission was detected at 405 nm. FRET efficiency was obtained from the quenching of donor fluorescence with E = (1 − Ida/Id), where Ida and Id are the fluorescence intensities in the presence and absence of quencher, respectively. As a reference for measuring the FRET efficiency, analogous peptoids with only a donor (no quencher) were also synthesized.
Circular dichroism (CD) measurements were carried out either with a Jasco 710 spectropolarimeter or with an Aviv circular dichroism spectrometer 410. A 0.1 cm path length quartz cell was used for far-UV CD. Step resolution, scan speed, response time, and bandwidth for Jasco 710 were 2 nm, 20 nm/min, 4 s, and 1.0 nm, respectively. All CD measurements were averaged over four scans. CD was measured at 25 μM in 5 mM Tris-HCl buffer (pH 7.5) with 30% acetonitrile (v/v). To see the effect of zinc, 250 μM ZnCl2 was added into the samples.
Perkin-Elmer Lambda 35 UV/vis spectrometer was used to monitor the change of absorbance for cobalt(II) d–d orbital transition at 300∼800 nm. The peptoids were titrated with Co(II)Cl2 in the presence of 30% acetonitrile (v/v) and 50 mM Tris-HCl (pH 7.5).
All buffers were degassed under in-house vacuum and purged with nitrogen to prevent unwanted disulfide formation. All titration samples were incubated for 30 min at room temperature before measurement of the fluorescence and CD. In order to ensure that the thiols in the two-helix bundle remain reduced, we treated peptoid 2 with Ellman's reagent [5,5′-dithiobis(2-nitrobenzoate), DTNB]. DTNB reacts only with free thiols. We found that the thiols in 2 were labeled with this reagent, indicating that there is no formation of the disulfide bond (Supporting Information Figure S17).
Analytical Gel Filtration
A Bio-Sil SEC-125 (300 × 7.8 mm, Bio-Rad, Hercules, CA) column was used for the gel filtration. The flow rate was 1 mL/min. Elution was monitored by absorbance at 214 nm. The buffer for this gel filtration was 5 mM Tris-HCl (pH 7.5) with 150 mM NaCl. Aliquots (20 μL) of 50 μM peptoids were injected onto the gel-filtration column.
Equilibrium Denaturant Titration
The data for denaturant-dependent FRET efficiency (E) were fitted by an apparent two-state transition from folded to unfolded state:
| (1) |
where [D] is the denaturant concentration, an and au are the FRET efficiencies of folded and unfolded state extrapolated to zero denaturant, ΔGu(H2O) is the free energy of unfolding in water, m is the denaturant dependence of free energy per mole of denaturant, R is the gas constant, and T is the temperature in kelvins.
Data Analysis for Zinc-Binding Affinities
The apparent zinc-binding dissociation constant (kd) was calculated by use of the following single zinc-binding model:
| (2) |
where [ZnCl2]t and [P]0 are the total concentrations of ZnCl2 and peptoid, respectively, and Emax and Emin are the FRET efficiencies of zinc-bound and free state, respectively.
For the competition assay between peptoid and EGTA for zinc, a 1:1 mixture of peptoid and zinc was titrated with EGTA (peptoid · Zn + EGTA ⇌ peptoid + EGTA · Zn). The EGTA-dependent data was fit by the following equations:
| (3) |
| (4) |
where K is the equilibrium constant for the exchange reaction. kd(EGTA · Zn) was calculated by use of the published pK (= 9.47, 8.85, 2.66, and 2.00) and log β (= 12.7) values.53 kd(EGTA · Zn) is 4.5 × 10−10 M. kd(peptoid · Zn) was estimated from K and kd(EGTA · Zn).
Supplementary Material
Acknowledgments
We thank Novartis (formerly Chiron) for providing research facilities and equipment. We also thank Tom James, Brian Shoichet, Kristin Brinner, Michael Connolly, Thomas Horn, and Deb Charych for valuable comments and assistance. B.-C.L. was supported by a Chancellor's graduate research fellowship at UCSF. R.N.Z., T.K.C., and B.-C.L. are supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract DE-AC02-05CH11231, and K.A.D. is supported by NIH Grant GM34993.
Footnotes
Supporting Information Available: Complete citation for ref 15; molecular weight of each peptoid determined by mass spectrometry; concentration-dependent FRET efficiency for self-association of peptoids; analytical gel filtration on a Bio-Sil SEC-125 column for standard molecules and peptoids 2_Q, 3_Q, 8_Q, and 9; CD spectra of 2_FQ and 3_FQ at various concentrations of peptoid in the absence and presence of 5 mol equiv of ZnCl2 relative to the amount of peptoid; CD at 220 nm for 2_FQ and 3_FQ; effect of acetonitrile and ZnCl2 on the secondary structure of 3_FQ; equilibrium GdnHCl titration of 2_FQ, 8_FQ, and 1_FQ; ZnCl2 titration of 9_FQ in the absence and presence of acetonitrile; effect of acetonitrile on zinc-binding affinities; effect of zinc on FRET efficiency of 13_FQ; competition assay for zinc binding between peptoids and FluoZin-3 or FluoZin-1; FRET self-association assay for 2_F + 2_Q, 8_F + 8_Q, and 9_F + 9_Q; weak correlation between the FRET efficiency of zinc-free/bound states and the apparent zinc-binding dissociation constants; Hill plot for zinc binding; Job plot for zinc binding; Co(II)Cl2 titration for 2, 3, 8, and 9; extinction coefficients for bound Co(II) in 2, 3, 8, and 9; reaction of 2 with DTNB; and amine submonomers. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Goodman CM, Choi S, Shandler S, DeGrado WF. Nat Chem Biol. 2007;3:252–262. doi: 10.1038/nchembio876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kirshenbaum K, Zuckermann RN, Dill KA. Curr Opin Struct Biol. 1999;9:530–535. doi: 10.1016/S0959-440X(99)80075-X. [DOI] [PubMed] [Google Scholar]
- 3.Barron AE, Zuckermann RN. Curr Opin Struct Biol. 1999;9:681–687. doi: 10.1016/s1367-5931(99)00026-5. [DOI] [PubMed] [Google Scholar]
- 4.Hill DJ, Mio MJ, Prince RB, Hughes TS, Moore JS. Chem Rev. 2001;101:3893–4012. doi: 10.1021/cr990120t. [DOI] [PubMed] [Google Scholar]
- 5.Raguse TL, Lai JR, LePlae PR, Gellman SH. Org Lett. 2001;3:3963–3966. doi: 10.1021/ol016868r. [DOI] [PubMed] [Google Scholar]
- 6.Lee BC, Zuckermann RN, Dill KA. J Am Chem Soc. 2005;127:10999–11009. doi: 10.1021/ja0514904. [DOI] [PubMed] [Google Scholar]
- 7.Goodman JL, Petersson EJ, Daniels DS, Qiu JX, Schepartz A. J Am Chem Soc. 2007;129:14746–14751. doi: 10.1021/ja0754002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zuckermann RN, Kerr JM, Kent SBH, Moos WH. J Am Chem Soc. 1992;114:10646–10647. [Google Scholar]
- 9.Murphy JE, Uno T, Hamer JD, Cohen FE, Dwarki V, Zuckermann RN. Proc Natl Acad Sci USA. 1998;95:1517–1522. doi: 10.1073/pnas.95.4.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Horn T, Lee BC, Dill KA, Zuckermann RN. Bioconjugate Chem. 2004;15:428–435. doi: 10.1021/bc0341831. [DOI] [PubMed] [Google Scholar]
- 11.Jang H, Fafarman A, Holub JM, Kirshenbaum K. Org Lett. 2005;7:1951–1954. doi: 10.1021/ol050371q. [DOI] [PubMed] [Google Scholar]
- 12.Yoo B, Kirshenbaum K. J Am Chem Soc. 2005;127:17132–17133. doi: 10.1021/ja055105m. [DOI] [PubMed] [Google Scholar]
- 13.Holub JM, Jang H, Kirshenbaum K. Org Biomol Chem. 2006;4:1497–1502. doi: 10.1039/b518247f. [DOI] [PubMed] [Google Scholar]
- 14.Miller SM, Simon RJ, Ng S, Zuckermann RN, Kerr JM, Moos WH. Drug Dev Res. 1995;35:20–32. [Google Scholar]
- 15.Zuckermann RN, et al. J Med Chem. 1994;37:2678–2685. doi: 10.1021/jm00043a007. [DOI] [PubMed] [Google Scholar]
- 16.Alluri PG, Reddy MM, Bachhawat-Sikder K, Olivos HJ, Kodadek T. J Am Chem Soc. 2003;125:13995–14004. doi: 10.1021/ja036417x. [DOI] [PubMed] [Google Scholar]
- 17.Liu B, Alluri PG, Yu P, Kodadek TA. J Am Chem Soc. 2005;127:8254–8255. doi: 10.1021/ja0515295. [DOI] [PubMed] [Google Scholar]
- 18.Hara T, Durell SR, Myers MC, Appella DH. J Am Chem Soc. 2006;128:1995–2004. doi: 10.1021/ja056344c. [DOI] [PubMed] [Google Scholar]
- 19.Masip I, Cortés N, Abad MJ, Guardiola M, Pérez-Payá E, Ferragut J, Ferrer-Montiel A, Messeguer A. Bioorg Med Chem. 2005;13:1923–1929. doi: 10.1016/j.bmc.2005.01.024. [DOI] [PubMed] [Google Scholar]
- 20.Nguyen JT, Turck CW, Cohen FE, Zuckermann RN, Lim WA. Science. 1998;282:2088–2092. doi: 10.1126/science.282.5396.2088. [DOI] [PubMed] [Google Scholar]
- 21.Kruijtzer JA, Nijenhuis WA, Wanders N, Gispen WH, Liskamp RM, Adan RA. J Med Chem. 2005;48:4224–4230. doi: 10.1021/jm0490033. [DOI] [PubMed] [Google Scholar]
- 22.de Haan EC, Wauben MH, Grosfeld-Stulemeyer MC, Kruijtzer JA, Liskamp RM, Moret EE. Bioorg Med Chem. 2002;10:1939–1945. doi: 10.1016/s0968-0896(01)00434-5. [DOI] [PubMed] [Google Scholar]
- 23.Patch JA, Barron AE. J Am Chem Soc. 2003;125:12092–12093. doi: 10.1021/ja037320d. [DOI] [PubMed] [Google Scholar]
- 24.Wu CW, Seurynck SL, Lee KYC, Barron AE. Chem Biol. 2003;10:1057–1063. doi: 10.1016/j.chembiol.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 25.Uktu Y, Dehan E, Ouerfelli O, Piano F, Zuckermann RN, Pagano M, Kirshenbaum K. Mol Biosyst. 2006;2:312–317. doi: 10.1039/b603229j. [DOI] [PubMed] [Google Scholar]
- 26.Schröder T, Schmitz K, Niemeier N, Balaban TS, Krug HF, Schepers U, Bräse S. Bioconjugate Chem. 1997;18:342–354. doi: 10.1021/bc0602073. [DOI] [PubMed] [Google Scholar]
- 27.Kirshenbaum K, Barron AE, Goldsmith RA, Armand P, Bradley EK, Truong KT, Dill KA, Cohen FE, Zuckermann RN. Proc Natl Acad Sci USA. 1998;95:4303–4308. doi: 10.1073/pnas.95.8.4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Armand P, Kirshenbaum K, Goldsmith RA, Farr-Jones S, Barron AE, Truong KT, Dill KA, Mierke DF, Cohen FE, Zuckermann RN, Bradley EK. Proc Natl Acad Sci USA. 1998;95:4309–4314. doi: 10.1073/pnas.95.8.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu CW, Sanborn TJ, Huang K, Zuckermann RN, Barron AE. J Am Chem Soc. 2001;123:6778–6784. doi: 10.1021/ja003154n. [DOI] [PubMed] [Google Scholar]
- 30.Wu CW, Kirshenbaum K, Sanborn TJ, Patch JA, Huang K, Dill KA, Zuckermann RN, Barron AE. J Am Chem Soc. 2003;125:13525–13530. doi: 10.1021/ja037540r. [DOI] [PubMed] [Google Scholar]
- 31.Armand P, Kirshenbaum K, Falicov A, Dunbrack RL, Jr, Dill KA, Zuckermann RN, Cohen FE. Folding Des. 1997;2:369–375. doi: 10.1016/S1359-0278(97)00051-5. [DOI] [PubMed] [Google Scholar]
- 32.Sanborn TJ, Wu CW, Zuckermann RN, Barron AE. Biopolymers. 2002;63:12–20. doi: 10.1002/bip.1058. [DOI] [PubMed] [Google Scholar]
- 33.Coleman JE. Annu Rev Biochem. 1992;61:897–946. doi: 10.1146/annurev.bi.61.070192.004341. [DOI] [PubMed] [Google Scholar]
- 34.Berg JM, Godwin HA. Annu Rev Biophys Biomol Struct. 1997;26:357–371. doi: 10.1146/annurev.biophys.26.1.357. [DOI] [PubMed] [Google Scholar]
- 35.Cousins RJ, Liuzzi JP, Lichten LA. J Biol Chem. 2006;281:24085–24089. doi: 10.1074/jbc.R600011200. [DOI] [PubMed] [Google Scholar]
- 36.Hutchinson EG, Thornton JM. Protein Sci. 1994;3:2207–2216. doi: 10.1002/pro.5560031206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeGrado WF, Wasserman ZR, Lear JD. Science. 1989;243:622–628. doi: 10.1126/science.2464850. [DOI] [PubMed] [Google Scholar]
- 38.Bryson JW, Betz SF, Lu HS, Suich DJ, Zhou HX, O'Neil KT, DeGrado WF. Science. 1995;270:935–941. doi: 10.1126/science.270.5238.935. [DOI] [PubMed] [Google Scholar]
- 39.Klemba M, Regan L. Biochemistry. 1995;34:10094–10100. doi: 10.1021/bi00031a034. [DOI] [PubMed] [Google Scholar]
- 40.Ladurner AG, Fersht AR. J Mol Biol. 1997;273:330–337. doi: 10.1006/jmbi.1997.1304. [DOI] [PubMed] [Google Scholar]
- 41.Viguera AR, Serrano L. Nat Struct Biol. 1997;4:939–946. doi: 10.1038/nsb1197-939. [DOI] [PubMed] [Google Scholar]
- 42.Scalley-Kim M, Minard P, Baker D. Protein Sci. 2003;12:197–206. doi: 10.1110/ps.0232003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang L, Rivera EV, Benavides-Garcia MG, Nall BT. J Mol Biol. 2005;353:719–729. doi: 10.1016/j.jmb.2005.08.038. [DOI] [PubMed] [Google Scholar]
- 44.Myers JK, Pace CN, Scholtz JM. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berg JM, Merkle DL. J Am Chem Soc. 1989;111:3759–3761. [Google Scholar]
- 46.Walkup GK, Imperiali B. J Am Chem Soc. 1996;118:3053–3054. [Google Scholar]
- 47.Ghering AB, Shokes JE, Scott RA, Omichinski JG, Godwin HA. Biochemistry. 2004;43:8346–8355. doi: 10.1021/bi035673j. [DOI] [PubMed] [Google Scholar]
- 48.Monsigny M, Mayer R, Roche AC. Carbohydr Lett. 2000;4:35–52. [PubMed] [Google Scholar]
- 49.Kitov PI, Bundle DR. J Am Chem Soc. 2003;125:16271–16284. doi: 10.1021/ja038223n. [DOI] [PubMed] [Google Scholar]
- 50.Rosenberg RC, Root CA, Wang RH, Cerdonio M, Gray HB. Proc Natl Acad Sci USA. 1973;70:161–163. doi: 10.1073/pnas.70.1.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burkoth TS, Fafarman AT, Charych DH, Connolly MD, Zuckermann RN. J Am Chem Soc. 2003;125:8841–8845. doi: 10.1021/ja0352101. [DOI] [PubMed] [Google Scholar]
- 52.Mezo AR, Cheng RP, Imperiali BJ. J Am Chem Soc. 2001;123:3885–3891. doi: 10.1021/ja004292f. [DOI] [PubMed] [Google Scholar]
- 53.Fahrni CJ, O'Halloran TV. J Am Chem Soc. 1999;121:11448–11458. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.