

Abstract
Lymphangioleiomyomatosis (LAM) is a potentially fatal lung disease characterized by nodules of proliferative smooth muscle-like cells. The exact nature of these LAM cells and their proliferative stimuli are poorly characterized. Herein we report the novel findings that the lymphangiogenic vascular endothelial growth factors (VEGF) C and D induce LAM cell proliferation through activation of their cognate receptor VEGF-R3 and activation of the signaling intermediates Akt/mTOR/S6. Furthermore, we identify expression of the proteoglycan NG2, a marker of immature smooth muscle cells, as a characteristic of LAM cells both in vitro and in human lung tissue. VEGF-C-induced LAM cell proliferation was in part a result of autocrine stimulation that resulted from cross talk with lymphatic endothelial cells. Ultimately, these findings identify the lymphangiogenic VEGF proteins as pathogenic growth factors in LAM disease and at the same time provide a novel pharmacotherapeutic target for a lung disease that to date has no known effective treatment.
The understanding of lymphatic biology has expanded rapidly with the discovery of the lymphangiogenic proteins vascular endothelial growth factors (VEGF) C and D and their cognate receptor VEGF-R3.1,2 They are members of the VEGF family of growth factors, having 40% homology with VEGF-A, and their importance in lymph vasculature biology is well demonstrated by the finding that VEGF-C null mice die as a result of abnormal lymphatic development.3 Increasingly it has become clear that the biological role of these VEGF proteins have expanded beyond effects on the lymphatic endothelium alone to include the peri-lymphatic milieu and the investing immature vascular smooth muscle or pericytes.4 Furthermore, they have now been implicated in both pathogenic and non-pathogenic human processes including cancer growth and metastasis, wound healing, and immune regulation.
The lymphatic endothelial cell (LEC) is distinct from blood vessel endothelium with respect to their biological function, structure, and protein repertoire.5 As such they represent a unique cell that may play a distinctive role in the pathogenesis of human disease. Like blood vessel endothelium they share their basement membrane with surrounding immature smooth muscle cells called pericytes. It is clear that along with VEGF, pericyte-endothelial cell interactions play an important role in modulating vascular biology.4
Until recently the lymphatic vasculature has been considered an innocent bystander in the pathogenesis of pulmonary disease. Historically, one such disease has been pulmonary lymphangioleiomyomatosis (LAM), a progressive and fatal lung disease that almost exclusively affects women in their reproductive years.6,7 Pathologically, LAM is characterized by proliferation of abnormal smooth muscle-like cells that form lung nodules distributed in a peri-lymphatic manner. The etiology of LAM has been linked to mutations in the TSC2 gene; however, the nature and site-of-origin of LAM cells remains speculative.8 Based on the LAM cells proximity to lymphatic vessels, we hypothesized that the LEC and lymphangiogenic proteins VEGF-C and/or -D might play a role in their proliferation; and that these smooth muscle- like LAM cells represented perivascular mural cells that were proliferating through a mechanism involving LEC cross talk.
Using both in vitro primary LAM-derived cells (LDC)9 and immunohistochemistry of human lung LAM tissues, we report that VEGF-C and -D induce LDC proliferation through activation of their cognate receptor VEGF-R3 and subsequently phosphatidylinositol 3-kinase (PI3K)/mTOR/S6 signaling. Of note, this VEGF-R3 proliferative signaling pathway did not appear to be inhibited by functional tuberin. Furthermore, LDC proliferation was induced by cross talk between LEC and LDC and mediated through LDC production of VEGF-C. Taken together, these results identify the lymphangiogenic VEGF proteins as novel pathogenic growth factors in LAM disease and the lymphatic endothelium as a modifying factor in LAM cell proliferation.
Materials and Methods
Chemicals and Reagents
Recombinant human VEGF-C, -D and mutated VEGF-C (Cys156Ser, a VEGF-R3 specific agonist), platelet-derived growth factor (PDGF), non-conjugated or biotin conjugated antibodies to VEGF-R1, VEGF-R2 or VEGF-R3, respectively, and the VEGF-C/D-binding chimeric protein VEGF-R3/Fc (human VEGF-R3 extracellular domain fused to the carboxy-terminal Fc region of human IgG1) were purchased from R&D Systems (Minneapolis, MN). The VEGF-R3 inhibitor, MAZ-51 was purchased from Alexis Biochemicals (San Diego, CA). Wortmannin, rapamycin, and antibodies to β-actin and α-smooth muscle actin were purchased from Sigma (St. Louis, MO). Monoclonal antibody to VEGF-R3 (M-20) was purchased from Santa Cruz Biotechnology, (Santa Cruz, CA). Antibodies to total and phospho-mTOR (ser 2448), Akt, phospho-Akt (ser473), S6-ribosomal protein, phospho-S6 (ser 235/236) (2F9), P70S6 Kinase, phospho-p70 S6 kinase (Thr389), ERK-2, phospho-ERK (Thr202/Tyr204) (D13.14), Tuberin TSC2, and Hamartin TSC1 were obtained from Cell Signaling Technologies (Danvers, MA). Antibodies to Prox-1, NG2, and phospho-tyrosine (4G10) were purchased from Millipore-Upstate (Lake Placid, NJ). Phycoerythrin and streptavidin conjugated goat anti-mouse secondary antibodies were purchased from Jackson Immunoresearch Laboratories (West Grove, PA).
Cells and Cell Culture
Primary LAM-derived smooth muscle-like cells (clone 5/50 and 12/89) 9 passages 3 to 10 were grown in DMEM/F12 medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; Mediatech, Herndon, VA), and constituents as previously described.10 These cells have been well characterized and were derived from nodules of separate patients with LAM that express tuberin but have one allele of the TSC2 gene that is mutated. All presented data are from either of the two LDC clones and are representative of the similar findings obtained by using either clone.
Primary adult human microvascular endothelial cells and pulmonary arterial smooth muscle cells (PASM) were obtained from Cambrex (East Rutherford, NJ) and grown in cell specific growth factor-supplemented nutrient media as per the manufacturer’s instructions (Cambrex, EBM-2). The microvascular endothelial cells all expressed the LEC marker, Prox-1,11 and thus served as a model of LEC for co-culture experiments.
For expression of wt-TSC2, LDC were seeded in six well plates at 2 × 105 per well and grown in full growth media until 70% to 80% confluent. Cells were then transfected with lentivirus carrying green fluorescent protein tagged wt-TSC2 (human) or the GFP alone (kind gifts of V. Krymskaya), by using methods as previously described.10 Forty-eight hours post-transfection, cells were serum starved for 24 to 48 hours before all experiments; growth factor stimulation at 200 ng/ml for time intervals as indicated, in the presence or absence of various inhibitors.
Immunoblotting and Immunoprecipitation
LAM cells (2 × 105) were grown in six well-plates until 70% confluent, serum starved for 24 hours, and lysates were obtained after stimulation with VEGF-C (200 ng/ml) in the absence or presence of various inhibitors (200 nmol/L Wortmannin, 200 nmol/L Rapamycin or 10 μmol/L of MAZ51) for different time intervals as indicated. Cells were rinsed twice with ice-cold PBS/sodium orthovanadate (Na3VO4, 10 mmol/L) and then lysed with radioimmunoprecipitation assay buffer (20 mmol/L Tris pH-7.4; 150 mmol/L NaCl; 5 mmol/L EDTA; 1% Triton-X 100; 0.5% sodium deoxycholate; 10% glycerol; 25 mmol/L NaF; 1 mmol/L phenylmethylsulfonyl fluoride; and 1 mmol/L Na3VO4) with phosphatase and protease inhibitors (Sigma). Lysate protein concentration was measured by using the Bio-rad protein assay kit (Bio-Rad Laboratories, Hercules, CA) with bovine serum albumin (BSA) as standard. Lysate proteins (10 to 15 μg) were separated by SDS-polyacrylamide gel electrophoresis under reducing conditions, transferred to a polyvinylidene difluoride membrane (Immobilon-P, Millipore, Billerica, MA). The membrane was blocked in 5% nonfat dry milk, and then probed with appropriate specific primary antibodies followed by horseradish peroxidase-conjugated secondary antibodies and signals were detected by using enhanced chemiluminescence method (Amersham, Piscataway, NJ).
For immunoprecipitation of VEGF-R3, LDC were stimulated with VEGF-C or VEGF-D (200 ng/ml) for 5 minutes and protein lysates were prepared by using the lysis buffer as described above. Pre-cleared lysates were incubated with 2 μg of anti-VEGF-R3 antibody bound to 30 μl of protein-A Sepharose beads (Amersham, Piscataway, NJ). The beads were washed, resuspended in Laemmli sample buffer, boiled at 95°C for 5 minutes, resolved on SDS-polyacrylamide gel electrophoresis under reducing conditions, proteins transferred to polyvinylidene difluoride membranes and immunoblotted with anti-phosphotyrosine antibody (4G10).
Flow Cytometry
Cultured cells were grown to 80% confluency, trypsinized, washed in PBS, resuspended in PBS at a density of 5 × 105 cells/ml/200 μl volume, then incubated with either antibody isotype-matched control IgG (represented by a shaded profile) or appropriate primary antibodies (represented by an un-shaded profile) for 20 minutes at 4°C and secondary antibody labeled with goat anti-mouse IgG conjugated with phycoerythrin or streptavidin for 15 minutes. Finally, labeled cells were suspended in PBS and analyzed on a FACSCalibur flow cytometer (Becton Dickinson FACSort, San Jose, CA).
Adhesion Assay
Assays were performed as previously described12 with some minor modifications. After coating 96-well microtiter plates (ICN, Linbro/Titertek, Aurora, OH) with VEGF-C or VEGF-D at 4°C overnight, wells were blocked with 3% BSA (Sigma) for 30 minutes at 37°C. After trypsinization, cells were incubated with or without appropriate antibodies (20 μg/ml) for 30 minutes on ice and 5 × 104 cells, suspended in serum free media, were seeded to each well. After 3 hours, adherent cells were fixed and stained with 1% formaldehyde, 0.5% crystal violet, 20% methanol for 30 minutes, and the number of adherent cells was evaluated by measuring absorbance at 595 nm in a microplate reader (SpectraMax 190, Molecular Devices, Sunnyvale, CA).
Migration Assay
Assays were performed as previously described12 by using 8 μm pore size polycarbonate Transwell inserts (Corning Costar, Cambridge, MA) coated with relevant ligand or 1% BSA as a binding control. Cells, 5 × 104, were incubated in the presence or absence of inhibitors for 30 minutes on ice and then seeded into the top chamber and 1% fetal calf serum (FCS) to the bottom chamber, to serve as a chemoattractant. Cells were allowed to migrate for 4 hours at 37°C. Cells that migrated and adhered to the bottom surface of the Transwell membrane were fixed, stained as per the manufacturers instructions (Diff-Quik, Dade Behring Inc, Newark, DE), and mounted onto glass slides. Cells were counted in 10 high power (×100) fields for each condition.
Proliferation Assay
Cell proliferation was determined by manual cell counting as well as by BrdU assay. For cell counting, trypsinized cells were resuspended in serum free medium (SFM) and 10 μl of cells were loaded into a hemacytometer. Twenty cell counting areas were assessed and the results were expressed as a mean ± SD. BrdU assays were performed by using the cell proliferation Biotrak enzyme-linked immunosorbent assay (ELISA) System (RPN 250, Amersham Biosciences, Piscataway, NJ) as per the manufacturer’s instructions. Briefly, after serum starvation over night, cells were treated with inhibitors or antibodies as indicated, and 5 × 104 cells/well were seeded in 96-well plates precoated with appropriate substrate. After incubation at 37°C for 24 hours, cells were resuspended in the presence of 10 μmol/L BrdU for 120 minutes, then washed, fixed, blocked, and incubated with horseradish peroxidase-conjugated antibody to detect incorporated BrdU. Proliferation was quantified by measuring absorbance at 450 nm in a microplate reader (SpectraMax 190, Molecular Devices).
Co-culture experiments were performed in Transwell culture wells. LDC and LEC were grown in 10 cm culture plates until 75% to 80% confluent, trypsinized, and 5 × 104 cells were seeded into separate Transwell plates; LDC on the upper chamber of the membrane was inserted in one plate and the LEC on the lower culture well was inserted in a separate Transwell plate. After 24 hours in full growth medium (FGM), cells were serum starved overnight and the Transwell insert with LDC was transferred to the Transwell with LEC and co-cultured for 24 hours in SFM. Subsequently, LDC were trypsinized, resuspended in BrdU labeling media, added to 96 well plates, incubated for 120 minutes, centrifuged, and optical density was measured as noted above. For comparison, similar experiments were performed with LDC alone grown on Transwell inserts in FGM and SFM. Experiments were performed at least 3 times and data are presented as mean ± SD.
Immunohistochemistry and Immunofluorescence Analysis
LDC were cultured on a 2-well Chamber Coverglass System (Nalgene Nunc International, Naperville, IL) for 24 to 48 hours and then fixed with 4% methanol-free formaldehyde and 0.5% Triton X-100-PBS. Following, blocking with 1% BSA, cells were stained with anti-smooth muscle α-actin 1:50 (Sigma), stained cells were subsequently analyzed by confocal microscopy (Zeiss/SM510, Thorwood, NY). Experiments were repeated four times and representative images reported. Institutional review board approval was obtained for all immunohistochemistry involving human lung tissue. Lung tissue specimens were analyzed from both patients who clinically had chylothoraces and those patients who did not. For the detection of NG2 and Prox-1, LAM tissue was formalin-fixed, paraffin-embedded, and after routine processing, sections that included preheated 1 mmol/L EDTA, pH8.0, antigen retrieval for 30 minutes were incubated in 1:200 NG2 or 1:100 Prox-1 antibodies for 30 minutes. Detection was completed by the use of a biotin-free polymer, Rabbit MACH3 (Biocare Medicals, Walnut Creek, CA) for 10 minutes. Nova Red (Vector Laboratories, Burlingame, CA) and modified Schmidts’s Hematoxylin counterstain was used as the chromogen and sections were mounted with a permanent mounting media.
VEGF-C ELISA
Supernatant from LEC or LDC grown in various conditions as indicated and in the presence or absence of specific growth factors (as a matrix coating the culture well) were collected and the concentration of soluble VEGF-C was determined by ELISA as per the manufacturers instructions (Zymed Laboratories, San Francisco, CA). Briefly, 100 μl of collected supernatant was added to a 96-well plate precoated with rabbit anti-human VEGF-C and incubated at 37°C for 1 hour. After washing wells with buffer solution, 100 μl of horseradish peroxidase-conjugated secondary antibody was added to each well and incubated at 4°C for 30 minutes, then 100 μl of tetramethylbenzidine substrate was added to each well and incubated for 10 minutes and absorbance was measured at 450 nm on a microplate reader.
Statistics
Data are presented as mean values ± SD from at least three separate experiments unless otherwise stated. All immunoblots were performed at least three times and representative examples are displayed.
Results
VEGF-C and VEGF-D Induce Human LDC Proliferation
Lung tissue from patients with LAM is characterized by nodules of proliferating smooth muscle-like cells in close proximity with lymphatic vessels. Since VEGF-C and -D are central mitogenic regulators of the peri-lymphatic milieu, we hypothesized that these growth factors played a role in LAM cell proliferation. To test this hypothesis in vitro, we used LDC, an established cell model of primary smooth muscle-like cells isolated from human lung LAM nodules.9,10 First, we confirmed these cells’ previously published characteristics and population uniformity; including the expression of desmin (Figure 1A, left), α-smooth muscle actin (Figure 1A, middle and right), TSC2 expression, and constitutive hyperphosphorylation of mTOR and S6 ribosomal protein (data not shown).
Figure 1.

VEGF-C and -D induce LDC cell proliferation. A: Primary LDC characteristics: expression of desmin (left) measured by flow cytometry (shaded area: fluorescence measurement by using isotype antibody; line: protein expression by using specific antibody) and α smooth muscle actin (original magnification: ×40, left; ×100, right) by immunocytochemistry. B: Adhesion assay by using primary human LDC plated on varying concentrations of VEGF-C or -D. After cell fixation and staining with crystal violet, absorbance at 595 nm was measured. C: Transwell LDC migration assay by using varying concentrations of VEGF-C or -D coating the membrane insert and 1% FCS as a chemotactic stimulus. The number of cells fixed to the undersurface of the membrane was counted in 10 hpf in 3 separate wells per condition. D: Proliferation of LDC measured as the extent of BrdU uptake in LDC grown for 24 hours on varying concentrations of growth factor as indicated. E: Proliferation assay of PASM as described above.
Next we tested the effect of VEGF-C/D on LDC adhesion and migration by using serum-starved LDC grown in wells coated with various concentrations of VEGF-C or -D. Figure 1, B and C, respectively, show that LDC adhere and migrate in a dose-dependent fashion when in contact with VEGF-C or -D substrate. Furthermore, VEGF-C and -D induced LDC proliferation (Figure 1D) in a dose-dependent fashion and to a similar extent as PDGF, an established proliferative stimulus.13 In contrast, normal primary human PASM did not proliferate in the presence of VEGF-C or -D (Figure 1E).
VEGF-C or -D Induced LDC Proliferation is Mediated by Activation of VEGF-R3
VEGF-R3 is the cognate receptor for VEGF-C and -D and after the binding of these proteins, the receptor heterodimerizes and undergoes autotyrosine phosphorylation. We investigated the extent of VEGF-R3 expression on LDC and PASM by fluorescence-activated cell sorting (FACS) analysis. Figure 2A shows that VEGF-R3 is expressed on LDC but not PASM. This finding is supported by previously published data14 showing strong expression of VEGF-R3 by immunohistochemistry in human LAM lung tissue. In contrast, neither VEGF-R1 nor -R2 were expressed on either LDC or PASM in vitro (data not shown). Of note, we analyzed other in vitro LAM cell-models that have been previously studied including ELT3 (rat uterine leiomyoma15) and human lung fibroblasts16 and found they did not express VEGF-R3 (data not shown).
Figure 2.

VEGF-C and -D induced LDC proliferation is mediated by VEGF-R3. A: Flow cytometry analysis of VEGF-R3 expression in primary human LDC (left) and PASM (right). The shaded area represents fluorescence measurement with isotype antibody, the line shows VEGF-R3 expression using specific antibody. B: Proliferation assay of LDC grown on either VEGF-C (diamonds) or VEGF-D (triangles) or PDGF (circles) in the presence or absence of MAZ-51 (10 μmol/L). C: Western blot analysis of lysates from LDC treated with VEGF-C or -D for 5 minutes in the presence or absence of VEGF-R3 inhibitor. Lysates were initially immunoprecipitated with VEGF-R3 antibody (R3-IP Ab) and then immunoblotted with the phospho-tyrosine antibody (4G10). Activated VEGF-R3 has 3 processed forms as demonstrated.
Because VEGF-R3 is the cognate receptor for VEGF-C and -D, we hypothesized that LAM cell proliferation induced by these growth factors might result from activation of VEGF-R3. Figure 2B shows that VEGF-C or -D induced dose-dependent proliferation of LDC, which was blocked in the presence of the indoline compound MAZ-51, which specifically blocks the tyrosine kinase activity of VEGF-R3.17 As expected, PDGF-induced LDC proliferation was not blocked by VEGR-R3 inhibition. Immunoblotting of immunoprecipitated VEGF-R3 from LDC lysates (Figure 2C) showed that in the presence of VEGF-C or -D, VEGF-R3 is phosphorylated (all three processed forms) and that R3 inhibitor blocks activation. In contrast, PASM cells showed no expression of VEGF-R3 and appropriately no receptor phosphorylation when stimulated with VEGF-C. These findings confirm that VEGF-C and -D induced LDC proliferation is at least, in part, a result of activation of their cognate receptor, VEGF-R3.
VEGF-C Induced LDC Proliferation is Transduced through PI3K/mTOR/S6 Activation
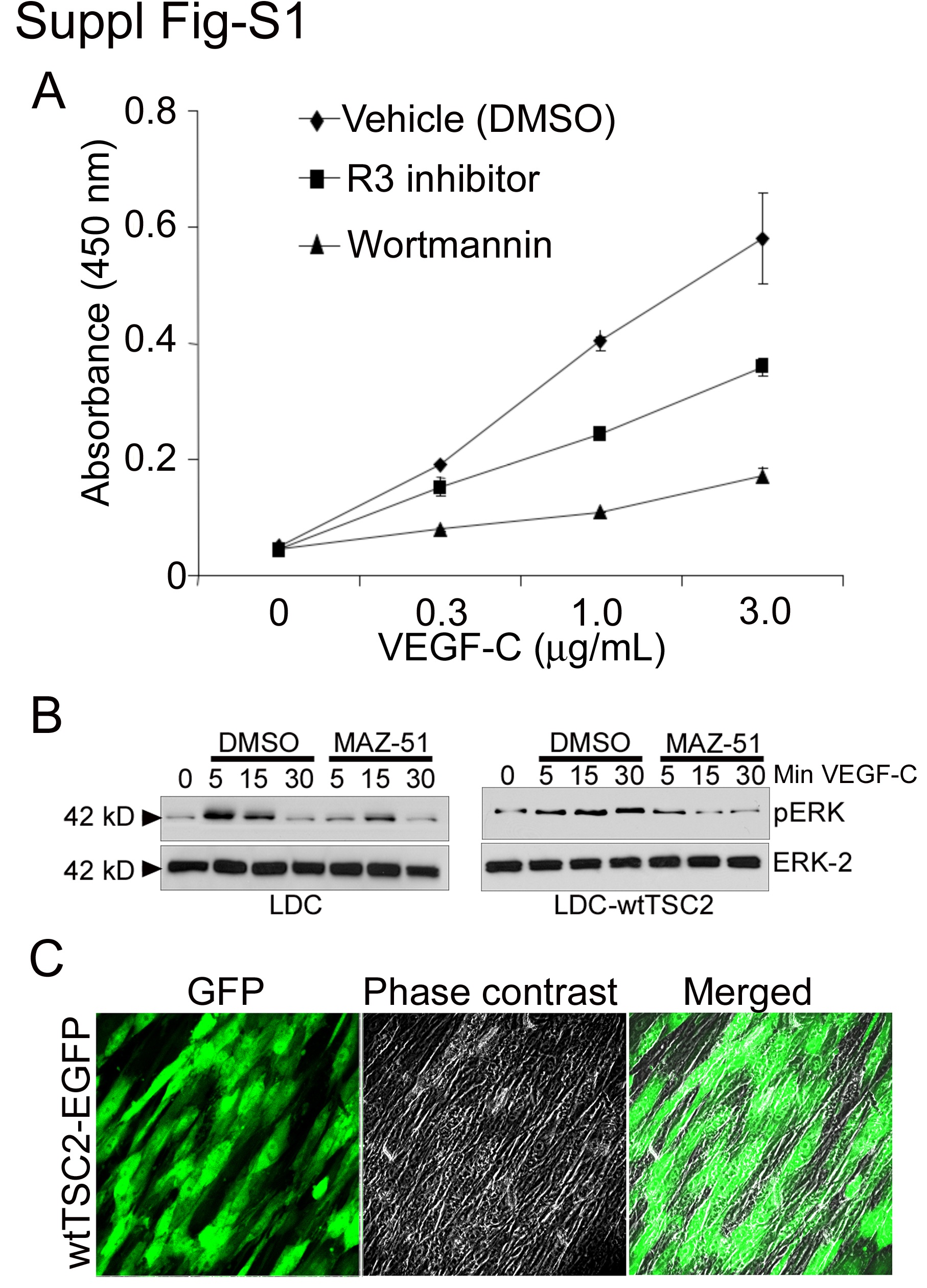
We next sought to determine the signaling intermediates that were activated after VEGF-R3 activation. Because wortmannin reduced VEGF-C or -D induced LDC proliferation (see Supplemental Figure S1A at http://ajp.amjpathol.org), we investigated whether PI3K/Akt-dependent activation of mTOR was involved.18 We performed Western blot analysis on protein lysates from LDC stimulated with VEGF-C (Cys156Ser), which is a mutated form of VEGF-C that specifically activates VEGF-R3.19 Figure 3A shows immunoblots of lysates from VEGF-C stimulated LDC in the absence (dimethyl sulfoxide) or presence of wortmannin (left), rapamycin (middle), and MAZ-51 (right). VEGF-C treatment of LDC induced phosphorylation of Akt, mTOR, S6 kinase (S6K), S6 ribosomal protein (Figure 3A), and Erk (see Supplemental Figure S1B at http://ajp.amjpathol.org) with maximal phosphorylation after 15 to 30 minutes. As expected, phosphorylation of these downstream signaling intermediates was inhibited by the pharmacological inhibitors wortmannin or rapamycin. Activation of these signaling intermediates was also inhibited by MAZ-51 suggesting they are involved in LDC-VEGF-R3 signaling. The ribosomal protein S6 has been shown to regulate LAM cell proliferation,9 and is found constitutively hyperphosphorylated, as a result of loss of TSC2 function in LAM cells. Because mTOR is an upstream activator of S6, through S6K, we next investigated the effect of VEGF-C on S6 activation in LDC. Similar to Akt and mTOR, VEGF-C induced phosphorylation of S6K and S6 at 15 to 30 minutes, which was inhibited by MAZ-51. Taken together these results suggest that VEGF-C, through activation of VEGF-R3, signals LDC proliferation at least in part through the Akt/mTOR/S6 pathway.
Figure 3.

VEGF-C induces PI3K/mTOR/S6 signaling. A: Western blots to detect phospho-Akt (pAkt), phospho-mTOR (p-mTOR), phospho-S6K (pS6K), and phospho-S6 (pS6) in lysates from LDC treated with the VEGF-R3 specific agonist VEGF-C (Cys156Ser), over time in the presence or absence of wortmannin (left), rapamycin (middle), or MAZ-51 (right); total Akt, mTOR, S6K, or S6 levels served as protein loading controls. B: Immunocytochemistry of LDCs labeled for pS6 in cells expressing only GFP (top) or GFP tagged wt-TSC2 (bottom). C: Western blots to detect pAkt, p-mTOR, pS6K, and pS6 in lysates from LDC expressing GFP tagged wt-TSC2 (left four lanes) or GFP alone (right four lanes) stimulated with VEGF-R3 specific agonist VEGF-C (Cys156Ser), over time; total Akt, mTOR, S6K, or S6 levels served as protein loading controls. Bottom two panels: Western blot for TSC2 expressing GFP tagged wt-TSC2 (left four lanes) or GFP alone (right four lanes), β-actin served as protein loading control. D: Western blots to detect pAkt, p-mTOR, and pS6 in lysates from LDC expressing GFP tagged wt-TSC2 stimulated with VEGF-R3 specific agonist VEGF-C (Cys156Ser) in the absence or presence of MAZ-51 over time; total Akt, mTOR, S6K, or S6 levels served as protein loading controls.
VEGF-C Signaling through Akt/mTOR is Minimally Modified by Functional TSC2
To determine whether functional tuberin altered VEGF-C/D induced LDC proliferation, we transfected LDC (see Supplemental Figure S1C at http://ajp.amjpathol.org), which express a mutant inactive form of tuberin, with wild-type TSC2 (wt-TSC2/EGFP, Figure 3B, bottom) or empty vector (EGFP, Figure 3B, top) and then determined phosphorylation of signaling proteins by immunoblot analysis. Figure 3B shows the characteristic hyperphosphorylation of S6 in LDC, which was diminished by wt-TSC2 expression, but not affected with transfection of EGFP alone.9,10 VEGF-C induced phosphorylation of Akt, mTOR, S6K, S6 (Figure 3C), and Erk MAP kinase (see Supplemental Figure S1B at http://ajp.amjpathol.org) is still apparent in the presence of functional TSC2. However, the extent and timing of maximal phosphorylation is altered. In addition, Figure 3D shows that VEGF-C induced activation of Akt, mTOR, or S6 in wt-TSC2 transfected LDC can still be inhibited by the VEGF-R3 inhibitor MAZ-51. Taken together these findings suggest that although the VEGF-C-induced mTOR proliferation signaling pathway appears to be modified by functional tuberin it is not significantly inhibited.
Human LAM Cell Nodules Express VEGF-R3 and NG2
It has previously been suggested that lymphangiogenesis is an etiological factor in LAM cell proliferation.14,20 To further investigate this and also to validate the importance of our in vitro findings, we performed immunohistochemistry of human LAM lung tissue by using antibodies to VEGF-R3 and Prox-1 on five separate lung tissue samples. Figure 4 shows representative pictures from two of these samples. Figure 4A demonstrates that there is expression of VEGF-R3 on cells within LAM nodules, which is more clearly demonstrated at a higher magnification (Figure 4B). This finding is consistent with previous reports.14 In contrast, lung tissue from the same sample that was not involved by nodular proliferation showed little expression of VEGF-R3 (Figure 4C). Furthermore, we did not find diffuse expression of the LEC specific marker, Prox-1, on LAM cells in lung nodules but rather discretely in endothelial cells (ECs) that lined lymphatic vessels (Figure 4D). These findings were true whether the LAM tissue was from patients with a clinical history of chylothoraces (data not shown). Taken together, our findings suggest that there is robust expression of VEGF-R3 in LAM cells intimately distributed next to lymph channels appropriately lined by Prox-1 positive LECs.
Figure 4.

VEGF-R3 is expressed in human LAM nodules. A: Photomicrograph of human LAM lung tissue stained for expression of VEGF-R3 (original magnification, ×20). B: Picture focuses on the LAM cells within the proliferative nodule outlined by the box in A (original magnification, ×100). C: Image of surrounding normal lung stained for VEGF-R3 (original magnification, ×40). D: Image of a LAM nodule surrounding a lymphatic vessel that is stained for Prox-1 (original magnification, ×100). Arrows indicate LECs that are Prox-1 positive.
Because Prox-1 negative LAM cells proliferate in close proximity to lymphatic vessels, we hypothesized that they may be immature smooth muscle cells, resembling pericytes. Using immunocytochemistry, Figure 5A (top, left) shows that in vitro, LDC abundantly express NG2 on their cell surface. This finding was confirmed by both flow cytometry (Figure 5A, bottom, right) and immunoblot of LDC lysates (Figure 5A, top, right). In contrast, expression of NG2 was not apparent in PASM measured by Western blot (top, right) or flow cytometry (bottom, left). To confirm these in vitro findings, we performed immunohistochemistry of human LAM lung tissue. Figure 5B shows significantly increased immunostaining with the pericyte marker NG2 in human lung LAM nodules (arrows and box). The nodule outlined by the box is displayed at higher magnification in Figure 5C. Robust staining of NG2 on cells within LAM nodules from two separate patients is demonstrated in Figure 5, D and E, respectively. There was no reactivity of isotype antibody (Figure 5F) or there was any NG2 staining in bronchiolar airway smooth muscle cells (Figure 5G), which are mature and therefore are not expected to express NG2. Taken together these results serve not only to further characterize markers of these unique LAM cells but also supports the notion that less-mature smooth muscle-like cells reside within LAM nodules distributed in a peri-lymphatic fashion.
Figure 5.

LDC and human LAM nodules express NG2. A: In vitro expression of NG2 in primary cultures human LDC determined by immunocytochemistry (top, left), Western blot (top, right), or flow cytometry (bottom, right). For comparison, expression of NG2 in normal PASM is also shown by Western blot (top, right) and flow cytometry (bottom, left). The shaded area in flow histogram represents isotype antibody; the line represents NG2 expression using specific antibody. B: Photomicrograph (original magnification, ×20; left) of human lung LAM nodules (arrow) stained for NG2, within otherwise normal appearing lung parenchyma. C: Image focuses on LAM cells (arrows) within the larger proliferative LAM nodule outlined by the box in B (original magnification, ×40). D and E: Image of LAM nodules from two separate patients, respectively (original magnification, ×100). F: Image of a LAM tissue nodule and surrounding normal lung stained with an isotype control antibody (original magnification, ×40). G: Image of unaffected lung tissue stained for NG2, from the same patient in B showing an airway and surrounding smooth muscle (indicated by the arrow) (original magnification, ×40).
Cross Talk with ECs Promotes LDC Proliferation through Autocrine VEGF-C Stimulation
Pericytes and ECs function through cross talk that can modify their respective biological functions, common extracellular environment, and ultimately vessel biology.4 Therefore, we hypothesized that LAM cell proliferation would be increased through interactions with LEC. First, we identified microvascular endothelial cells to serve as a model of LEC. Figure 6A (top) demonstrates that these ECs uniformly expressed the LEC-specific marker Prox-1. This finding was confirmed by Western blot (Figure 6A, bottom), which showed the presence of Prox-1 in these ECs but not LDC. We then used a Transwell culture system to determine whether a soluble factor was responsible for LDC proliferation when grown in co-culture (Co-Cx) with LEC. Using BrdU uptake (Figure 6B, top) and manual cell counting (Figure 6B, bottom) as measures of cell proliferation, we found that LDC grown in SFM in the presence of LEC had increased proliferation to an extent comparable with LDC alone, grown in FGM. This finding suggested that a soluble factor(s) was responsible for this proliferative response.
Figure 6.

Cross talk with endothelium increases LDC proliferation through autocrine stimulation by VEGF-C. A: Immunocytochemistry of primary microvascular endothelial cells (LEC) labeled to detect expression of Prox-1, a LEC-specific marker (top); immunoblot comparing Prox-1 expression in lysates from LEC and LDC (bottom). B: Proliferation assay, measuring BrdU uptake (top), or cell number counts (bottom) of LDC grown in Transwell plates in SFM (black bars) or FGM (stippled bars) or in co-culture with LEC in serum free conditions (Co-Cx, diagonal bars). C: Western blot for VEGF-C in lysates from LDC and LEC grown in serum free conditions (top). β-actin serves as a protein loading control; ELISA measuring VEGF-C concentration in supernatant from LDC or LEC grown in SFM (dark bars) or FGM (stippled bars) (bottom). D: ELISA measuring VEGF-C concentration in the SFM and FGM that used to grow LDC and LEC cells (first two columns); supernatant from LDC grown in either FGM (black bar) or SFM after stimulation with VEGF-C (stippled bar); or SFM after co-culture with LEC in the absence (diagonal bars) or presence (brick bars) of the VEGF-C “sponge,” VEGF-R3/Fc (R3/Fc). E: Proliferation assay measuring BrdU uptake, of LDC grown in SFM (black bars) or in co-culture with LEC in the absence (stippled bar) or presence (diagonal bar) of R3/Fc.
Because LDC have previously been shown to express VEGF-C,14 we hypothesized that this growth factor was one of the soluble stimulatory proteins. We showed by Western blot (Figure 6C, top) and ELISA (Figure 6C, bottom) that the primary LDC, but not LEC, used in our experiments expressed and secreted VEGF-C. Figure 6C (bottom) shows that when grown in the presence of SFM neither LDC nor LEC secreted VEGF-C. In contrast, when grown in FGM, LDC but not LEC were stimulated to secrete VEGF-C. Neither the SFM nor FGM used to grow these cells contained VEGF-C (Figure 6D). Furthermore, when LDC were grown on VEGF-C substrate (Figure 6D, stippled bars) or grown in Co-Cx with LEC (diagonal bars) they secreted levels of VEGF-C equivalent to that of LDC grown in FGM (black bars). Figure 6D also demonstrates that VEGF-R3/Fc, when added to co-cultured cells, effectively bound VEGF-C resulting in decreased levels in the Co-Cx supernatant. In separate experiments (Figure 6E), we found that Co-Cx promotes an increase in LDC proliferation measured by BrdU uptake (stippled bar) and cell counting (data not shown), which was significantly decreased in the presence of VEGF-R3/Fc (diagonal bar). Taken together these results suggest that LDC proliferation is mediated at least in part through cross talk with LEC and the resultant autocrine secretion of VEGF-C.
Discussion
LAM is a devastating disease, characterized by nodules of proliferative smooth muscle-like cells that progressively destroy lung parenchyma. The etiology of LAM cells and the molecular mechanisms underlying this lung disease remain elusive. In this report, we have identified a novel proliferative stimulus for LAM cells, namely the lymphangiogenic growth factors VEGF-C and -D.
VEGF-C and -D form a subgroup of the VEGF growth factor family, which can induce cell migration and proliferation through activation of their cognate receptor, VEGF-R3.21 Although initially described as activating ECs to induce lymphangiogenesis, increasingly the profile of VEGF-C/D’s cell expression, receptor activation, and cellular effects have diversified.22
We have shown that LDCs express VEGF-R3. This might appear to contradict previously reported findings,14,20 in which immunohistochemistry of human LAM tissue demonstrated LAM cell clusters that expressed VEGF-R3 on cells located only along the perimeter of the clusters but not within the cluster itself. This finding was interpreted as VEGF-R3-expressing LEC “coating” the LAM cell cluster after its fragmentation from parenchymal LAM nodules into lymphatics. However, in light of our findings, other interpretations are plausible. Namely, that the cells at the cluster periphery are not LEC but rather LAM cells that preferentially highly express VEGF-R3. As opposed to the centrally located cells the peripheral LAM cells are in immediate contact with the pericellular milieu and would require high receptor expression to bind ligands. Certainly this is the case for other biological proteins such as integrins and actin at the leading edge of migrating cells.23 This not withstanding, when taken together with Kumasaka et al.’s20 findings, our combined results describe a novel concept in LAM biology; the notion that there is an intimate relationship of LAM cells to the LEC. Our findings indicate for the first time that this cross talk is biologically important and involves autocrine VEGF-C activation of VEGF-R3.
There are a number of potential sources of VEGF-C and -D in LAM disease. First, it is clear from our study and others14 that the LAM cell itself produces VEGF, which can, as we have demonstrated, act in an autocrine fashion. The second potential source is the alveolar pneumocyte. Although the most common lung tissue abnormalities associated with LAM are proliferative smooth muscle cell nodules and cysts, there may also be changes of multifocal micronodular pneumocyte hyperplasia.24 In this setting the pneumocyte, which expresses VEGF-C and -D under normal conditions,1,25 may overexpress these growth factors and act as a paracrine stimulus for LAM cells. Finally, soluble VEGF produced at distant pathological sites may act as a proliferative stimulus. There is evidence to suggest that LAM represents a metastatic site of a systemic disorder and thus VEGF production by the “primary” site may act as a source of soluble VEGF.20,26 The study by Seyama et al,27 which showed that patients with LAM have elevated serum levels of VEGF-D, supports this possibility.
In comparison with VEGF-R2, the cognate receptor for VEGF-A, the signaling proteins involved in transducing cell proliferation after VEGF-R3 activation are not well characterized.28 In this study we have shown for the first time that VEGF-C and -D induce proliferation of primary LDC through activation of VEGF-R3 and subsequently the signaling intermediates Akt/mTOR/S6 and Erk (Figure 7). Of note, other receptor tyrosine kinases such as PDGF-R also transduce their effects on LAM cells through PI3K/Akt/mTOR suggesting that this proliferative signaling mechanism is part of a common final pathway for growth factor-induced LAM cell proliferation.8 This is supported by recent clinical trials29 demonstrating regression of angiomyolipomas in patients treated with the mTOR inhibitor, sirolimus (rapamycin). The pro-proliferative ribosomal protein S6 is activated by mTOR and is typically under the inhibitory control of TSC2/TSC1 complex. In the case of LAM, TSC2 is mutated, reversing its inhibitory effect and resulting in S6 hyperphosphorylation and increased cell proliferation. Our results suggest that in the case of VEGF-R3 signaling, TSC2 does not appear to be entirely inhibitory but rather acts in a modulatory fashion affecting the time course and extent of activation of signaling proteins. This TSC2-“bypass” does not appear to be unique to VEGF because other growth factors have been reported to signal in a similar fashion.13
Figure 7.

Proposed VEGF-C/D cell signaling pathway to transduce LAM cell proliferation. After VEGF-R3 binding and activation, Akt is phosphorylated and as is in turn mTOR, which by itself or through S6, induces cell proliferation. TSC2 does not appear to have a significant inhibitory effect on VEGF-R3 signaling but does play a modulatory role, and Akt may also have a direct modulatory effect on TSC2. Erk is also used by VEGF-R3 to induce LAM proliferation. LDC-LEC cross talk serves as an activating mechanism for LAM-VEGF-R3 through autocrine production of VEGF-C/D. The potential mechanisms of LDC-LEC cross talk resulting in release of VEGF-C from LDC include soluble factor production and/or cell-cell contact.
The origin and nature of the LAM cell is poorly understood but is characterized as smooth muscle like. Consequently, there is no well-established in vitro cell model. As a result, a number of cell types, each with their own inherent limitations, have been used including transfected fibroblasts,16 renal angiomyolipoma cells,30 and cells derived from Eker rat uterine leiomyomas.15 The limitations of primary human LAM cells used in our studies include the following: first, like all tissue derived cells, in vivo characteristics are lost both as a result of the isolation procedure and through variable differentiation over time in culture; second, in vitro cell protein expression may differ from that in vivo and in the case of NG2, may be misleading since it can be expressed in some non-pericyte cells.4
This not withstanding, a number of points suggest that our findings may be relevant and applicable to the in vivo biology of LAM. First, these primary LAM cells are species (human) and organ-origin (lung) specific and relevant. Second, they were the only cell types of the previously used LAM cell models (noted above) that expressed VEGF-R3, a protein consistently shown to be present in human LAM lung. Third, LAM cells were derived from human LAM tissue nodules whose peri-vascular distribution supports the notion that LAM cell proliferation may be a result of LEC cross talk. Fourth, similar to VEGF-R3, expression of NG2 was found both in vitro and in vivo. Fifth, there is some evidence that other members of the perivascular epithelioid cell tumor family (such as angiomyolipomas [AML], lesions often co-existent with LAM) may be of pericyte origin.31 Although separately none of these points are definitive, taken together they suggest that LAM cells may be vascular mural smooth muscle cells that undergo abnormal proliferation; and, that at least in part the proliferative stimulus might arise from LEC cross talk.
Pericyte-endothelial cell cross talk is increasingly being recognized as playing an essential role in static and dynamic vessel function.32 We found for the first time that through EC cross talk LDC proliferation is increased in an autocrine VEGF-C-dependent manner (Figure 7). The soluble factor(s) that induce VEGF-C secretion from LDC is unknown at this time. Furthermore, whether this factor originates from the LDC or LEC or both remains to be determined. Our results are consistent with previous findings where secretion of growth factors, including VEGF, has been demonstrated in other pericyte-EC co-cultures.33 Cross talk can also be transduced through, direct cell-cell contact involving cadherins34 and presumably, this may also be playing a role in LAM disease. Recently, others have shown that matrix modulators such as matrix metalloproteases,16,35 also play a role in LAM pathogenesis. In the case of VEGF-C and -D further studies are required to investigate these other potential mechanisms in the context of autocrine VEGF stimulation. Recent work also shows that LAM cells within body fluids express VEGF-R3, which presumably gain access to the various body compartments through lymphatic or blood vessel infiltration.20 Whether LAM cells are definitively pericytes remains to be determined; however, our results do serve to further characterize these poorly understood and unique cells.
Previous studies have shown that LAM lung disease may occur sporadically or in association with tuberous sclerosis complex (TSC), an autosomal dominant disorder characterized by multiorgan hamartomas. Approximately 40% of patients with TSC who have no pulmonary symptoms, have been found to have lung parenchymal abnormalities consistent with LAM.36,37 In contrast, all of the patients with TSC who are found to have lung disease also have renal AML.36 These findings support the hypothesis that LAM nodules are a result of distant metastases from the AML lesions. However, only 60% of sporadic LAM cases have co-incident AML, challenging this hypothesis.38,39,40 It is important to consider though that both AML and LAM lesions are classified within the broad category of perivascular epithelioid cell tumors. These lesions are characterized by their peri-vascular location, positive desmin, and HMB-45 staining (common to LAM cells) and their ability, albeit it variably, to metastasize to various tissue sites. Thus, one might speculate that sporadic LAM represents simply a primary lung PEComa, separate from clear cell “sugar” tumor, which may metastasize at a frequency that differs from that of AML. Soluble and matrix-associated growth factors, such as VEGF, would have their proliferative effect both locally and in distant tissue sites. The evidence from the literature is that the growth factors implicated in LAM are extensive,13,41,42 but the combination of our in vitro and in vivo findings suggests that the lymphangiogenic VEGF proteins play an important role. In this context and at this time, the role of TSC mutations, and their protein products hamartin and tuberin in VEGF-C and -D-induced LAM cell proliferation is unclear.
In conclusion, we have identified a novel proliferative stimulus for LDC, namely the lymphangiogenic growth factors VEGF-C and -D. Primary lung LDC adhere, migrate, and proliferate in the presence of both growth factors through activation of their cognate receptor, VEGF-R3. LDC appear to have an immature smooth muscle cell phenotype (NG2 positive) suggesting they are perivascular mural cells and through cross talk with LECs are stimulated to produce VEGF-C, which exerts its proliferative effect in an autocrine manner. After activation of VEGF-R3, the proliferative effect of VEGF-C and -D is transduced through the intermediate signaling proteins PI3K/Akt/mTOR and subsequently S6. Taken together these results suggest a novel pathogenic mechanism for LAM through the lymphangiogenic proteins VEGF-C and -D and identify VEGF-R3 as a novel potential pharmacotherapeutic target.
Supplementary Material
Acknowledgments
We thank Dr. Vera P. Krymskaya (University of Pennsylvania) for providing primary LDCs and lentivirus to express GFP-wtTSC2, and Ms. Linda Murphy and Dr. Wilma L. Lingle (Mayo Clinic, Rochester) for helping us with immunohistochemistry.
Footnotes
Address reprint requests to Nicholas E. Vlahakis, M.D., Thoracic Disease Research Unit, Mayo Clinic College of Medicine, 200 First St. SW, Rochester, MN 55905. E-mail: vlahakis.nicholas@mayo.edu.
Supported by a LAM Foundation grant (N.E.V.), an American Lung Association Research grant RG-1018-N (N.E.V.), a Brewer Foundation grant (N.E.V.) and the Mayo Foundation.
R.B.I. and S.O. contributed equally to this work.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
References
- Alitalo K, Carmeliet P. Molecular mechanisms of lymphangiogenesis in health and disease. Cancer Cell. 2002;1:219–227. doi: 10.1016/s1535-6108(02)00051-x. [DOI] [PubMed] [Google Scholar]
- Jussila L, Alitalo K. Vascular growth factors and lymphangiogenesis. Physiol Rev. 2002;82:673–700. doi: 10.1152/physrev.00005.2002. [DOI] [PubMed] [Google Scholar]
- Karkkainen MJ, Haiko P, Sainio K, Partanen J, Taipale J, Petrova TV, Jeltsch M, Jackson DG, Talikka M, Rauvala H, Betsholtz C, Alitalo K. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat Immunol. 2004;5:74–80. doi: 10.1038/ni1013. [DOI] [PubMed] [Google Scholar]
- Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res. 2005;97:512–523. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- Podgrabinska S, Braun P, Velasco P, Kloos B, Pepper MS, Skobe M. Molecular characterization of lymphatic endothelial cells, Proc Natl Acad Sci USA. 2002;99:16069–16074. doi: 10.1073/pnas.242401399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glassberg MK. Lymphangioleiomyomatosis. Clin Chest Med. 2004;25:573–582, vii. doi: 10.1016/j.ccm.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Ryu JH, Moss J, Beck GJ, Lee JC, Brown KK, Chapman JT, Finlay GA, Olson EJ, Ruoss SJ, Maurer JR, Raffin TA, Peavy HH, McCarthy K, Taveira-Dasilva A, McCormack FX, Avila NA, Decastro RM, Jacobs SS, Stylianou M, Fanburg BL. The NHLBI lymphangioleiomyomatosis registry: characteristics of 230 patients at enrollment. Am J Respir Crit Care Med. 2006;173:105–111. doi: 10.1164/rccm.200409-1298OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay G. The LAM cell: what is it, where does it come from, and why does it grow? Am J Physiol Lung Cell Mol Physiol. 2004;286:L690–L693. doi: 10.1152/ajplung.00311.2003. [DOI] [PubMed] [Google Scholar]
- Goncharova EA, Goncharov DA, Spaits M, Noonan DJ, Talovskaya E, Eszterhas A, Krymskaya VP. Abnormal growth of smooth muscle-like cells in lymphangioleiomyomatosis: role for tumor suppressor TSC2. Am J Respir Cell Mol Biol. 2006;34:561–572. doi: 10.1165/rcmb.2005-0300OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncharova EA, Goncharov DA, Eszterhas A, Hunter DS, Glassberg MK, Yeung RS, Walker CL, Noonan D, Kwiatkowski DJ, Chou MM, Panettieri RA, Jr, Krymskaya VP. Tuberin regulates p70 S6 kinase activation and ribosomal protein S6 phosphorylation: a role for the TSC2 tumor suppressor gene in pulmonary lymphangioleiomyomatosis (LAM). J Biol Chem. 2002;277:30958–30967. doi: 10.1074/jbc.M202678200. [DOI] [PubMed] [Google Scholar]
- Wigle JT, Harvey N, Detmar M, Lagutina I, Grosveld G, Gunn MD, Jackson DG, Oliver G. An essential role for Prox1 in the induction of the lymphatic endothelial cell phenotype. EMBO J. 2002;21:1505–1513. doi: 10.1093/emboj/21.7.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlahakis NE, Young BA, Atakilit A, Sheppard D. The lymphangiogenic vascular endothelial growth factors VEGF-C and -D are ligands for the integrin alpha9beta1. J Biol Chem. 2005;280:4544–4552. doi: 10.1074/jbc.M412816200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irani C, Goncharova EA, Hunter DS, Walker CL, Panettieri RA, Krymskaya VP. Phosphatidylinositol 3-kinase but not tuberin is required for PDGF-induced cell migration. Am J Physiol Lung Cell Mol Physiol. 2002;282:L854–L862. doi: 10.1152/ajplung.00291.2001. [DOI] [PubMed] [Google Scholar]
- Kumasaka T, Seyama K, Mitani K, Sato T, Souma S, Kondo T, Hayashi S, Minami M, Uekusa T, Fukuchi Y, Suda K. Lymphangiogenesis in lymphangioleiomyomatosis: its implication in the progression of lymphangioleiomyomatosis. Am J Surg Pathol. 2004;28:1007–1016. doi: 10.1097/01.pas.0000126859.70814.6d. [DOI] [PubMed] [Google Scholar]
- Astrinidis A, Cash TP, Hunter DS, Walker CL, Chernoff J, Henske EP. Tuberin, the tuberous sclerosis complex 2 tumor suppressor gene product, regulates Rho activation, cell adhesion and migration. Oncogene. 2002;21:8470–8476. doi: 10.1038/sj.onc.1205962. [DOI] [PubMed] [Google Scholar]
- Zhe X, Yang Y, Jakkaraju S, Schuger L. Tissue inhibitor of metalloproteinase-3 downregulation in lymphangioleiomyomatosis: potential consequence of abnormal serum response factor expression. Am J Respir Cell Mol Biol. 2003;28:504–511. doi: 10.1165/rcmb.2002-0124OC. [DOI] [PubMed] [Google Scholar]
- Kirkin V, Mazitschek R, Krishnan J, Steffen A, Waltenberger J, Pepper MS, Giannis A, Sleeman JP. Characterization of indolinones which preferentially inhibit VEGF-C- and VEGF-D-induced activation of VEGFR-3 rather than VEGFR-2. Eur J Biochem. 2001;268:5530–5540. doi: 10.1046/j.1432-1033.2001.02476.x. [DOI] [PubMed] [Google Scholar]
- Tee AR, Fingar DC, Manning BD, Kwiatkowski DJ, Cantley LC, Blenis J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci USA. 2002;99:13571–13576. doi: 10.1073/pnas.202476899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joukov V, Kumar V, Sorsa T, Arighi E, Weich H, Saksela O, Alitalo K. A recombinant mutant vascular endothelial growth factor-C that has lost vascular endothelial growth factor receptor-2 binding, activation, and vascular permeability activities. J Biol Chem. 1998;273:6599–6602. doi: 10.1074/jbc.273.12.6599. [DOI] [PubMed] [Google Scholar]
- Kumasaka T, Seyama K, Mitani K, Souma S, Kashiwagi S, Hebisawa A, Sato T, Kubo H, Gomi K, Shibuya K, Fukuchi Y, Suda K. Lymphangiogenesis-mediated shedding of LAM cell clusters as a mechanism for dissemination in lymphangioleiomyomatosis. Am J Surg Pathol. 2005;29:1356–1366. doi: 10.1097/01.pas.0000172192.25295.45. [DOI] [PubMed] [Google Scholar]
- Otrock ZK, Makarem JA, Shamseddine AI. Vascular endothelial growth factor family of ligands and receptors: review. Blood Cells Mol Dis. 2007;38:258–268. doi: 10.1016/j.bcmd.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Tammela T, Enholm B, Alitalo K, Paavonen K. The biology of vascular endothelial growth factors. Cardiovasc Res. 2005;65:550–563. doi: 10.1016/j.cardiores.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Vicente-Manzanares M, Choi CK, Horwitz AR. Integrins in cell migration–the actin connection. J Cell Sci. 2009;122:199–206. doi: 10.1242/jcs.018564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui K, W KR, Hilbert SL, Yu ZX, Takeda K, Travis WD, Moss J, Ferrans VJ. Hyperplasia of type II pneumocytes in pulmonary lymphangioleiomyomatosis. Arch Pathol Lab Med. 2000;124:1642–1648. doi: 10.5858/2000-124-1642-HOTIPI. [DOI] [PubMed] [Google Scholar]
- Baluk P, Tammela T, Ator E, Lyubynska N, Achen MG, Hicklin DJ, Jeltsch M, Petrova TV, Pytowski B, Stacker SA, Yla-Herttuala S, Jackson DG, Alitalo K, McDonald DM. Pathogenesis of persistent lymphatic vessel hyperplasia in chronic airway inflammation. J Clin Invest. 2005;115:247–257. doi: 10.1172/JCI22037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks DM, Pacheco-Rodriguez G, DeCastro RM, McCoy JP, Jr, Wang JA, Kumaki F, Darling T, Moss J. Molecular and genetic analysis of disseminated neoplastic cells in lymphangioleiomyomatosis. Proc Natl Acad Sci USA. 2004;101:17462–17467. doi: 10.1073/pnas.0407971101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyama K, Kumasaka T, Souma S, Sato T, Kurihara M, Mitani K, Tominaga S, Fukuchi Y. Vascular endothelial growth factor-D is increased in serum of patients with lymphangioleiomyomatosis. Lymphat Res Biol. 2006;4:143–152. doi: 10.1089/lrb.2006.4.143. [DOI] [PubMed] [Google Scholar]
- Cross MJ, Dixelius J, Matsumoto T, Claesson-Welsh L. VEGF-receptor signal transduction. Trends Biochem Sci. 2003;28:488–494. doi: 10.1016/S0968-0004(03)00193-2. [DOI] [PubMed] [Google Scholar]
- Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, Schmithorst VJ, Laor T, Brody AS, Bean J, Salisbury S, Franz DN. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358:140–151. doi: 10.1056/NEJMoa063564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Astrinidis A, Howard S, Henske EP. Estradiol and tamoxifen stimulate LAM-associated angiomyolipoma cell growth and activate both genomic and nongenomic signaling pathways. Am J Physiol Lung Cell Mol Physiol. 2004;286:L694–L700. doi: 10.1152/ajplung.00204.2003. [DOI] [PubMed] [Google Scholar]
- Lantuejoul S, Isaac S, Pinel N, Negoescu A, Guibert B, Brambilla E. Clear cell tumor of the lung: an immunohistochemical and ultrastructural study supporting a pericytic differentiation. Mod Pathol. 1997;10:1001–1008. [PubMed] [Google Scholar]
- Gerhardt H, Betsholtz C. Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res. 2003;314:15–23. doi: 10.1007/s00441-003-0745-x. [DOI] [PubMed] [Google Scholar]
- Tallquist MD, Klinghoffer RA, Heuchel R, Mueting-Nelsen PF, Corrin PD, Heldin CH, Johnson RJ, Soriano P. Retention of PDGFR-beta function in mice in the absence of phosphatidylinositol 3′-kinase and phospholipase Cgamma signaling pathways. Genes Dev. 2000;14:3179–3190. doi: 10.1101/gad.844700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhardt H, Wolburg H, Redies C. N-cadherin mediates pericytic-endothelial interaction during brain angiogenesis in the chicken. Dev Dyn. 2000;218:472–479. doi: 10.1002/1097-0177(200007)218:3<472::AID-DVDY1008>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Fleming MV, Stetler-Stevenson WG, Liotta LA, Moss J, Ferrans VJ, Travis WD. Immunohistochemical study of matrix metalloproteinases (MMPs) and their tissue inhibitors (TIMPs) in pulmonary lymphangioleiomyomatosis (LAM). Hum Pathol. 1997;28:1071–1078. doi: 10.1016/s0046-8177(97)90061-7. [DOI] [PubMed] [Google Scholar]
- Costello LC, Hartman TE, Ryu JH. High frequency of pulmonary lymphangioleiomyomatosis in women with tuberous sclerosis complex. Mayo Clin Proc. 2000;75:591–594. doi: 10.4065/75.6.591. [DOI] [PubMed] [Google Scholar]
- Moss J, Avila NA, Barnes PM, Litzenberger RA, Bechtle J, Brooks PG, Hedin CJ, Hunsberger S, Kristof AS. Prevalence and clinical characteristics of lymphangioleiomyomatosis (LAM) in patients with tuberous sclerosis complex. Am J Respir Crit Care Med. 2001;164:669–671. doi: 10.1164/ajrccm.164.4.2101154. [DOI] [PubMed] [Google Scholar]
- Bernstein SM, Newell JD, Jr, Adamczyk D, Mortenson RL, King TE, Jr, Lynch DA. How common are renal angiomyolipomas in patients with pulmonary lymphangiomyomatosis? Am J Respir Crit Care Med. 1995;152:2138–2143. doi: 10.1164/ajrccm.152.6.8520787. [DOI] [PubMed] [Google Scholar]
- Chu SC, Horiba K, Usuki J, Avila NA, Chen CC, Travis WD, Ferrans VJ, Moss J. Comprehensive evaluation of 35 patients with lymphangioleiomyomatosis. Chest. 1999;115:1041–1052. doi: 10.1378/chest.115.4.1041. [DOI] [PubMed] [Google Scholar]
- Kerr LA, Blute ML, Ryu JH, Swensen SJ, Malek RS. Renal angiomyolipoma in association with pulmonary lymphangioleiomyomatosis: forme fruste of tuberous sclerosis? Urology. 1993;41:440–444. doi: 10.1016/0090-4295(93)90504-4. [DOI] [PubMed] [Google Scholar]
- Evans SE, Colby TV, Ryu JH, Limper AH. Transforming growth factor-beta 1 and extracellular matrix-associated fibronectin expression in pulmonary lymphangioleiomyomatosis. Chest. 2004;125:1063–1070. doi: 10.1378/chest.125.3.1063. [DOI] [PubMed] [Google Scholar]
- Inoue Y, King TE, Jr, Barker E, Daniloff E, Newman LS. Basic fibroblast growth factor and its receptors in idiopathic pulmonary fibrosis and lymphangioleiomyomatosis. Am J Respir Crit Care Med. 2002;166:765–773. doi: 10.1164/rccm.2010014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}