

Abstract
Polyvalent interactions allow biological structures to exploit low-affinity ligand–receptor binding events to affect physiological responses. We describe here the use of bacteriophage Qβ as a multivalent platform for the display of polycationic motifs that act as heparin antagonists. Point mutations to the coat protein allowed us to generate capsids bearing the K16M, T18R, N10R, or D14R mutations; because 180 coat proteins form the capsid, the mutants provide a spectrum of particles differing in surface charge by as much as +540 units (K16M vs. D14R). Whereas larger poly-Arg insertions (for example, C-terminal Arg8) did not yield intact virions, it was possible to append chemically synthesized oligo-Arg peptides to stable wild-type (WT) and K16M platforms. Heparin antagonism by the particles was evaluated by using the activated partial thrombin time (aPTT) clotting assay; this revealed that T18R, D14R, and WT-(R8G2)95 were the most effective at disrupting heparin-mediated anticoagulation (>95 % inhibition). This activity agreed with measurements of ζ potential (ZP) and retention time on cation exchange chromatography for the genetic constructs, which distribute their added positive charge over the capsid surface (+180 and +360 for T18R and D14R relative to WT). The potent activity of WT-(R8G2)95, despite its relatively diminished overall surface charge is likely a consequence of the particle’s presentation of locally concentrated regions with high positive charge density that interact with heparin’s extensively sulfated domains. The engineered cationic capsids retained their ability to inhibit heparin at high concentrations and showed no anticlotting activity of the kind that limits the utility of antiheparin polycationic agents that are currently in clinical use.
Keywords: bacteriophage Q beta, genetic engineering, hematologic agents, heparin, polycations, viruses
Introduction
Polyvalent interactions are essential for triggering numerous biological functions at all levels.[1, 2] Cell–cell communication, antibody–antigen recognition, and cellular uptake[3] are a few examples in which multiple simultaneous ligand–receptor recognition events combine to initiate and effect subsequent biological cascades. The ability to manipulate these polyvalent interactions is a prerequisite for using the associated biological cascades in mechanistic studies, diagnostics, and/or therapeutic agents. Viruses have received much attention in this type of endeavor:[4,5] their polyvalent and proteinaceous nature, along with their ease of production and manipulation makes them attractive agents for targeting physiological function.[6, 7]
Bacteriophage Qβ is an icosahedral RNA virus comprised of 180 copies of a 14.1 kDa, 132 amino acid coat protein.[8] The virus is readily expressed and purified from E. coli in high yields and shows good stability to extremes of pH, temperature, and a variety of solvents.[9] Prior studies have shown that the coat protein can tolerate some genetic insertions,[10] and our own investigations have successfully introduced point mutants as well as unnatural amino acids.[11] We have also shown that various ligands can be chemically conjugated to solvent-exposed amines on the outer surface of the capsid[11, 12] with concomitant effects on in vivo behavior such as charge-induced changes in circulation lifetime.[13]
Here we describe the polyvalent display of cationic motifs on Qβ that results in particles that act as potent heparin antagonists. Heparin is a sulfated glycosaminoglycan composed of repeating D-glucosamine/L-uroninc acid disaccharide subunits arranged in linear chains.[14] With chain lengths of up to 100 disaccharides and an average of 2.7 sulfates per subunit, heparin is the most negatively charged biopolymer known. The proteoglycan heparan sulfate, a close relative of heparin, is present at many cell surfaces and participates in numerous and varied physiological events including inflammation,[15] cell–cell signalling,[16] metastasis,[17] and multiple aspects of developmental biology.[18] Heparin is used routinely as an anticoagulant because it stimulates inhibition of thrombin by antithrombin,[20] especially following medical procedures in which postoperative thrombosis is of concern.[19] However, complications such as uncontrolled bleeding from heparin overdose are widely known,[21] and are presumably exacerbated by heparin’s heterogeneity and variations in individual responses.[22] These significant concerns continue to fuel research into novel well-defined compounds with specific antiheparin activity.[23–26]
Results and Discussion
Polycationic particles via mutation
The N and C termini of the Qβ coat protein are somewhat solvent exposed. In infectious Qβ, several of the capsid proteins bear a large C-terminal extension domain that is used to target a receptor on the E. coli host for cell entry.[27] This observation has prompted others to successfully use the C terminus for genetically appended additions to the capsid; this is exemplified by incorporating C-terminal peptide extensions (11–24 amino acids) after the stop codon to yield mixed particles containing up to 48 % of the read-through coat protein.[10] In an effort to prepare well-defined cationic particles with high loadings, we added oligo-Arg sequences within the open-reading frame of the Qβ coat protein at the N or C terminus. However, inserting Arg8, Arg5, or Arg2 peptides in this manner provided no intact particles, although expression of the mutated coat protein in E. coli was not severely diminished (see Figure S1 in the Supporting Information); this suggests that proper protein folding and/or particle assembly was inhibited by the added residues. No improvement was observed when a Gly2 or Gly5 spacer was introduced at the C terminus before the oligo-Arg insertions.
As an alternative, we targeted the loop formed by amino acids Asn10 through Thr18, which comprises the most solvent exposed and conformationally flexible part of the Qβ structure (Figure 1; note that as the N-terminal Met is generally cleaved from the Qβ coat protein, our nomenclature begins with Ala1).[8, 28] We have previously attached a variety of molecules to Lys16 within this loop, showing that intact capsids can tolerate the addition of functional groups in this vicinity.[11] We have also expressed large quantities of the K16M point mutant as a highly robust particle. However, the replacement of D14GKQT18 with Arg5 again gave rise to good coat protein expression but no isolable assembled particles.
Figure 1.

A) Crystal structure of wild-type bacteriophage Qβ, highlighting the peptide loops that are composed of coat protein residues 10–18 (red). This region is further magnified in B) dull green N10; bright green I11; white G12, G15; blue K13, K16; red D14; orange Q17; purple T18.
Point mutations proved to be more tractable. While modest at the single-protein level, such changes can dramatically alter particle properties because 180 of these sites are assembled on a compact topology in the final icosahedral structure. For example, mutation of Asp14 to Arg yields the sequence G12KR14GKQ, which falls into the pattern of XBBXBX (B=basic amino acid) that has been found by Cardin and Weintraub to associate strongly with glycosaminoglycans.[29] The polyvalent presentation of this construct in the assembled virion would therefore be expected to provide 180 potential heparin-binding sites per particle. This D14R mutant protein was found to assemble into isolable virus-like particles (Figure S2), although at significantly decreased yield compared to WT (5 vs. 100 mg per liter of liquid culture). Because a carboxylate sidechain is replaced with a guanidinium group in this construct, the D14R particle should exhibit a change in surface charge of +360 units relative to WT, and +540 units relative to the readily expressed and purified K16M mutant, assuming full ionization of surface residues.[30]
For comparison purposes, we also generated the N10R and T18R point mutants, each of which should bear a +180 surface charge relative to WT. These two constructs were isolated as icosahedral particles in yields similar to WT (Figures S3 and S4). Interestingly, combinations of the stable single point mutations were not well tolerated: the N10R/T18R double mutant did not yield intact particles, whereas D14R/T18R provided low yields of a very unstable capsid (Figure S5). The poor recovery and delicate nature of the latter structure made further characterization of this particle impractical.
Polycationic particles via bioconjugation
Our unsuccessful attempts to produce viable polycationic particles by distributing multiple arginine residues evenly over the capsid surface (for example, N10R/T18R) or at locally concentrated positions (for example, oligo-Arg loop insertions) meant that chemical modification of stable particles would be necessary to achieve higher arginine loadings. We have previously employed such derivatization reactions to decorate Qβ and other virus-like particles with a variety of molecules, including negatively charged oligonucleotides,[31] metal complexes,[12] and carbohydrates.[32] Thus, amine residues on the outer capsid surface of Qβ (most likely to be accessible are Lys2, Lys13, Lys16, and the N terminus) were targeted for the polyvalent display of oligo-Arg peptides 1 and 2 (Figure 2), which were synthesized by using standard Fmoc chemistry (Figure S6 and the Experimental Section).
Figure 2.

The azide oligo-Arg peptide R8G2 1 and alkyne oligo-Arg peptide R5 2 were synthesized by using standard Fmoc chemistry
Peptides 1 and 2 were conjugated to Qβ by using the two-step sequence that is shown in Figure 3. The K16M and WT Qβ particles were treated with N-hydroxysuccinimides 3 and 4 to give the polyvalent azide 5 and alkyne 6, respectively. Conjugation to the complementary peptides 1 and 2 by using precatalyst 7 in efficient copper-catalyzed azide–alkyne cycloaddition (CuAAC) “click” reactions[33] provided K16M-(R5)50 (8) and WT-(R8G2)95 (9). All virus particles were purified by sucrose gradient centrifugation and ultracentrifugation, and were shown to be intact by size-exclusion FPLC (Figure 4) and transmission electron microscopy (data not shown). In addition to verifying the exclusive presence of complete viral capsids, the FPLC traces also provided information on particle aggregation. As shown in Figure 4, WT-(R8G2)95 exhibited a higher tendency to form aggregates than K16M-(R5)50 at the same concentration.
Figure 3.

Attachment of oligo-Arg peptides to WT and K16M Qβ.
Figure 4.

Size-exclusion FPLC of 100 μL of 0.6 mg mL−1 A) K16M-(R5)50 (8) and B) WT-(R8G2)95 (9) in 0.1 M KPi pH 7 buffer, showing intact particles in each case. The elution volumes of the dispersed particles are different due to different column packing conditions.
The presence of covalently attached oligo-Arg peptides on particles 8 and 9 was confirmed and quantitated by using mass spectrometry, as shown in Figure 5. A single lysine per subunit of K16M was shown to be labeled with azide linker 3, and approximately 28 % of these (50 out of 180) were addressed with alkyne 2 (Figure 5B). Similarly, WT was labeled with four alkyne-containing linkers 4, followed by CuAAC reaction to capture one or two of these with azide 1 (Figure 5C). The data show that the resulting particle 9 was composed of subunits bearing zero, one, or two appended R8G2 chains in the approximate ratio of 11:6:2, giving rise to an average overall loading of 95 ± 5 chains per particle, assuming no change in detection efficiency by MALDI for the derivatized subunits. By taking into account the capping of amine residues performed in the chemical manipulations, K16M-(R5)50 and WT-(G2R8)95 should possess surface charges of −110 and +40 relative to WT, respectively (Table 1); this allows for an intriguing decoupling of overall charge from the presence of chains that bear high local concentrations of arginine residues.
Figure 5.

MALDI-MS of coat proteins from A) WT, B) K16M-(R5)50, and C) WT-(R8G2)95, following disassembly and denaturation of the capsids with 5.5 M urea, DTT reduction, and cysteine capping with iodoacetamide.
Table 1.
Activity and characterization of antiheparin virus-like particles.
| Particle | Maximum charge relative to WT[a] | Clotting time [s][b] | Heparin inhibition [%][c] | Retention time, [NaCl] at elution[d] | ZP [mV][e] |
|---|---|---|---|---|---|
| No heparin | n/a | 54 ± 2 | 100 | n/a | n/a |
| Heparin only | n/a | 118 ± 4 | 0 | n/a | n/a |
| K16M | −180 | 117 ± 1 | 2 ± 2 | – | −0.8 ± 0.6 |
| WT | 0 | 73 ± 2 | 70 ± 3 | – | 0.6 ± 0.5 |
| N10R | 180 | 86 ± 3 | 50 ± 5 | 60.3 min, 0.31M | 0.5 ± 0.4 |
| T18R | 180 | 57 ± 1 | 95 ± 2 | 42.3 min, 0.21M | 0.7 ± 0.5 |
| D14R | 360 | 54 ± 1 | 100 ± 2 | 63.3 min, 0.33M | 2.9 ± 1.2 |
| WT-(R8G2)95 | 40[f] | 55 ± 1 | 92 ± 2 | – | −0.6 ± 0.3 |
| K16M-(R5)50 | −110[g] | 67 ± 1 | 80 ± 2 | – | −11.5 ± 2.0 |
The acylation or replacement of an amine-containing residue is counted as −1; the addition of a positively-charged residue counts as +1.
Determined by the aPTT assay as described in the text. Error limits are the standard deviation from a minimum of three independent experiments.
Inhibition of heparin’s effect on blood plasma clotting (100 % = clotting time in the absence of heparin; 0 % = clotting time in the presence of heparin but no candidate inhibitor).
Retention time on cation exchange chromatography (HiTrap SP HP column, 10 mM KPi, pH 7, eluted with NaCl gradient by increasing by 12.5 mM min−1). A dash indicates that the particle eluted in the void volume without added NaCl, and therefore does not interact with the cation exchange resin.
Zeta potential determined in 25 mM KPi, pH 7.
Includes the removal of 4 positive charges per subunit by acylation of sidechain and N-terminal amino groups (−720) plus the addition of a maximum of 760 positive charges by attachment of 95 G2R8 chains.
Includes the removal of one positive charge per subunit by K16M substitution (−180), another by N-acylation (−180), and the addition of 250 charges by attachment of 50 R5 chains.
Ion exchange chromatography and ζ potentials
To evaluate the surface charge state of the resulting particles, we measured their retention time in cation exchange chromatography and their ζ potential (ZP) and compared these values to the maximum expected change in surface charge brought about by genetic and chemical manipulation (Table 1). Notably, prior measurements estimated a pI of 5.3 for WT,[9] but under our conditions (25 mM potassium phosphate, pH 7) WT exhibited a ZP of 0.6 ± 0.5 mV.[34]
The results from the two experimental measurements correlate reasonably well with the expected charge for the particles. K16M, which is expected to have the least amount of surface positive charge, has a negative ZP and does not bind to the cation exchange column. WT, N10R, and T18R are predicted to be moderate in charge and were found to exhibit similar and more positive ZP values, with two of the three interacting fairly strongly with the cation exchange resin. D14R, which should be the most positively charged particle, does indeed exhibit the greatest cation exchange retention time and ZP. Neither of the chemically derivatized particles bound to the ion exchange resin; this is consistent with their resemblance in total charge to their parent K16M and WT platforms. The WT-(R8G2)95 particle exhibited a small and negative ZP that was not very different from that of the WT capsid itself, which it should most closely approximate in overall surface charge. K16M-(R5)50, which is expected to be negatively charged with respect to WT, did exhibit a negative ZP, although the magnitude of this value was unexpectedly large. Because the measurement of ZP correlates the motion of the particle in an applied electric field with its charge, detailed comparisons between the genetically encoded point mutants and the chemically modified capsids bearing longer and more flexible oligo-Arg molecules are likely inappropriate.
Coagulation assays
The chemically and genetically modified particles were tested for their ability to inhibit heparin-mediated anticoagulation activity by using the activated partial thrombin time (aPTT) assay.[35] Premixed solutions of heparin (10 ng, ca. 0.05 mU) and virus (15 μg) were added to normal human plasma and aPTT reagent in HEPES-buffered saline (HBS). Coagulation was initiated by adding CaCl2 to a final concentration of 3.9 mM, and the clotting time was recorded (Table 1). Under these conditions the average time for coagulation in the absence of heparin or virus was 54 ± 1 s, and adding heparin significantly extended this time to 118 ± 5 s. The addition of engineered Qβ particles to the heparin solutions gave a variety of outcomes within this range. K16M showed no effect, as might be expected for the particle bearing the least number of surface positive charges; even with 50 μg of added virus (5000-fold mass excess), the clotting times exceeded 100 s (data not shown). In contrast, the wild-type particle from which K16M is derived exhibited moderate heparin inhibition (clotting time 73 ± 2 s), and the cationic capsids T18R or D14R were very potent inhibitors (clotting times 57 ± 1 and 54 ± 1 s, respectively, even with as little as 5 μg (2 pmol) of nanoparticles). N10R exhibited much poorer antiheparin activity (clotting time 86 ± 3 s) than T18R, in spite of the expectation that their overall surface charge densities should be similar. The chemically constructed particles 8 and 9 were also strong inhibitors, with the WT-(R8G2)95 structure showing greater activity (clotting time 55 ± 1 s) than K16M-(R5)50 (clotting time 67 ± 1 s).
The strong heparin inhibition exhibited by WT-(R8G2)95, K16M-(R5)50, and D14R is consistent with charge complementarity in the first two cases and the expected presence of a known glycosaminoglycan recognition motif in the third. However, the potent activity of T18R was a surprise because it possesses one fewer net positive charge per subunit than D14R and presumably does not conform to the heparin-binding sequence in the exposed Asn10–Thr18 loop. Equally intriguing is the relative inactivity of N10R compared to T18R because both replace a neutral solvent-exposed amino acid with Arg. It seems likely that the details of the three-dimensional structure and dynamics of the accessible loop are important in ways not fully appreciated, which may well be dependent on the specific display platform.
Indeed, K16M-(R5)50 and WT-(R8G2)95 show far greater heparin antagonism than would be expected based on their cation exchange interaction and ZP. The binding of heparin to biological targets and synthetically-tailored molecules is believed to occur largely through electrostatic interactions (for example, FGF binding[36]). We surmise that the display of highly charged oligopeptides on solvent-accessible loops enables efficient local interactions with heparin chains, with the particle having longer cationic oligo-Arg chains at higher density being the more effective.
Protamine sulfate, a commercial antiheparin agent, is the benchmark for comparison in the aPTT assay. Protamine is a 4–10 kDa peptide isolated from salmon sperm; commercial preparations favor ~4.5 kDa fragments that contain approximately 70 % Arg.[37] This compound is currently the only approved drug in North America for treating heparin overdose. As shown in Figure 6, 1 μg of Protamine (0.2 nmol) readily passivated heparin activity in the aPTT assay. A similar result was observed for oligo-Arg peptide 1 (1 μg, 0.66 nmol), and as mentioned above, for 5 μg of the WT-(R8G2)95 nanoparticle (9). On a molar basis (0.66 nmol 1 vs. 2 pmol 9), the polyvalent peptide conjugate is therefore approximately 330-fold more potent than the free peptide at achieving heparin inactivation. On a per-peptide basis, these values represent a 3.5-fold enhancement in activity (660 pmol 1 vs. 190 pmol 1 displayed on the nanoparticle surface).
Figure 6.
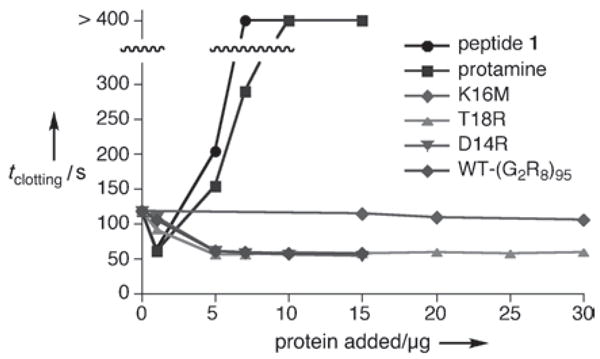
Concentration dependence of added virus and peptide on coagulation time in the aPTT assay. The data points for protamine and 1 at >400 s represent experiments in which clotting was not observed within that time.
Although it is commonly used in patients, protamine has several severe drawbacks related to toxicity.[38] Figure 6 highlights a key clinical risk: while 1 μg of protamine inhibited heparin activity, samples containing 5 μg or more showed increasing coagulation times in a sharp dose-dependent manner. This acute heparin-like anticoagulant activity is well known, and multiple points along the coagulation cascade are suspected targets;[39] in the aPTT assay, thrombin inhibition is the likely mechanism.[40] The clinical consequence of administering protamine to parients is careful titration and constant adjustment of the heparin/protamine ratio.[41] Such complications are not specific to protamine, but instead appear to be a general feature of cationic polymers.[42] Indeed, we found that more than 1 μg of 1 in the aPTT assay also strongly inhibited clotting. In contrast, none of the virus-based heparin inhibitors showed any change in activity with three-fold increases in concentration, and T18R in six-fold excess yielded no adverse effects, presumably because the particle concentrations were still low. The solution offered here by the display of accessible heparin-binding motifs on a structurally defined and stable scaffold differs from other preparations such as PEGylated[43] or truncated[44] protamine or structural analogues of protamine,[23, 24] all of which rely on cationic polymers that are likely to have drawbacks similar to the parent material.
Conclusions
The efforts described here to create polycationic Qβ particles for heparin antagonism have yielded several important results. The formation of intact capsids was strongly dependent on the position and nature of mutation: T18R, N10R, and D14R substitutions were all tolerated, but more ambitious cationic insertions (double mutants, oligo-Arg) did not yield assembled particles. Despite this limitation, the polyvalent scaffold afforded by Qβ allows the effect of a single stable point mutation to be magnified 180-fold in the final assembled virion; the genetically encoded structures described here differ in surface charge by as much as +540 units with only two point mutations (K16M cf. D14R). For comparison, a recent and excellent report described extensive mutational efforts to generate “supercharged” green-fluorescent proteins ranging from −30 to +48 relative to the wild-type structure.[45] For the alteration of bulk properties, the nature of virus-type scaffolds offers a considerable advantage, allowing one to make substantial changes (for example, to surface charge) with minimal mutagenesis.
Whereas genetic insertion of poly-Arg moieties proved intractable, stable virions provided polyvalent platforms for the construction of penta- and octa-Arg-bearing particles in different densities. The chemically and genetically engineered viruses were then tested for their ability to act as heparin antagonists in the aPTT clotting assay. In general, increasing either the theoretical positive charge on the particle surface or the number of appended oligo-Arg chains led to greater disruption of heparin anticoagulant activity; this is likely due to binding of the polycationic particles to the polyanionic glycosaminoglycan. On a per-particle basis, the activity of the engineered virus-like particles was substantially superior to that of Protamine, with the added benefit of no ill-effects at higher particle concentrations. In contrast, Protamine and naked Arg peptides showed acute heparin-like anticoagulant activity at higher concentrations. Tests of the virus-like particles against low-molecular-weight heparin and synthetic heparin pentasaccharide (both of which are receiving increasing attention along with the still widely prescribed unfractionated heparin), as well as in vivo experiments, are in progress.
Experimental Section
Mutagenesis, protein expression, and virus purification
1) WT and K16M. Expression and purification of these constructs in the pQE-60 vector transformed into M15 E. coli Met auxotroph cells proceeded as previously described.[11] 2) Arginine point mutants. The WT coat protein (0.4 kbp) was cloned into the IPTG-inducible pET-11d vector (5.7 kbp) by using the NcoI and BamHI restriction sites. Point mutations were introduced in whole-plasmid PCR reactions by using primers whose termini contained restriction sites for subsequent circularization, thereby reforming the whole plasmid containing the mutated gene. The D14R and T18R mutants were generated by using the forward primer TC AAT CCC CGC GGG GTA AAT CCC ACT AAC GGC G, which contains a silent SacII site for subsequent ligation (priming region underlined, restriction site italicized). Reverse primers were TAC CCC GCG GGG ATT GAG GAC CAG AGT TTG TTT TCC TCG TTT CCC GAT GTT ACC TAA AGT for D14R, and TAC CCC GCG GGG ATT GAG GAC CAG TCG TTG TTT TCC ATC TTT CCC GAT G for T18R (mutations in boldface). The N10R mutant was generated by using the forward primer G ACC ATG GCA AAA TTA GAG ACT GTT ACT TTA GGT CGA ATC GGG AAA GAT GGA AAA CAA AC, and the reverse primer A TAC CAT GGT ATA TCT CCT TCT TAA AG, both containing the NcoI restriction sites. Thus, PCR amplification, digestion with the appropriate enzyme, circularization via ligation, and subsequent transformation into chemically competent E. coli BL21 DE3 pLysS cells (Invitrogen) yielded the desired constructs. IPTG-induced expression of the Qβ coat protein and purification of the resulting assembled virions proceeded as previously described.[11] Protein concentrations were determined by using the Modified Lowry Protein Assay (Pierce, Rockford, IL, USA).
Azide and alkyne-derivatized Qβ
Compounds 3[46] and 4[46] were prepared as previously described. WT bearing alkynes or K16M bearing azides at surface-exposed amine residues were prepared by incubating a 2 mg mL−1 solution of protein with 6 mM 3 or 4 in 0.1 M KPi buffer (pH 7) with 20 % DMSO overnight at room temperature. The derivatized virions 5 and 6 were separated from excess reagent by ultracentrifugation by using a 10–40 % sucrose gradient, then isolating the protein bands corresponding to intact virions, and concentrating by subsequent ultracentrifugation and solvation in 0.1 M KPi pH 7 buffer. Size-exclusion FPLC (Superose-6 column) was used to verify the recovery of intact particles in 70–80 % yield.
Peptide synthesis
Peptide synthesis was performed by following standard Fmoc chemistry by using amino acids (Fmoc-Arg(Pbf)-OH and Fmoc-Gly-OH), resins (NovaSynTGR for R8G2 and Rink Amide MBHA for R5, 0.5 mmol scale) and HCTU from Novabiochem (Gibbstown, NJ, USA). During synthesis, after the addition of the final Arg, 7-heptynoic acid or 5-azido-pentanoic acid were coupled to the elongated peptides to yield azide or alkyne-terminated 1 or 2, respectively. Peptides were cleaved from the resin with TFA/2 % tri-isopropylsilane for 3 h at room temperature, after which they were precipitated with Et2O (50 mL), filtered over a glass frit, washed with Et2O, eluted with H2O/20 % MeCN/0.1 % TFA, and the eluant was finally lyophilized. The crude peptides were purified on reversed-phase HPLC, and fractions that corresponded to the separated peptides were collected, lyophilized, and confirmed by MALDI-MS (1: m/z calcd: 1506.8 Da [M+H]+, found: 1508.3 Da; 2: m/z calcd: 907.1 Da [M+H]+, found: 906.9 Da).
Copper-catalyzed azide–alkyne cycloaddition (CuAAC)
CuAAC reaction to generate WT-(R8G2)95 was conducted under a N2 atmosphere in a glove box by making a solution containing 0.3 mgmL−1 5, 33 μM 1, 0.17 mM Cu(MeCN)4(OTf) (added as a 100 mM stock solution in acetonitrile), and 0.34 mM sulfonated bathophenanthroline in 0.1 M Tris-HCl pH 8 (final volume 3 mL). K16M-(R5)50 was similarly prepared by mixing a solution containing 1.2 mg mL−1 6, 0.5 mM 2, 0.5 mM Cu(MeCN)4(OTf), and 1.0 mM sulfonated bathophenanthroline in 0.1M Tris pH 8 (final volume 1 mL). The reactions were agitated by gentle tumbling overnight at room temperature under N2, and the resulting conjugates were purified and characterized as above.
MALDI-MS of Qβ coat protein
A 10 M solution of urea (60 μL), and a 1 M solution of DTT (10 μL) were added to the virus-like particle (20 μg) in 0.1 M KPi pH 7 (40 μL), and the mixture was placed at 37 °C for 1 h. A 100 mg mL−1 aqueous solution of iodoacetamide (100 μL) was then added, and the mixture was left at 37 °C for an additional hour. A 1 M aqueous solution of DTT (50 μL) was added at room temperature, the mixture was allowed to stand for 10 min, and the solution was then evaporated to dryness by using a vacuum microcentrifuge. The samples were resuspended in 0.2 % TFA in H2O (50 μL) and purified with ZipTips (Millipore) by using the following solutions in the manufacturer-specified protocol: wetting solution 50 % MeCN in H2O, equilibration solution 0.2 % TFA in H2O, wash solution 0.1% TFA/5 % MeOH in H2O, elution solution 5 μL of 0.1 % TFA/65 % MeCN in H2O. The resulting solutions of purified peptides (0.5 μL) were spotted onto the MALDI-MS plate with a saturated solution of sinapinic acid (0.5 uL in 1:1 MeCN/H2O).
aPTT Assay
The desired amount of virus capsid in 0.1M KPi pH 7 was added to a 5 μg mL−1 solution of porcine heparin (sodium salt, 13.5–15 kDa, 174 kU g−1, EMD Biosciences, 10 μL of), and the volume was adjusted to 25 μL with 0.1 M KPi pH 7 buffer. Normal human plasma (40 μL, George King Biomedical, Overland Park, KS, USA) was then added, and the resulting solution (64 μL) was added to Platelin LS (50 μL; aPTT reagent, Trinity Biotech, Bray, Ireland). The final phosphate concentration was found by control experiments to avoid precipitation of calcium phosphate. The assay was conducted by using the STart4 Viscosity-based Detection System (Diagnostica Stago, Parsippany, NJ, USA): the mixture of reagents was placed into the cell and allowed to warm to 37 °C for 3 min, after which the reaction was initiated by the addition of 21.3 mM CaCl2 in HBS (35 μL, composed of 50 m M HEPES, 100 mM NaCl, and 0.02 % NaN3, pH 7.4), and the resulting clotting times were recorded. Protamine sulfate for control reactions was obtained from MP Biomedicals (Solon, OH, USA).
ζ potential measurements
Virus samples were made to 0.01 mg/mL in 25 mM KPi pH 7 buffer, with a final volume of 1.5 mL. Measurements were conducted at 25 °C by using a Brook-haven Instruments Zeta-PALS analyzer. A minimum of eight measurements were recorded and averaged for each sample. The averages from two independent samples were combined to yield the final result for each particle.
Ion exchange chromatography
Cation exchange chromatography was performed by using a 1 mL HiTrap SP HP column (GE Healthcare) under the control of an AKTA Explorer FPLC system. The solutions used were A) 10 mM KPi pH 7 and B) 0.5M NaCl. After injection of the sample and equilibration with A, a linear gradient was applied that increased the concentration of B from 0–100 % over 40 mL at 0.5 mLmin−1. Protein elution was monitored by detecting UV absorption, with a 260 nm/280 nm ratio of ~2 indicative of intact Qβ.
Supplementary Material
Acknowledgments
This work was supported by The Skaggs Institute for Chemical Biology, the TSRI Summer Internship Program (C.E.), the NIH (R01 CA112075, RR021886, HL082588), and the Canadian Institutes of Health Research (postdoctoral fellowship to A.K.U.).
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- 1.Mammen M, Choi SK, Whitesides GM. Angew Chem. 1998;110:2908–2953. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 1998;37:2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 2.Carlson CB, Mowery P, Owen RM, Dykhuizen EC, Kiessling LL. ACS Chem Biol. 2007;2:119–127. doi: 10.1021/cb6003788. [DOI] [PubMed] [Google Scholar]
- 3.Kawamura KS, Sung M, Bolewska-Pedyczak E, Gariepy J. Biochemistry. 2006;45:1116–1127. doi: 10.1021/bi051338e. [DOI] [PubMed] [Google Scholar]
- 4.Petty NK, Evans TJ, Fineran PC, Salmond GPC. Trends Biotechnol. 2006;24:7–15. doi: 10.1016/j.tibtech.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 5.Pattenden LK, Middelberg APJ, Niebert M, Lipin DI. Trends Biotechnol. 2005;23:523–529. doi: 10.1016/j.tibtech.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 6.Agadjanian H, Weaver JJ, Mahammed A, Rentsendorj A, Bass S, Kim J, Dmochowski IJ, Margalit R, Gray HB, Gross Z, Medina-Kauwe LK. Pharm Res. 2006;23:367–377. doi: 10.1007/s11095-005-9225-1. [DOI] [PubMed] [Google Scholar]
- 7.Young LS, Searle PF, Onion D, Mautner V. J Pathol. 2006;208:299–318. doi: 10.1002/path.1896. [DOI] [PubMed] [Google Scholar]
- 8.Golmohammadi R, Fridborg K, Maija B, Valegard K, Liljas L. Structure. 1996;4:543–554. doi: 10.1016/s0969-2126(96)00060-3. [DOI] [PubMed] [Google Scholar]
- 9.Overby LR, Barlow GH, Doi RH, Jacob M, Spiegelman S. J Bacteriol. 1966;91:442–448. doi: 10.1128/jb.91.1.442-448.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vasiljeva I, Kozlovska T, Cielens I, Strelnikova A, Kazaks A, Ose V, Pumpens P. FEBS Lett. 1998;431:7–11. doi: 10.1016/s0014-5793(98)00716-9. [DOI] [PubMed] [Google Scholar]
- 11.Strable E, Prasuhn DE, Jr, Udit AK, Brown SD, Link AJ, Ngo J, Lander G, Quispe J, Potter CS, Carragher B, Tirrell DA, Finn MG. Bioconjugate Chem. 2008;19:866–875. doi: 10.1021/bc700390r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prasuhn DE, Jr, Yeh RM, Obenaus A, Manchester M, Finn MG. Chem Commun (Cambridge) 2007:1269–1271. doi: 10.1039/b615084e. [DOI] [PubMed] [Google Scholar]
- 13.Prasuhn DE, Jr, Singh P, Strable E, Brown S, Manchester M, Finn MG. J Am Chem Soc. 2008;130:1328–1334. doi: 10.1021/ja075937f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Capila I, Linhardt RJ. Angew Chem. 2002;114:426–450. doi: 10.1002/1521-3773(20020201)41:3<390::aid-anie390>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2002;41:390–412. [Google Scholar]
- 15.Parish CR. Nat Rev Immunol. 2006;6:633–643. doi: 10.1038/nri1918. [DOI] [PubMed] [Google Scholar]
- 16.Lin XH, Perrimon N. Matrix Biol. 2000;19:303–307. doi: 10.1016/s0945-053x(00)00073-1. [DOI] [PubMed] [Google Scholar]
- 17.Mellor P, Harvey JR, Murphy KJ, Pye D, O’Boyle G, Lennard TWJ, Kirby JA, Ali S. British J Cancer. 2007;97:761–768. doi: 10.1038/sj.bjc.6603928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.H3cker U, Nybakken K, Perrimon N. Nat Rev Mol Cell Biol. 2005;6:530–541. doi: 10.1038/nrm1681. [DOI] [PubMed] [Google Scholar]
- 19.Kiil J, Axelsen F, Kiil J, Andersen D. Lancet. 1978;311:1115–1116. doi: 10.1016/s0140-6736(78)90297-0. [DOI] [PubMed] [Google Scholar]
- 20.Pike RN, Buckle AM, Le Bonniec BF, Church FC. FEBS J. 2005;272:4842–4851. doi: 10.1111/j.1742-4658.2005.04880.x. [DOI] [PubMed] [Google Scholar]
- 21.Warkentin TE, Levine MN, Hirsh J, Horsewood P, Roberts RS, Gent M, Kelton JG. New Eng J Med. 1995;332:1330–1335. doi: 10.1056/NEJM199505183322003. [DOI] [PubMed] [Google Scholar]
- 22.Maddineni J, Walenga JM, Jeske WP, Hoppensteadt DA, Fareed J, Wahi R, Bick RL. Clin Appl Thromb/Hemostasis. 2006;12:267–276. doi: 10.1177/1076029606291434. [DOI] [PubMed] [Google Scholar]
- 23.Schick BP, Maslow D, Moshinski A, San Antonio JD. Blood. 2004;103:1356–1363. doi: 10.1182/blood-2003-07-2334. [DOI] [PubMed] [Google Scholar]
- 24.Choi S, Clements DJ, Pophristic V, Ivanov I, Vemparala S, Bennet JS, Klein ML, Winkler JD, Degrado WF. Angew Chem. 2005;117:6843–6847. doi: 10.1002/anie.200501279. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:6685–6689. doi: 10.1002/anie.200501279. [DOI] [PubMed] [Google Scholar]
- 25.Mecca T, Consoli GML, Geraci C, La Spina R, Cunsolo F. Org Biomol Chem. 2006;4:3763–3768. doi: 10.1039/b608887b. [DOI] [PubMed] [Google Scholar]
- 26.Shenoy S, Sobel M, Harris RH. Curr Pharm Des. 1999;5:965–986. [PubMed] [Google Scholar]
- 27.Weber K, Konigsberg W. In: RNA Phages. Zinder ND, editor. Cold Springs Harbor Laboratory Press; New York: 1975. pp. 51–84. [Google Scholar]
- 28.Natarajan P, Lander GC, Shepherd CM, Reddy VS, Brooks CL, Johnson JE. Nat Rev Microbiol. 2005;3:809–817. doi: 10.1038/nrmicro1283. [DOI] [PubMed] [Google Scholar]
- 29.Cardin AD, Weintraub HJ. Arterioscler Thromb Vasc Biol. 1989;9:21–32. doi: 10.1161/01.atv.9.1.21. [DOI] [PubMed] [Google Scholar]
- 30.Cardin and Weintraub also identified XBBBXXBX as an active sequence. If Gly15 of Qβ is ignored due to its buried position –-an assumption supported by both its X-ray crystal structure and solvent-accessibility profile –- then the N10R/D14R double mutant would provide the corresponding pattern in reverse: Q17K[G]R14KGIR10G. Unfortunately, particles could not be isolated from E. coli cells expressing this protein.
- 31.Strable E, Johnson JE, Finn MG. Nano Lett. 2004;4:1385–1389. [Google Scholar]
- 32.Kaltgrad E, Sen Gupta S, Punna S, Huang C-Y, Chang A, Wong C-H, Finn MG, Blixt O. ChemBioChem. 2007;8:1455–1462. doi: 10.1002/cbic.200700225. [DOI] [PubMed] [Google Scholar]
- 33.Gupta SS, Kuzelka J, Singh P, Lewis WG, Manchester M, Finn MG. Bioconjugate Chem. 2005;16:1572–1579. doi: 10.1021/bc050147l. [DOI] [PubMed] [Google Scholar]
- 34.ZP values are derived from the electrophoretic mobility of the particle. Thus, variables such as solution conditions (for example, ionic strength) and particle topology can complicate comparisons of ZPs among different structures. This problem is further exacerbated with viruses because the encapsidated genetic material (DNA, RNA) can affect the measured surface potentials of the particles in variable ways. As such, ZPs are most useful for ascertaining relative differences in surface potential between topologically similar particles under identical conditions. Good discussions of these matters can be found in Schaldach CM, Bourcier WL, Shaw HF, Viani BE, Wilson WD. J Colloid Interface Sci. 2006;294:1–10. doi: 10.1016/j.jcis.2005.06.082.Penrod SL, Olson TM, Grant SB. J Colloid Interface Sci. 1995;173:521–523. For the particles in this study, 25 mM KPi pH 7 was determined to be the optimal buffer based on measurement stability. When variations in buffer concentration were made to induce corresponding changes in the observed ZPs, large errors were observed.
- 35.Gale AJ, Heeb MJ, Griffin JH. Blood. 2000;96:585–593. [PubMed] [Google Scholar]
- 36.Jastrebova N, Vanwildemeersch M, Rapraeger AC, Giminez-Gallego G, Lindahl U, Spillmann D. J Biol Chem. 2006;281:26884–26892. doi: 10.1074/jbc.M600806200. [DOI] [PubMed] [Google Scholar]
- 37.Callanan MJ, Carroll WR, Mitchell ER. J Biol Chem. 1957;229:279–287. [PubMed] [Google Scholar]
- 38.Horrow JC. Anesth Analg. 1985;64:348–361. [PubMed] [Google Scholar]
- 39.Velders AJ, Wildevuur CRH. Ann Thorac Surg. 1986;42:168–171. doi: 10.1016/s0003-4975(10)60512-7. [DOI] [PubMed] [Google Scholar]
- 40.Cobel-Geard RJ, Hassouna HI. Am J Hematol. 1983;14:227–233. doi: 10.1002/ajh.2830140304. [DOI] [PubMed] [Google Scholar]
- 41.Shigeta O, Kojima H, Hiramatsu Y, Jikuya T, Terada Y, Atsumi N, Sakakibara Y, Nagasawa T, Mitsui T. J Thorac Cardiovasc Surg. 1999;118:354–360. doi: 10.1016/S0022-5223(99)70227-8. [DOI] [PubMed] [Google Scholar]
- 42.DeLucia A, III, Wakefield TW, Andrews PC, Nichol BJ, Kadell AM, Wrobleski SK, Downing J, Stanley JC. J Vasc Surg. 1993;18:49–58. doi: 10.1067/mva.1993.42736. [DOI] [PubMed] [Google Scholar]
- 43.Chang LC, Lee HF, Chung MJ, Yang VC. Bioconjugate Chem. 2005;16:147–155. doi: 10.1021/bc0499735. [DOI] [PubMed] [Google Scholar]
- 44.Liang JF, Zhen L, Chang LC, Yang VC. Biochemistry. 2003;68:116–120. doi: 10.1023/a:1022109905487. [DOI] [PubMed] [Google Scholar]
- 45.Lawrence MS, Phillips KJ, Liu DR. J Am Chem Soc. 2007;129:10110–10112. doi: 10.1021/ja071641y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Q, Chan T, Hilgraf R, Fokin V, Sharpless K, Finn M. J Am Chem Soc. 2003;125:3192–3193. doi: 10.1021/ja021381e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.