

Abstract
Leptospira interrogans is a pathogenic spirochete that causes disease in both humans and animals. LigB (Leptospiral immunoglobulin-like protein B) contributes to the binding of Leptospira to extracellular matrix proteins such as fibronectin (Fn), fibrinogen, laminin, and collagen. A high affinity Fn-binding region of LigB has been recently localized to LigBCen2, which contains the partial eleventh and full twelfth immunoglobulin-like repeats (LigBCen2R) and 47 amino acids of the non-repeat region (LigBCen2NR) of LigB. In this study, LigBCen2NR was shown to bind to the N-terminal domain (NTD) of Fn (KD = 379 nm) by an enzyme-linked immunosorbent assay and isothermal titration calorimetry. Interestingly, this sequence was not observed to adopt secondary structure by far UV circular dichroism or by differential scanning calorimetry, in agreement with computer-based secondary structure predictions. A low partition coefficient (Kav) measured with gel permeation chromatography, a high hydrodynamic radius (Rh) measured with dynamic light scattering, and the insensitivity of the intrinsic viscosity to guanidine hydrochloride treatment all suggest that LigBCen2NR possesses an extended and disordered structure. Two-dimensional 15N-1H HSQC NMR spectra of intact LigBCen2 in the absence and presence of NTD are consistent with these observations, suggesting the presence of both a β-rich region and an unstructured region in LigBCen2 and that the latter of these selectively interacts with NTD. Upon binding to NTD, LigBCen2NR was observed by CD to adopt a β-strand-rich structure, suggestive of the known β-zipper mode of NTD binding.
Leptospira interrogans is a pathogenic spirochete that causes leptospirosis throughout the world, especially in developing countries but also in regions of the United States where it has reemerged (1). Weil's syndrome, a severe form of this disease, is an acute febrile illness associated with multiorgan damage, including liver failure (jaundice), renal failure (nephritis), pulmonary hemorrhage, and meningitis (1), and has a 15% mortality rate if not treated (2). The molecular pathogenesis of leptospirosis is poorly understood, and the bacterial virulence factors involved are largely unknown. Recently, several potential Leptospira virulence factors have been described, including sphingomyelinases, serine proteases, zinc-dependent proteases, and collagenase (3); LipL32 (4); lipopolysaccharide (5); a novel factor H, laminin, and Fn-binding protein (Lsa24 or Len) (6–8); Loa 22 (9); and Lig (Leptospiral immunoglobulin-like) proteins (10–12).
Lig proteins, including LigA, LigB, and LigC, contain multiple immunoglobulin-like repeat domains (13 in LigA, 12 in LigB and LigC) (10–12). Interestingly, the first 630 residues, from the N terminus to the first half of the seventh immunoglobulin-like domain, are conserved between LigA and LigB, but the rest of the immunoglobulin-like domains are variable (10–12) between the two proteins. Also, a non-immunoglobulin-like repeat region found on the C-terminal tail of LigB is not found in LigA (10–12). Lig proteins are categorized as microbial surface components recognizing adhesive matrix molecules (MSCRAMMs)2 due to their ability to bind to eukaryotic cells (13) through their interactions with extracellular matrix components, including fibronectin (Fn), laminin, collagens, elastin, and tropoelastin (13, 14, 45). Previously, a high affinity Fn-binding region was localized to LigBCen2, which includes the partial eleventh and complete twelfth immunoglobulin-like repeat region and the first 47 amino acids of the non-repeat regions of LigB (15). LigBCen2 was shown to bind to both the N-terminal domain (NTD) and the gelatin binding domain (GBD) of Fn. The addition of calcium induces a conformational change in LigBCen2 and enhances binding between LigBCen2 and the NTD of Fn (15).
The first step in the process of bacterial infection is cellular adhesion, mediated by bacterial adhesins interacting with various components of the extracellular matrix (16). Known interaction modes between Fn and bacterial Fn-binding proteins include the β-zipper (17, 18) and the cationic cradle (19). It was recently discovered that the Fn-binding domains in certain Fn-binding proteins are disordered and extended but gain structure upon binding to the NTD of Fn (20–22).
We have performed a fine-mapping study of the NTD-binding site on LigBCen2 and identified this site as LigBCen2NR, a portion of the non-repeat region (amino acids 1119–1165). The addition of NTD promotes the folding of LigBCen2NR from a disordered and extended structure to a folded structure. This finding is notable, since LigBCen2NR is located in the non-immunoglobulin-like region of LigB, as compared with other Fn-binding proteins, such as Staphylococcus aureus FnbpA and FnbpB (23), Streptococcus dysgalactiae FnBB (17), and Streptococcus pyogenes SfbI and SfbII (24). Thus, the binding mode appears to be similar to the known β-zipper mechanism but unique in sequence-specific interactions. This finding provides the fundamental groundwork for the development of a therapeutic agent to target this interaction in order to prevent or treat Leptospira infection.
MATERIALS AND METHODS
Reagents and Antibodies
Rabbit anti-GST antibody and horseradish peroxidase-conjugated goat anti-rabbit antibody were ordered from Molecular Probes, Inc. (Eugene, OR) and Zymed Laboratories Inc. (San Diego, CA), respectively. NTD or GBD of Fn, aldolase, bovine serum albumin, ovalbumin, chymotrypsinogen A, ribonuclease A, aprotinin, insulin chain B, sodium chloride, sodium phosphate monobasic, and sodium phosphate dibasic were purchased from Sigma.
Plasmid Construction and Protein Purification
The construction, expression, and purification of LigCon (amino acids 1–630) were performed as described previously (12). Constructs for the expression of histidine-tagged or GST-fused LigBCen2 (amino acids 1014–1156) and GST were generated using the vector pQE30 (Qiagen, Alencia, CA) and/or pGEX-4T-2 (GE Healthcare), respectively, as previously described (14). Constructs for the expression of histidine-tagged or GST-fused LigBCen2R (amino acids 1014–1123) and LigBCen2NR (amino acids 1119–1165) were generated using the vector pQE30 and pGEX-4T-2 (Fig. 1). To perform the PCRs, the following forward and backward primers were utilized (14): LigBCen2R forward primer 5′-GGATCCACTGCGACTTACAAT-3′ and backward primer 5′-GTCGACCGTGTCCGTTTTGTT-3′; LigBCen2NR forward primer 5′-CGGGATCCAACAAAACGGACACG-3′ and backward primer 5′-CGGTCGACATTGGAACTATTAAT-3′. Primers were engineered to introduce a BamHI site at the 5′-end of each fragment and a stop codon followed by a SalI site at the 3′-end of each fragment. PCR products were sequentially digested with BamHI and SalI and then ligated into pQE30 or pGEX-4T-2 cut with BamHI and SalI, respectively. In this study, we purified the soluble form of the histidine tag or GST fused with LigBCen2, LigBCen2R, or LigBCen2NR from Escherichia coli, as previously described (25). Tris buffer (25 mm Tris and 150 mm sodium chloride at pH 7.0) containing 100 μm calcium chloride was used in all experiments, since we have previously shown that calcium enhances the binding of LigBCen2 to NTD (15) and that both LigBCen2R and LigBCen2NR bind calcium (data not shown).
FIGURE 1.

A schematic diagram showing the location of the truncated LigBCen2 protein, including the LigBCen2R and LigBCen2NR constructs used in this study.
NMR Sample Preparation and Experiments
E. coli harboring the expression plasmid for His-tagged LigBCen2 were cultured in M9 minimal medium. The recombinant LigBCen2 (retaining the His6 tag) was labeled with 15NH4Cl (Cambridge Isotopes, Cambridge, MA), expressed, and purified, as previously described (26). The purified 15N-labeled LigBCen2 was dialyzed against Tris buffer with 100 μm calcium chloride at pH 6.0 and concentrated to 0.95 mm (0.3 ml) using the Amicon Ultra centrifugal filter (Millipore, Billerica, MA). For some spectra, 15N-labeled LigBCen2 was also mixed with 1.44 mm (unlabeled) NTD.
NMR spectra were recorded at 25 °C on a Varian Inova 600-MHz spectrometer equipped with a triple resonance (hydrogen, carbon, and nitrogen) z axis pulse-field gradient probe. Two-dimensional 15N-1H fast HSQC spectra were recorded with spectral widths of 2.4 kHz in t1 (400 real + imaginary data points) and 10 kHz in t2 (2048 real + imaginary data points) (27), with 16 or 32 transients per free induction decay for LigBCen2 in the absence or presence of NTD and a recycle delay of 1.0 s. NMR data were processed and analyzed using the software tools nmrPipe, nmrDraw, and Pipp (28, 29). Data were apodized using a phase-shifted sine bell function and zero-filled prior to Fourier transformation.
Surface Plasmon Resonance (SPR)
Association and dissociation rate constants for LigBCen2/NTD binding were measured by SPR analysis performed with a Biacore 2000 instrument (GE Healthcare) at 26.7 °C. 1 μm His-tagged LigBCen2 in Tris buffer containing 100 μm CaCl2 was immobilized on an nitrilotriacetic acid chip (GE Healthcare) conjugated with 500 μm nickel sulfate. Serial concentrations (100, 200, 400, and 800 nm) of NTD were injected into the flow cells at a flow rate of 5 μl/min over the immobilized LigBCen2. All experiments are duplicated. All sensogram data have been corrected by subtracting data from a control cell injected with Tris buffer containing 100 μm CaCl2. Kinetic parameters were obtained by fitting the data to the one-step biomolecular association reaction model (1:1 Langmuir model) with the curve-fitting BIAevaluation software, version 3.0.
ELISA Fn Binding Assays
To determine the binding of GST-LigBCen2, GST-LigBCen2R, GST-LigBCen2NR, or GST-LigCon (negative control) to the NTD of Fn, 1 μm NTD was coated on microtiter plate wells, incubated at 4 °C for 16 h, and blocked with blocking buffer (100 μl/well) containing 3% bovine serum albumin in Tris buffer with 100 μm calcium chloride at room temperature for 2 h. Then serial concentrations (as indicated by Fig. 3A) of GST-LigBCen2, GST-LigBCen2R, GST-LigBCen2NR, or GST-LigCon in 100 μl of Tris buffer with 100 μm calcium chloride were added to the microtiter plate wells for 1 h at 37 °C. To detect the binding of GST-LigBCen2, GST-LigBCen2R, GST-LigBCen2NR, or GST-LigCon, rabbit anti-GST (1:200) and horseradish peroxidase-conjugated goat anti-rabbit IgG (1:1,000) were used as primary and secondary antibodies, respectively. After washing the plates three times with TBST (0.05% Tween 20, 100 μm calcium chloride in Tris buffer), 100 μl of TMB (KPL, Gaithersburg, MD) was added to each well and incubated for 5 min. The reaction was stopped by adding 100 μl of 0.5% hydrofluoric acid to each well. Each plate was read at 630 nm using an ELISA plate reader (Bioteck EL-312, Winooski, VT). Each value represents the mean ± S.E. of three trials in triplicate samples. Statistically significant (p < 0.05) differences are indicated by an asterisk.
FIGURE 3.

Determination of binding constant and thermodynamics of the LigBCen2NR/NTD interaction by ELISA and ITC. A, binding of serial concentrations of LigBCen2NR to immobilized NTD by ELISA. Serial concentrations of GST-LigBCen2R, GST-LigBCen2NR, GST-LigBCen2 (positive reference), or GST-LigCon (negative reference) were added to 1 μm NTD- or bovine serum albumin-coated wells (negative control; data not shown). B, determination of the binding affinity by ITC. The cell contained 1 ml of NTD, and the syringe contained 250 μl of LigBCen2NR. Top, heat differences obtained from 25 injections of LigBCen2NR; bottom, integrated curve with experimental data (♦) and the best fit (line). The thermodynamic parameters are shown in Table 1.
Isothermal Titration Calorimetry (ITC)
The experiments were carried out with a CSC 5300 microcalorimeter (Calorimetry Science Corp., Lindon, UT) at 25 °C, as described previously (14). In a typical experiment, the cell contained 1 ml of a solution of NTD, and the syringe contained 250 μl of a solution of LigBCen2R or LigBCen2NR. The concentrations of LigBCen2R, LigBCen2NR, and NTD are described in Table 1. Both solutions were in Tris buffer with 100 μm calcium chloride. The titration was performed in 25 injections of 10 μl with a stirring speed of 250 rpm, and the delay time between the injections was 5 min. Data were analyzed using Titration Binding Work version 3.1 software (Calorimetry Science), fitting them to an independent binding model.
TABLE 1.
Thermodynamic parameters for the interaction of NTD and LigBCen2R or LigBCen2NR
| Macromolecule | LigB residues | [LigB] | [NTD] | ΔH | TΔS | ΔG | Kd | n |
|---|---|---|---|---|---|---|---|---|
| μm | μm | kcal mol−1 | kcal mol−1 | kcal mol−1 | nm | |||
| LigBCen2NR | 1119–1165 | 362 | 61 | 13.13 ± 1.55 | 22.25 | −9.12 | 379 ± 16 | 0.94 ± 0.01 |
| LigBCen2R | 1014–1123 | 118 | 5.9 | NFa | NF | NF | NF | NF |
a NF, non-fittable.
Prediction of Protein Disorder
The LigBCen2NR folding prediction was carried out using PONDR (Predictor of Naturally Disordered Regions), a software package containing VL-XT (refers to the merger of three predictors, one trained on variously characterized long disordered regions and two trained on x-ray-characterized terminal disordered regions), XL1-XT (the XL1-XT predictor is a feedforward neural network optimized to predicted regions greater than 39 amino acids), and VL3 (the VL3 predictor is a feedforward neural network that was trained on regions of 152 long regions of disorder that were characterized by various methods), which predict naturally disordered regions (30–32). PONDR can be used as a Web service for remote and automatic data processing. The analyses were performed using default values.
Differential Scanning Calorimetry (DSC)
The excess heat capacity Cp(T) of LigBCen2, LigBCen2R, or LigBCen2NR was measured using a DSC Q1000 microcalorimeter (Waters, New Castle, DE). Degassed samples containing 3 μm LigBCen2, LigBCen2R, LigBCen2NR, or Tris buffer with 100 μm calcium chloride were heated at a 10-K/h scan rate. Cp(T) data were recorded, corrected for buffer base line, and normalized to the amount of the samples. The TA Universal Analysis software (Waters) was used for the data analysis and display. All calorimetric experiments in this study were repeated three times to ensure reproducibility.
Gel Permeation Chromatography
LigBCen2, LigBCen2R, and LigBCen2NR were analyzed for their partition coefficient (Kav) and effective radii (Stokes radii, RS) in Tris buffer with 100 μm calcium chloride using a Superdex 200 HR 10/30 column (GE Healthcare) attached to a fast protein liquid chromatography (GE Healthcare) system. Protein samples were preequilibrated with Tris buffer with 100 μm calcium chloride and eluted with the same buffer at a flow rate of 0.5 ml/min. The column was calibrated using a low molecular weight gel filtration calibration kit (GE Healthcare). The standard globular proteins contained in the kit were ribonuclease A (13,700 Da), chymotrypsinogen (25,000 Da), ovalbumin (43,000 Da), and albumin (67,000 Da). Blue dextran 2000 (2,000,000 Da) (Amersham Biosciences) and aldolase (158,000 Da) were used to indicate the void volume (Vo) and the total volume (Vt), respectively. The elution volume (Ve) of each sample was measured. To define the relationships between the elution volumes of protein samples and their respective molecular weight, the Kav value for each protein was calculated using Equation 1,
 |
where Vo and Vt for the column used were 7.96 and 23.56 ml, respectively. Kav values of standard and LigBCen2, LigBCen2R, or LigBCen2NR were plotted against the logarithm of the protein molecular weights to fit Equation 2 as follows (33),
where a and b are constants. RS values of the proteins were determined using sample elution volumes and standard curves, as described in the calibration kit (GE Healthcare).
Dynamic Light Scattering (DLS)
One mg/ml LigBCen2, LigBCen2R, or LigBCen2NR was dialyzed against prefiltered (0.22-μm Millipore filters) Tris buffer with 100 μm calcium chloride. The samples were placed in a 1-ml plastic cuvette. The standard globular proteins, including albumin (67,000 Da), ovalbumin (43,000 Da), chymotrypsinogen A (25,000 Da), ribonuclease A (13,700 Da), aprotinin (6,500 Da), or insulin chain B (3,400 Da), were used to generate the calibration curve of globular proteins. The automated measurements were collected with a Zetasizer Nano ZS instrument (Malvern Instruments Ltd., Worcestershire, UK), using a 2-min equilibrium delay at each measurement. The data were adjusted using the method of cumulants to obtain the hydrodynamic radii (Rh). The logarithms of the Rh values of standards and LigBCen2, LigBCen2R, or LigBCen2NR were plotted against the logarithm of the protein molecular weights to fit the equation,
where a and b are constants.
Intrinsic Viscosity Measurements
The viscosities of the LigBCen2, LigBCen2R, and LigBCen2NR were measured using a Cannon-Ubbelohde Semi-Micro Dilution Viscometer (catalog number 25 9722-H50; Cannon Instrument Co., State College, PA) with a viscometer constant, 0.002044 mm2/s2, at 25 °C. Before measuring the viscosities, 1 ml of each protein at concentrations of 0.5, 0.75, 1, and 1.5 mg/ml were dialyzed overnight against Tris buffer with 100 μm calcium chloride, with or without 6 m guanidine hydrochloride, and the same buffer was used as a reference solution. The specific viscosity (ηsp) was determined as previously described (34). Specific viscosity/concentration values of untreated or 6 m guanidine hydrochloride-treated LigBCen2, LigBCen2R, or LigBCen2NR were plotted against the concentration of proteins, and the intrinsic viscosity [η] was calculated using the following equation,
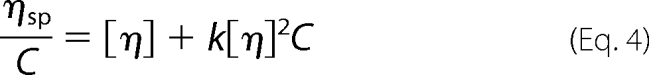 |
where c represents protein concentration and k is a dimensionless constant. The values of [η] expected for a denatured protein shown in Table 3 were obtained from the following equation (34),
where n represents the number of residues in the protein.
TABLE 3.
Intrinsic viscosity of recombinant LigBCen2, LigBCen2R, and LigBCen2NR
| Measured [η] from viscometry |
Predicted [η] for denatured protein | ||
|---|---|---|---|
| 0 m GnHCl | 6 m GnHCl | ||
| LigBCen2 | 9.55 | 22.23 | 21.39 |
| LigBCen2R | 4.75 | 20.45 | 17.87 |
| LigBCen2NR | 10.76 | 10.98 | 10.20 |
CD Spectroscopy
CD analysis was performed on an Aviv 215 spectropolarimeter (Lakewood, NJ) under N2 atmosphere. CD spectra were measured at room temperature (25 °C) in a 1-cm path length quartz cell. Spectra of LigBCen2, LigBCen2R, or LigBCen2NR were recorded in Tris buffer with 100 μm calcium chloride at a protein concentration of 10 μm. Three spectra were recorded for each condition from 190 to 250 nm for far UV CD in 1-nm increments. In the thermal denaturation experiment, 10 μm LigBCen2, LigBCen2R, or LigBCen2NR were used, and data were collected at 2 °C/min increments from 20 to 100 °C, recording the ellipticity at 205 nm, with 30-s temperature equilibrations, followed by 30-s data averaging. In order to measure the melting point, the first order derivative of the melting curve was taken. Structural changes in LigBCen2R and LigBCen2NR upon binding to the NTD of Fn were examined by analyzing changes in the CD spectrum. LigBCen2R (10 μm) or LigBCen2NR (10 μm) was preincubated with or without the NTD (10 μm) of Fn for 1 h at 25 °C, and the far UV CD spectra were recorded as described above. The changes of the CD spectra upon NTD-LigBCen2R or NTD-LigBCen2NR interaction were determined by computationally subtracting the ellipticity of the NTD-LigBCen2R or NTD-LigBCen2NR complex from the added spectra of the free forms of interacting proteins. The deconvolution of the resulting spectra was performed as described above. In all CD experiments, the background spectrum of buffer without protein was subtracted from the protein spectra. CD spectra were initially analyzed by the software accompanying the spectrophotometer. Analysis of spectra to extrapolate secondary structures was performed by Dichroweb (35) (available on the World Wide Web), using the K2D and Selcon 3 analysis programs (36, 37).
Fluorescence Spectroscopy
Fluorescence emission spectra were measured on a Hitachi F4500 spectrofluorometer (Hitachi, San Jose, CA). All spectra were recorded in the correct spectrum mode of the instrument, using excitation and emission band-passes of 5 nm. The intrinsic tryptophan fluorescence of the protein was recorded by exciting the solution at 295 nm and measuring the emission between 305 and 400 nm. For the LigBCen2NR/NTD binding experiment, the emission spectra of untreated or LigBCen2NR (10 μm) treated with NTD (10 μm) were taken. LigBCen2NR contains no tryptophan; therefore, for the LigBCen2NR/NTD binding experiment, the spectrum of LigBCen2NR in isolation was essentially background and was subtracted from the spectrum for the mixture.
Matrix-assisted Laser Desorption Ionization-Time of Flight (MALDI-TOF) Mass Spectrometry
The molecular weights of samples of purified LigBCen2, LigBCen2R, and LigBCen2NR were analyzed using MALDI-TOF mass spectra recorded on an Applied Biosystems 4700 mass spectrometer (Applied Biosystems, Foster City, CA). Prepared samples included 10 μm LigBCen2, LigBCen2R, or LigBCen2NR in deionized water.
Statistical Analysis
Significance between samples was determined using Student's t test following logarithmic transformation of the data. Two-tailed p values were determined for each sample, and a p value of <0.05 was considered significant. Each data point represents the mean ± S.E. of samples tested in triplicate. An asterisk indicates that the result was statistically significant.
RESULTS
Identification of the Binding Sites of NTD on LigBCen2
In order to investigate the general structural properties of LigBCen2 and its interactions with Fn, uniformly 15N-labeled LigBCen2 was prepared, and two-dimensional 15N-1H chemical shift correlation NMR experiments were performed in the absence and presence of an equimolar amount of unlabeled NTD (Fig. 2, A–D).
FIGURE 2.

NMR provides evidence for distinct structured and disordered regions of LigBCen2. A, the 1H-15N HSQC of apo-LigBCen2 exhibits a set of well dispersed peaks, indicative of folded, β-rich residues, and a set of sharper peaks clustered toward the center, indicative of residues in an unstructured region. This is emphasized by the HSQC viewed at a higher contour level (B). The number of peaks indicative of folded and unfolded residues roughly matches the number of residues expected to adopt the Ig-like fold of repeat regions 11 and 12 and the number of residues included from the non-repeat region, respectively. C, when unlabeled NTD is added at an equimolar concentration, most of the sharp, clustered peaks either disappear or are greatly diminished in intensity, whereas the well dispersed peaks corresponding to folded protein remain largely unperturbed, as highlighted by the overlay of this spectrum with the apo-LigBCen2 spectrum (D). The disappearance of the peaks corresponding to the unstructured region(s) of the LigBCen2 construct suggests that the non-repeat sequence selectively binds to NTD.
The NMR spectrum of apo-LigBCen2 shows evidence for distinct structured and unstructured regions, with a set of highly resolved peaks dispersed across both dimensions of the spectrum and another set of peaks of higher intensity clustered toward the center of the spectrum (Fig. 2, A and B). The number of well dispersed peaks and their positions are consistent with the β-sheet-rich immunoglobulin-like fold predicted for repeat regions 11 and 12, which would place backbone N-H groups in unique chemical environments and would cause the corresponding 15N-1H correlation peaks to appear at widely varying positions in the NMR spectrum. In contrast, for a dynamically unstructured polypeptide, the corresponding 15N-1H correlation peaks are stronger due to increased motion and cluster to similar regions of the spectrum due to the more uniform chemical environment of the solvent. A set of such peaks is clearly present, as highlighted by the LigBCen2 15N-1H spectrum at a higher contour level (Fig. 2B). The number of peaks associated with folded and unfolded sets of N-H groups roughly matches the number of residues expected to adopt the β-sheet-rich fold of the twelfth and partial eleventh Ig-like repeats (LigBCen2R) and the number of residues included from the non-repeat region (LigBCen2NR), respectively.
When 1.44 mm unlabeled NTD was added to 0.95 mm 15N-LigBCen2, the well dispersed peaks corresponding to folded protein remained largely unperturbed, whereas most of the sharp, clustered peaks either disappeared or were greatly diminished in intensity (Fig. 2C), as highlighted by the overlay of this spectrum with the apo-LigBCen2 spectrum (Fig. 2D). Interestingly, application of a transverse relaxation optimized spectroscopy-based 15N-1H HSQC experiment useful for very large proteins (38) did not recover the lost peaks (data not shown). These results demonstrate that NTD does not bind to the structured region of LigBCen2. The nature and number of peaks that are affected by the addition of NTD suggest that NTD specifically binds to LigBCen2NR and results in chemical exchange in the intermediate rate regime, where the peaks whose chemical environment changes between conformational states are broadened beyond the level of detection.
In order to test the possibility that the peak disappearance is due to sampling between free and bound states, SPR was performed to measure the binding kinetics for the LigBCen2-NTD interaction at pH 6.0 and 26.7 °C to correspond to the NMR conditions (supplemental Fig. S1). Based on the rates obtained, (kon = 2.28 × 105 ± 0.23 × 105 s−1 m−1, koff = 2.47 × 10−2 ± 0.16 × 10−2 s−1), the kex should be 112 ± 11 s−1 at the conditions of the NMR sample (1.44 mm NTD, 0.95 mm LigBCen2, [NTD]free = 0.49 mm). Resonances undergoing exchange broadening due to ligand binding usually refocus at saturation. Given the unusually slow off-rate of binding, at saturating concentrations of NTD, the spectrum of LigBCen2 should show a single set of peaks reflecting the bound state. A saturation of >99% was predicted for the NMR sample, assuming simple two-state binding with a KD of 93 nm (15). Therefore, the line broadening observed for the NMR sample is not due to exchange between free and bound states. One possible explanation is the formation of a “fuzzy complex,” or a dynamic ensemble of conformational states, upon binding NTD. This has been previously observed for other intrinsically disordered proteins, including the NTD-binding SfbI of S. pyogenes (39, 40). Regardless of the origin of the peak disappearance, the NMR experiments indicate that NTD interacts specifically with the disordered region of LigBCen2 in the intact construct.
In order to evaluate the pH sensitivity of the LigBCen2-NTD interaction, the SPR measurements were performed at both pH 6.0 and 7.0. The dissociation constants calculated at each pH (KD = koff/kon = 109 ± 8 nm at pH 6.0, 95.5 ± 1.4 nm at pH 7.0) are in good agreement with each other, and the value at pH 7.0 is in excellent agreement with the previously reported KD of 93 ± 8 nm at pH 7.0 (15). The kinetic rates at each pH were also in close agreement with each other (kon = 2.72 ± 0.19 × 105 s−1 m−1, koff = 2.6 ± 0.22 × 10−2 s−1 at pH 7.0). All subsequent biophysical measurements were performed at pH 7.0.
In order to test the NMR-derived hypothesis that NTD interacts specifically with LigBCen2NR, various concentrations of GST-LigCon (a truncation that cannot interact with Fn, used as a negative control), GST-LigBCen2 (positive control), GST-LigBCen2R, GST-LigBCen2NR, and GST were added to an NTD-coated microtiter plate. As shown in Fig. 3A, LigBCen2NR binds NTD, but no NTD binding was observed for LigBCen2R or LigCon (Fig. 1) (13, 14). These results were also confirmed by ITC experiments. LigBCen2NR binding to NTD is an endothermic reaction (unfavorable enthalpy, favorable entropy) with a dissociation constant (KD) of 379 nm (Fig. 3B and Table 1). The favorable entropy suggests that the complex formation of LigBCen2 and NTD involves significant hydrophobic interactions. By contrast, ITC experiments demonstrate that LigBCen2R does not bind to NTD (Table 1). Both ELISA and ITC results support the NMR-derived hypothesis that LigBCen2NR, the non-immunoglobulin-like domain 47-residue region of LigBCen2, selectively binds to NTD.
LigBCen2NR Possesses a Disordered and Unfolded Structure
To further characterize the structural properties of LigBCen2NR and investigate the NMR-derived hypothesis that this region is unstructured, the amino acid sequence of LigBCen2NR was analyzed by PONDR, a software tool used to predict naturally disordered protein regions. More than half of the residues in LigBCen2NR (all but residues 1146–1164) were predicted to be disordered by VL-XT in the PONDR software package. In addition, a highly disordered structure was also predicted in LigBCen2NR by XL1-XT and VL3 in the PONDR software package (data not shown).
In order to confirm this sequence-based prediction, the secondary structure of LigBCen2, LigBCen2R, or LigBCen2NR was examined by far UV CD. As presented in Fig. 4A, both LigBCen2 and LigBCen2R contain significant β-sheet structure (40% in LigBCen2 and 55% in LigBCen2R). However, LigBCen2NR contains 65% random coil. Thus, compared with LigBCen2R, LigBCen2NR possesses a highly disordered and unfolded structure, in agreement with the NMR analysis of LigBCen2 presented above.
FIGURE 4.

Analysis of LigBCen2, LigBCen2R, or LigBCen2NR by far UV CD, gel permeation chromatography, DLS, and viscometry. A, far UV CD analysis of LigBCen2, LigBCen2R, and LigBCen2NR. The molar ellipticity, Φ, was measured from 190 to 250 nm for a 10 μm concentration of each protein in Tris buffer with 100 μm calcium chloride. B, gel permeation chromatography of standard molecular mass markers, LigBCen2, LigBCen2R, and LigBCen2NR. The partition coefficient (Kav) is plotted as a function of the molecular weight of each individual protein on a log scale. C, dynamic light scattering of standard molecular mass markers, LigBCen2, LigBCen2R, and LigBCen2NR. The hydrodynamic radius (Rh) is plotted as a function of the molecular weight of each individual protein on a log-log scale. For C and D, the molecular weights of the standards are indicated under “Materials and Methods,” and the molecular weights of LigBCen2, LigBCen2R, and LigBCen2NR are indicated in Table 2. D, determination of the intrinsic viscosity of proteins under native or denaturing conditions. The specific viscosity/concentration versus concentration is plotted for LigBCen2, LigBCen2R, and LigBCen2NR in Tris buffer with 100 μm calcium chloride and with or without 6 m guanidine hydrochloride. The intercept at the y axis is the intrinsic viscosity.
Gel Permeation Chromatography and Dynamic Light Scattering Indicate That LigBCen2NR Adopts an Extended Structure
Most proteins containing disordered structures are extended instead of globular and highly packed. Due to differences in the hydrodynamic properties (such as RS and the Kav) of globular and extended proteins, gel permeation chromatography is a powerful technique to determine if a protein is unfolded. The Kav values of LigBCen2, LigBCen2R, LigBCen2NR, and other globular protein standards were determined by the Ve value obtained from gel permeation chromatography and calculated by Equation 1 above. In addition, Kav was plotted against the logarithm of molecular weight, and a calibration curve of globular proteins was made by using the data from the globular protein standards. Compared with LigBCen2 and LigBCen2R, the value of Kav for LigBCen2NR was far from the calibration curve and less than expected for a globular protein with a similar molecular mass (Fig. 4B). These results indicate that LigBCen2NR is not a globular protein. Similarly, the RS value of LigBCen2NR revealed by gel permeation chromatography (1.93 nm) is significantly larger than expected for a globular protein (0.62 nm) (Table 2), suggesting that LigBCen2NR forms an extended structure.
TABLE 2.
Estimated radii of recombinant LigBCen2, LigBCen2R, and LigBCen2NR
| Calculated mass | Mass spectroscopy | Expected RS from GPC calibration curve | RS from GPC | Expected Rh from DLS calibration curve | Rh from DLS | |
|---|---|---|---|---|---|---|
| Da | Da | nm | nm | nm | nm | |
| LigBCen2 | 18,130.97 | 18,131.60 | 2.05 | 1.95 | 2.04 | 2.07 |
| LigBCen2R | 13,473.76 | 13,416.81 | 1.67 | 2.07 | 1.75 | 1.71 |
| LigBCen2NR | 5,956.57 | 5,929.34 | 0.62 | 1.93 | 0.93 | 1.97 |
The hydrodynamic radii Rh values of LigBCen2, LigBCen2R, LigBCen2NR, and globular protein standards were measured using dynamic light scattering to confirm the results from the gel permeation chromatography measurements. The Rh value of LigBCen2 or LigBCen2R was close to the value of the globular proteins possessing a similar molecular mass (Fig. 4C and Table 2). However, the Rh value of LigBCen2NR (1.97 nm) was significant larger than that of globular proteins with a similar molecular mass (0.94 nm) (Fig. 4C and Table 2). The distinctly larger Rh of LigBCen2NR suggested that it is a highly extended protein, consistent with the results obtained from gel permeation chromatography.
Intrinsic Viscosity Measurements Suggest that LigBCen2NR Is Extended and Denatured
Measurement of intrinsic viscosity is a well known method to determine if a protein is denatured because of the direct relationship between the number of residues of a denatured protein and its intrinsic viscosity, as shown in Equation 5. For extended or denatured proteins, intrinsic viscosity is also generally larger than for folded proteins (21).
To further examine if LigBCen2NR is denatured and extended, the viscosities of LigBCen2, LigBCen2R, and LigBCen2NR were measured. The specific viscosity over protein concentration was plotted against concentration, as shown in Fig. 4D. The data were fitted to Equation 4 to obtain intrinsic viscosities, and these values were compared with the values expected for denatured proteins, as calculated by Equation 5. As shown in Fig. 4D and Table 3, the intrinsic viscosity of untreated LigBCen2 or LigBCen2R measured by viscometry was smaller than that of LigBCen2 or LigBCen2R treated with guanidine hydrochloride. However, the intrinsic viscosities of both guanidine hydrochloride-treated and untreated LigBCen2NR were close to the intrinsic viscosity expected for a denatured protein, as calculated by Equation 5. Taken together, these results indicate that LigBCen2NR is denatured and extended.
CD and DSC Thermal Unfolding Experiments Further Indicate That LigBCen2NR Has an Unfolded Structure
In order to gain more insight on the conformation of LigBCen2, LigBCen2, LigBCen2R, and LigBCen2NR were subjected to thermal unfolding experiments using CD spectrophotometry and DSC. As shown in Fig. 5A, the melting points of LigBCen2 and LigBCen2R are 54.0 ± 0.5 and 58.1 ± 0.8 °C, respectively, as measured by monitoring the CD signal at 205 nm from 20 to 80 °C. The higher melting temperature for LigBCen2R indicates that the presence of LigBCen2NR reduces the thermal stability of LigBCen2R. The midpoint of the DSC transition curves for LigBCen2 (54.2 ± 0.4 °C) and LigBCen2R (57.3 ± 0.2 °C) are consistent with the CD-derived melting points (Fig. 5B). Compared with the results of LigBCen2 and LigBCen2R, no obvious cooperative unfolding transition of LigBCen2NR was observed (Fig. 5). Therefore, both thermal and chemical denaturation experiments indicate that LigBCen2NR has an unfolded structure.
FIGURE 5.

Thermal unfolding transitions of LigBCen2, LigBCen2R, or LigBCen2NR as measured by CD and DSC. A, thermal unfolding of 10 μm LigBCen2, LigBCen2R, or LigBCen2NR was observed by CD, measuring ellipticity at 205 nm from 20 to 100 °C. A transition point was identified for LigBCen2 and LigBCen2R but not for LigBCen2NR. The melting temperatures were determined by the location of the peak in the derivative of the ellipticity curve, as shown in the lower panel. The transition midpoints of LigBCen2 and LigBCen2R are 54.0 ± 0.5 and 58.1 ± 0.8 °C, respectively. B, thermal unfolding transitions of LigBCen2, LigBCen2R, and LigBCen2NR measured by DSC. Molar heat capacity (kcal/(mol·K)) was plotted against temperature (°C) from 20 to 80 °C for 3 μm LigBCen2, LigBCen2R, or LigBCen2NR. The dotted line indicates that the midpoint temperatures of the transitions for LigBCen2 and LigBCen2R are 54.2 ± 0.4 and 57.3 ± 0.2 °C, respectively.
The Conformation of LigBCen2NR Changes upon the Addition of NTD
In order to determine if the binding of NTD affects the structure of LigBCen2NR, the CD spectra of NTD, LigBCen2NR (Fig. 6A), and NTD mixed with LigBCen2R or LigBCen2NR were recorded (Fig. 6, B and C). The NTD spectrum and either the LigBCen2R or the LigBCen2NR spectrum were added as a comparison to assess conformational changes upon binding. Consistently, the far UV CD spectrum of the NTD presented on Fig. 6A is similar to that published previously (20, 21, 41), since a minimum in intensity was located around 215 nm, and two peaks were located around 200 nm and 230 nm, respectively due to tyrosine side chains (42, 43). As shown in Fig. 6, A and B, the spectra of apo-LigBCen2NR and complexed NTD-LigBCen2NR after subtraction of the NTD CD spectrum are significantly different, but the spectrum of apo-LigBCen2R cannot be distinguished from a mixture of NTD and LigBCen2R after subtraction of the NTD CD spectrum. Therefore, conformational changes occur upon binding of NTD to LigBCen2NR. In order to clarify whether NTD or LigBCen2NR changes conformation after binding, the intrinsic fluorescence spectra of unbound or LigBCen2NR-bound NTD were measured. In a previous study, it was shown that conformational changes in NTD are easily detected by intrinsic tryptophan fluorescence due to the presence of seven tryptophan residues in NTD that are sensitive to small changes in environment (20). Furthermore, because of the absence of tryptophan in LigBCen2NR, the intrinsic fluorescence spectrum of NTD-bound LigBCen2NR reflects only the conformation of NTD. The addition of LigBCen2NR did not significantly change the fluorescence spectrum of NTD (supplemental Fig. S2), which suggests that binding of LigBCen2NR does not significantly alter the chemical environment of the tryptophan residues of NTD. This observation, which suggests that NTD does not undergo significant conformational rearrangement upon binding, has been reported previously for binding of a domain of FnbA from S. dysgalactiae to NTD, and it is thought to be generally true for many NTD-binding MSCRAMMs that NTD remains conformationally unchanged upon binding (20). Thus, the difference between the CD spectrum of the LigBCen2-NTD complex and the sum of the spectra of the individual components shown in Fig. 6C is considered to be due to conformational changes in LigBCen2NR and not NTD.
FIGURE 6.

Modification of the far UV CD spectrum of LigBCen2NR upon NTD binding. Shown are far UV spectra of the apo forms and mixtures of 10 μm NTD and 10 μm either LigBCen2NR (A) or LigBCen2R (B). The ellipticities shown in A are those of uncomplexed LigBCen2NR (short dashed line), complexed NTD and LigBCen2NR after subtraction of the NTD CD spectrum (long and short dashed line), and the CD spectrum of 10 μm NTD (solid line). The ellipticities in B are those of LigBCen2R (short dashed line), a mixture of NTD and LigBCen2R after subtraction of the NTD CD spectrum (long and short dashed line), and the CD spectrum of 10 μm NTD (solid line). C, the far UV spectra of mixtures of NTD and LigBCen2NR (dashed line) or LigBCen2R (solid line) after subtraction of the CD spectra of both apo-NTD and apo-LigBCen2NR or LigBCen2R.
The CD spectra of free and complexed LigBCen2NR were analyzed by software to quantitate secondary structure before and after binding of NTD (Fig. 6A). The results of analysis show that LigBCen2NR gains secondary structure upon the addition of NTD because the percentage of random coil dramatically decreases (from 65 to 46%), whereas the β-sheet composition increases (from 28 to 48%). However, the α-helix composition changes little (from 7 to 6%). This indicates that binding of NTD may cause LigBCen2NR to adopt a β-sheet-rich structure.
DISCUSSION
MSCRAMM is a class of well known adhesin proteins that enable microbes to attach to host cells by binding to their extracellular matrices. LigB binds to Fn, fibrinogen, laminin, elastin, proelastin, and collagen and hence probably plays a substantial role in adhesion for pathogenic Leptospira (13, 14, 44, 45). We have shown that LigBCen2 strongly binds to the NTD and weakly binds to the GBD of Fn (14) (NTD, KD = 272 nm; GBD, KD = 1200 nm). LigBCen2 contains an immunoglobulin-like repeat region, LigBCen2R, and a non-repeat 47-residue region, LigBCen2NR. In this study, two-dimensional NMR spectroscopy revealed both folded and disordered regions of LigBCen2, demonstrated that the folded region does not bind to NTD, and indicated that the disordered region specifically interacts with NTD. Using ELISA and ITC, we found that LigBCen2NR indeed binds to NTD. However, the binding affinity of LigBCen2NR/NTD (KD = 379 nm) (Table 1) is 4-fold lower than LigBCen2/NTD (KD = 93 nm) (15). It is not presently clear why this is true, but it may suggest that residues near the interface between LigBCen2R and LigBCen2NR may play a substantial role in binding and that severing the two regions reduces their binding capacity. In protein binding studies of intrinsically disordered proteins, it is not uncommon for regions flanking binding elements to contribute to binding without directly contacting the binding partner (40). A similarly unexplained reduction in binding affinity has previously been observed for truncated forms of BBK32 from B. burgdorferi (21). Further study is needed to clarify this result.
The N-terminal domain of BBK32 (amino acids 56–205) from B. burgdorferi possesses Fn binding activity but lacks secondary structure (21). Additional Fn-binding proteins previously shown to be disordered include FNBD-D, the D1–D3 repeat region of FnbpA from S. aereus; FNBD-A and FNBD-B, the A1–A3 and B1–B3 repeat regions of FnbA and FnbB from S. dysgalactiae; and FNBD-P, the P1–P4 repeat regions of Sfb from S. pyogens (20). These Fn-binding regions change their conformation to a folded form upon binding to NTD (20, 21, 23). Similarly, upon binding to NTD, LigBCen2NR folds into a β-strand-rich structure, much like the D123 domain of FnbPA or the N-terminal of BBK32 (21). High resolution structures of the complex between the B3 region of FnbB and the first and the second type I module of Fn (17, 18) and the complex between the first or the fifth Fn binding region of FnbpA and the second to the fifth type I module of Fn (23, 39, 46) all indicate that a two β-strand complex, called a β-zipper, mediates those interactions. The binding of Sfb and BBK32 to NTD are also accomplished through the β-zipper interaction (23, 39, 46). The β-strand-rich conformation formed by LigBCen2NR after binding NTD observed via far UV CD spectroscopy suggests that the binding of LigBCen2NR to NTD might be also mediated by a β-zipper interaction. However, the entropy-driven interaction of LigBCen2NR and NTD is distinct from the enthalpy-driven binding of other bacterial proteins known to bind NTD via the β-zipper interaction (Table 1) (23, 39, 46). In addition, we are unable to identify substantial sequence similarity between LigBCen2NR and BBK32 or other Fn-binding proteins, so additional study is needed to further characterize the binding between LigBCen2NR and NTD.
The on-rate of LigBCen2-NTD binding obtained from SPR is on the order of 105 m−1 s−1, 3 orders of magnitude below rates expected for diffusion-limited two-state binding (108 to 109 m−1 s−1) (47). This indicates that additional processes occur either before or concurrently with association of LigBCen2 with NTD. The observed increase in secondary structure of LigBCen2NR upon binding may account for this.
The off-rate of the NTD-LigBCen2 complex is also strikingly slow, yielding a residence time on the order of 40 s. This may be an important feature for the role of LigB in infection. Recent experiments (48, 49) have suggested that a small koff of a drug-target complex is an important predictor, in some cases more than a small KD, of in vivo efficacy. This is because in vivo the concentration of ligand is often not constant but is instead influenced by absorption, distribution, metabolism, and excretion. In some cases, this leads to a greater dependence on koff of the overall duration of the complex. Since a bacterial adhesin is challenged by many of the same limitations to concentration as a drug, it is interesting to speculate that the slow off-rate reported here is evolutionarily optimized to avoid clearance, whereas the slow on-rate may be optimized to facilitate movement through the host.
We have demonstrated that the region of LigBCen2 that mediates its interaction with the NTD of Fn is localized to LigBCen2NR. LigBCen2NR has a disordered structure that dramatically increases its β-strand content after binding to NTD. Disordered proteins often alter their conformations when binding their partners (50). One possible role of protein disorder in protein-protein interactions is the large accessible surface area per residue contributed by the disordered protein (18, 51). The unfavorable decrease in the entropy of LigBCen2NR upon binding NTD due to the disorder-order transition is offset by a larger increase in entropy, possibly due to hydrophobic interactions, as demonstrated by ITC. Another advantage of protein disorder in binding is that coupled folding and binding usually contribute high specificity and low affinity to the interaction (50).
In conclusion, the moderate affinity and slow kinetics of the LigBCen2NR interaction with the NTD of Fn might aid in Leptospira infection of a host organism by increasing the time it remains adhered to cells while simultaneously facilitating efficient transmission within the host. These intriguing results motivate further study of the role this disordered region plays in the pathogenesis of leptospiral infection.
Supplementary Material
Acknowledgments
We thank Drs. Bhargavi Jayaraman and Charlene Mottler for help with ITC, Ellen Keene for help with CD, Dr. Robin Yates for help with fluorescence, Anthony Condo for help with DSC, and Drs. Moonsoo Jin and Roisin Owens for help with SPR. We thank Dr. Robert Sherwood for assistance with mass spectrometry, Dr. Sandra Dias for assistance with gel permeation chromatography, and Dr. Yuanming Zhang and Graham Kerslick for assistance with DLS. Thanks are also extended to Hung-Yen Lee for suggestions during the course of this study.
This work was supported, in whole or in part, by a National Institutes of Health biophysics training grant (to A. G.). This work was also supported in part by the Harry M. Zweig Memorial Fund for Equine Research, the New York State Science and Technology Foundation (CAT), the Biotechnology Research and Development Corporation (BRDC), and National Science Foundation Grant MCB-0641582 (to L. K. N.).
 The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
- MSCRAMM
- microbial surface component recognizing adhesive matrix molecules
- DLS
- dynamic light scattering
- DSC
- differential scanning calorimetry
- Fn
- fibronectin
- GBD
- gelatin binding domain
- ITC
- isothermal titration calorimetry
- MALDI-TOF
- matrix-assisted laser desorption ionization-time of flight
- NTD
- N-terminal domain
- SPR
- surface plasmon resonance
- GST
- glutathione S-transferase
- ELISA
- enzyme-linked immunosorbent assay.
REFERENCES
- 1.Faine S. B., Adher B., Bolin C., Perolat P. (eds) (1999) Leptospira and Leptospirosis, pp. 67–69MedSci, Medbourne, Australia [Google Scholar]
- 2.Segura E. R., Ganoza C. A., Campos K., Ricaldi J. N., Torres S., Silva H., Céspedes M. J., Matthias M. A., Swancutt M. A., López Liñán R., Gotuzzo E., Guerra H., Gilman R. H., Vinetz J. M. (2005) Clin. Infect. Dis. 40,343–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bulach D. M., Zuerner R. L., Wilson P., Seemann T., McGrath A., Cullen P. A., Davis J., Johnson M., Kuczek E., Alt D. P., Peterson-Burch B., Coppel R. L., Rood J. I., Davies J. K., Adler B. (2006) Proc. Natl. Acad. Sci. U.S.A. 103,14560–14565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang C. W., Wu M. S., Pan M. J., Hsieh W. J., Vandewalle A., Huang C. C. (2002) J. Am. Soc. Nephrol. 13,2037–2045 [DOI] [PubMed] [Google Scholar]
- 5.Werts C., Tapping R. I., Mathison J. C., Chuang T. H., Kravchenko V., Saint Girons I., Haake D. A., Godowski P. J., Hayashi F., Ozinsky A., Underhill D. M., Kirschning C. J., Wagner H., Aderem A., Tobias P. S., Ulevitch R. J. (2001) Nat. Immunol. 2,346–352 [DOI] [PubMed] [Google Scholar]
- 6.Barbosa A. S., Abreu P. A., Neves F. O., Atzingen M. V., Watanabe M. M., Vieira M. L., Morais Z. M., Vasconcellos S. A., Nascimento A. L. (2006) Infect. Immun. 74,6356–6364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stevenson B., Choy H. A., Pinne M., Rotondi M. L., Miller M. C., Demoll E., Kraiczy P., Cooley A. E., Creamer T. P., Suchard M. A., Brissette C. A., Verma A., Haake D. A. (2007) PLoS ONE 2,e1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Verma A., Hellwage J., Artiushin S., Zipfel P. F., Kraiczy P., Timoney J. F., Stevenson B. (2006) Infect. Immun. 74,2659–2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ristow P., Bourhy P., da Cruz McBride F. W., Figueira C. P., Huerre M., Ave P., Girons I. S., Ko A. I., Picardeau M. (2007) PLoS Pathog. 3,e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsunaga J., Barocchi M. A., Croda J., Young T. A., Sanchez Y., Siqueira I., Bolin C. A., Reis M. G., Riley L. W., Haake D. A., Ko A. I. (2003) Mol. Microbiol. 49,929–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palaniappan R. U., Chang Y. F., Hassan F., McDonough S. P., Pough M., Barr S. C., Simpson K. W., Mohammed H. O., Shin S., McDonough P., Zuerner R. L., Qu J., Roe B. (2004) J. Med. Microbiol. 53,975–984 [DOI] [PubMed] [Google Scholar]
- 12.Palaniappan R. U., Chang Y. F., Jusuf S. S., Artiushin S., Timoney J. F., McDonough S. P., Barr S. C., Divers T. J., Simpson K. W., McDonough P. L., Mohammed H. O. (2002) Infect. Immun. 70,5924–5930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin Y. P., Chang Y. F. (2008) J. Vet. Sci. 9,133–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin Y. P., Chang Y. F. (2007) Biochem. Biophys. Res. Commun. 362,443–448 [DOI] [PubMed] [Google Scholar]
- 15.Lin Y. P., Raman R., Sharma Y., Chang Y. F. (2008) J. Biol. Chem. 283,25140–25149 [DOI] [PubMed] [Google Scholar]
- 16.Patti J. M., Allen B. L., McGavin M. J., Höök M. (1994) Annu. Rev. Microbiol. 48,585–617 [DOI] [PubMed] [Google Scholar]
- 17.Schwarz-Linek U., Werner J. M., Pickford A. R., Gurusiddappa S., Kim J. H., Pilka E. S., Briggs J. A., Gough T. S., Höök M., Campbell I. D., Potts J. R. (2003) Nature 423,177–181 [DOI] [PubMed] [Google Scholar]
- 18.Bingham R. J., Rudiño-Piñera E., Meenan N. A., Schwarz-Linek U., Turkenburg J. P., Höök M., Garman E. F., Potts J. R. (2008) Proc. Natl. Acad. Sci. U.S.A. 105,12254–12258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kingsley R. A., Keestra A. M., de Zoete M. R., Bäumler A. J. (2004) Mol. Microbiol. 52,345–355 [DOI] [PubMed] [Google Scholar]
- 20.House-Pompeo K., Xu Y., Joh D., Speziale P., Höök M. (1996) J. Biol. Chem. 271,1379–1384 [DOI] [PubMed] [Google Scholar]
- 21.Kim J. H., Singvall J., Schwarz-Linek U., Johnson B. J., Potts J. R., Höök M. (2004) J. Biol. Chem. 279,41706–41714 [DOI] [PubMed] [Google Scholar]
- 22.Penkett C. J., Redfield C., Jones J. A., Dodd I., Hubbard J., Smith R. A., Smith L. J., Dobson C. M. (1998) Biochemistry 37,17054–17067 [DOI] [PubMed] [Google Scholar]
- 23.Meenan N. A., Visai L., Valtulina V., Schwarz-Linek U., Norris N. C., Gurusidappa S., Hook M., Speziale P., Potts J. R. (2007) J. Biol. Chem. 282,25813–25902 [DOI] [PubMed] [Google Scholar]
- 24.Schwarz-Linek U., Höök M., Potts J. R. (2004) Mol. Microbiol. 52,631–641 [DOI] [PubMed] [Google Scholar]
- 25.Palaniappan R. U., McDonough S. P., Divers T. J., Chen C. S., Pan M. J., Matsumoto M., Chang Y. F. (2006) Infect. Immun. 74,1745–1750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jayaraman B., Nicholson L. K. (2007) Biochemistry 46,12174–12189 [DOI] [PubMed] [Google Scholar]
- 27.Mori S., Abeygunawardana C., Johnson M. O., van Zijl P. C. (1995) J. Magn. Reson. B 108,94–98 [DOI] [PubMed] [Google Scholar]
- 28.Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) J. Biomol. NMR 6,277–293 [DOI] [PubMed] [Google Scholar]
- 29.Garrett D., Powers R., Gronenborn A., Clore M. (1991) J. Magn. Reson. (1969) 95,214–220 [DOI] [PubMed] [Google Scholar]
- 30.Li X., Romero P., Rani M., Dunker A. K., Obradovic Z. (1999) Genome Inform. Ser. Workshop Genome Inform. 10,30–40 [PubMed] [Google Scholar]
- 31.Romero, Obradovic, Dunker K. (1997) Genome Inform. Ser. Workshop Genome Inform. 8,110–124 [PubMed] [Google Scholar]
- 32.Romero P., Obradovic Z., Li X., Garner E. C., Brown C. J., Dunker A. K. (2001) Proteins 42,38–48 [DOI] [PubMed] [Google Scholar]
- 33.Ohno H., Blackwell J., Jamieson A. M., Carrino D. A., Caplan A. I. (1986) Biochem. J. 235,553–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanford C., Kawahara K., Lapanje S., Hooker T. M., Jr., Zarlengo M. H., Salahuddin A., Aune K. C., Takagi T. (1967) J. Am. Chem. Soc. 89,5023–5029 [DOI] [PubMed] [Google Scholar]
- 35.Lobley A., Whitmore L., Wallace B. A. (2002) Bioinformatics 18,211–212 [DOI] [PubMed] [Google Scholar]
- 36.Böhm G., Muhr R., Jaenicke R. (1992) Protein Eng. 5,191–195 [DOI] [PubMed] [Google Scholar]
- 37.Sreerama N., Woody R. W. (1994) J. Mol. Biol. 242,497–507 [DOI] [PubMed] [Google Scholar]
- 38.Pervushin K., Riek R., Wider G., Wüthrich K. (1997) Proc. Natl. Acad. Sci. U.S.A. 94,12366–12371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schwarz-Linek U., Pilka E. S., Pickford A. R., Kim J. H., Höök M., Campbell I. D., Potts J. R. (2004) J. Biol. Chem. 279,39017–39025 [DOI] [PubMed] [Google Scholar]
- 40.Tompa P., Fuxreiter M. (2008) Trends Biochem. Sci. 33,2–8 [DOI] [PubMed] [Google Scholar]
- 41.Odermatt E., Engle J., Richter H., Hörmann H. (1982) J. Mol. Biol. 159,109–123 [DOI] [PubMed] [Google Scholar]
- 42.Stevens E. S., Morris E. R., Charlton J. A., Rees D. A. (1987) J. Mol. Biol. 197,743–745 [DOI] [PubMed] [Google Scholar]
- 43.Welsh E. J., Frangou S. A., Morris E. R., Rees D. A., Chavin S. I. (1983) Biopolymers 22,821–831 [DOI] [PubMed] [Google Scholar]
- 44.Choy H. A., Kelley M. M., Chen T. L., Møller A. K., Matsunaga J., Haake D. A. (2007) Infect. Immun. 75,2441–2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin Y. P., Lee D. W., McDonough S. P., Nicholson L. K., Sharma Y., Chang Y. F. (2009) J. Biol. Chem. 284,19380–19391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raibaud S., Schwarz-Linek U., Kim J. H., Jenkins H. T., Baines E. R., Gurusiddappa S., Höök M., Potts J. R. (2005) J. Biol. Chem. 280,18803–18809 [DOI] [PubMed] [Google Scholar]
- 47.Eigen M., Hammes G. G. (1963) Adv. Enzymol. Relat. Areas Mol. Biol. 25,1–38 [DOI] [PubMed] [Google Scholar]
- 48.Berezov A., Zhang H. T., Greene M. I., Murali R. (2001) J. Med. Chem. 44,2565–2574 [DOI] [PubMed] [Google Scholar]
- 49.Copeland R. A., Pompliano D. L., Meek T. D. (2006) Nat. Rev. Drug Discov. 5,730–739 [DOI] [PubMed] [Google Scholar]
- 50.Dyson H. J., Wright P. E. (2005) Nat. Rev. Mol. Cell Biol. 6,197–208 [DOI] [PubMed] [Google Scholar]
- 51.Dunker A. K., Lawson J. D., Brown C. J., Williams R. M., Romero P., Oh J. S., Oldfield C. J., Campen A. M., Ratliff C. M., Hipps K. W., Ausio J., Nissen M. S., Reeves R., Kang C., Kissinger C. R., Bailey R. W., Griswold M. D., Chiu W., Garner E. C., Obradovic Z. (2001) J. Mol. Graph. Model. 19,26–59 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.