

Abstract
Patients with systemic lupus erythematosus develop accelerated atherosclerosis independent of traditional risk factors. The 3-hydroxy-3-methyl-glutaryl coenzyme A (HMG-CoA) reductase inhibitors are widely prescribed for hyperlipidemia, but they also exhibit anti-inflammatory actions that appear to be independent of their suppressive actions on plasma cholesterol levels. In this study, we analyzed the effect of the HMG-CoA reductase inhibitor simvastatin on disease manifestations in gld.apoE−/− mice that lack functional Fas ligand and apolipoprotein E and exhibit accelerated atherosclerosis and aggravated lupus-like features. Wild-type, gld, apoE−/−, and gld.apoE−/− mice were maintained on a high cholesterol Western diet and received daily simvastatin (0.125 mg/kg) or saline for 12 wk. Serum cholesterol levels were unaffected by simvastatin treatment, but atherosclerotic lesion area was reduced in both apoE−/− and gld.apoE−/− mice treated with simvastatin. Simvastatin also reduced the lymphadenopathy, renal disease, and proinflammatory cytokine production seen in gld.apoE−/−, but not gld, mice. The immunomodulatory effects in gld.apoE−/− mice were associated with enhanced STAT6 and decreased STAT4 induction in submandibular lymph node cells. Along with reductions in serum TNF-α and IFN-γ levels, there was also an increase in IL-4 and IL-10 transcript levels in lymph nodes. These data indicate that HMG-CoA reductase inhibitors ameliorate atherosclerosis and lupus-like autoimmunity independent of their cholesterol-lowering effects via a shift from a Th1 to a Th2 phenotype in the gld.apoE−/− model. Thus, the anti-inflammatory activities of statins may have utility for the treatment of both autoimmunity and atherosclerosis in patients with systemic lupus erythematosus.
Systemic lupus erythematosus (SLE)3 is a complex autoimmune disease involving multiple organs that is characterized by autoantibody production (1). In recent years, much attention has been given to the rising incidence of accelerated atherosclerosis in SLE, which precedes more advanced cardiovascular diseases in these patients (2–5). Accelerated atherosclerosis has also been shown to occur in other autoimmune diseases, including rheumatoid arthritis and systemic sclerosis (3, 6, 7). One factor that contributes to the initiation and progression of atherosclerosis is hypercholesterolemia, in particular increased low-density lipoprotein (LDL) (8). In this regard, it has been shown that both pediatric and adult SLE patients have abnormal lipid profiles, including decreased levels of high-density lipoprotein, and increased triglycerides and vLDL (9, 10).
Hypercholesterolemia is widely treated with statins, a class of drugs that inhibit 3-hydroxy-3-methyl-glutaryl coenzyme A (HMG-CoA) reductase, the first committed step of cholesterol synthesis. By decreasing plasma lipid levels, statin treatment decreases the risk for cardiovascular disease and stroke in hypercholesterolemic patients (11–13). Other studies have shown that in addition to lowering plasma cholesterol levels, HMG-CoA reductase inhibitors have immunomodulatory properties that are independent of their serum lipid-lowering properties (14). In this regard, pilot studies indicate that SLE patients treated with simvastatin exhibit decreased proteinuria, and rheumatoid arthritis patients treated with atorvastatin have reduced C-reactive protein levels as well as a clinical improvement of the disease (15). Other studies demonstrate that atorvastatin reduces disease activity in rheumatoid arthritis patients (16), and simvastatin inhibits the inflammatory components of multiple sclerosis (17).
Interestingly, lipid levels in rodents remain unaffected by statin treatment due to robust feedback regulation of hepatic HMG-CoA reductase (14, 18, 19). For this reason, mice and rats are good models to study the anti-inflammatory and immunomodulatory properties of these drugs in the absence of confounding metabolic effects. For example, mice with experimental autoimmune encephalomyelitis treated with lovastatin exhibit a decrease in duration and clinical severity of the disease (20). In addition, fluvastatin treatment in a model of acute peritoneal inflammation in rats inhibits adhesion and extravasation of leukocytes (21).
The goal of the current study was to examine clinically relevant doses of statin on disease in the apoE−/−, gld, and gld.apoE−/− mouse strains that are models of atherosclerosis, autoimmunity, and accelerated atherosclerosis associated with autoimmunity, respectively (22). Previous experiments with gld.apoE−/− mice have shown that this strain exhibits more atherosclerosis than apoE−/− mice and more autoimmunity and lymphoproliferation than gld mice, indicating positive feedback interactions between these disease processes (22). The current data suggest that statin treatment in gld.apoE−/− mice may lessen not only the severity of atherosclerosis, but also of SLE when it is associated with accelerated atherosclerosis.
Materials and Methods
Study protocol
The Jackson Laboratory B6Smn.C3-gld (stock 001021) is considered fully congenic on C57BL/6 background, as is the apoE−/− mouse. Therefore, the gld.apoE−/− mouse has a C57 background that complements the gld and apoE−/− single mutants, both on a C57 background. Mice on a C57BL/6 background with the genotypes apoE−/−, gld, gld.apoE−/−, and apoE+/+
gld+/+ (wild type; wt) were maintained on a Purina ProLab 3000 mouse diet (normal diet), and at 7 wk of age, mice from each genotype received Teklad (Harlan Teklad) adjusted calories Western diet: 21% (w/w) fat, 0.15% (w/w) cholesterol, and 19.5% (w/w) casein, without sodium cholate. At 7 wk of age, mice from each genotype received either saline vehicle (200 μl) or activated simvastatin (0.125 mg/kg) administered by daily i.p. injection for 12 wk. Because simvastatin has little or no inherent activity in the absence of lactone ring hydrolysis, an alkaline hydrolysis procedure was performed before administration to mice (23). All mouse experiments were conducted under protocols approved by the Institutional Animal Care and Use Committee of Boston University School of Medicine.
Quantitative analyses of atherosclerosis, hyperlipidemia, splenomegaly, and lymphadenopathy
After 12 wk on a high cholesterol Western diet, food was removed for an 8-h fast. Following the fast, the mice were weighed and sacrificed. Blood was drawn by cardiac puncture for determination of total plasma cholesterol, LDL, high-density lipoprotein, and triglyceride levels, which was performed by a clinical laboratory at Boston Medical Center. Spleen and submandibular lymph nodes were excised and weighed. The vasculature was perfused intracardially with 0.9% sodium chloride, and the aorta was isolated from the aortic arch to the iliac bifurcation. The adventitia was thoroughly stripped, and the aorta was opened longitudinally, pinned to a white silicone gel, and fixed in 10% neutral buffered Formalin for 24 h. After fixation, aortae were rinsed with PBS, stained with Oil Red O solution, and destained in 60% isopropyl alcohol. Aortae were photographed using an Olympus digital camera and lesion size measured using NIH Image on a Macintosh computer.
Autoantibodies
Serum levels of anti-nuclear Abs (ANA) were measured by immunofluorescence using HEp-2-coated slides (The Binding Site). Slides were incubated for 20 min with serial dilutions (1/40 to 1/2560) of mouse serum in PBS, washed in PBS, and then incubated with FITC-labeled goat anti-mouse IgG (whole molecule; Sigma-Aldrich). Slides were counterstained with Evans blue and viewed using fluorescent microscopy. The titer value is defined as the inverse value of the last positive dilution.
Urine and kidney analysis
Urine samples were obtained using metabolic cages from 19-wk-old wt, apoE−/−, gld, and gld.apoE−/− mice. Samples were diluted 1/10 and measured using a protein assay (Bio-Rad) at an absorbance of 595 nm. Calculations are shown as milligrams of protein per 24 h. Formalin-fixed kidney sections were stained with H&E. Stained sections were coded, then digitally photographed and analyzed by an investigator who was blinded to section identity using a stereo microscope (Nikon Opiphot) fitted with a digital camera (Diagnostic Instruments). Glomerular cross-sectional area of at least 25 glomeruli from five randomly photographed low-powered fields was measured in each animal using computer-assisted pixel counting (Photopaint 10; Corel). Mean glomerular tuft area (AG) for each animal was calculated from all available glomerular profiles, and tuft volume (VG) was calculated (24). VG = (B/k) × (AG)1.5, where B is the shape coefficient for an idealized glomerulus (B = 1.38) and k is a size distribution coefficient (k = 1.10).
Cytokine analysis
Circulating levels of TNF-α were quantified using the cytometric bead array mouse inflammation kit (BD Biosciences). Circulating levels of IFN-γ were quantified by ELISA (BD Biosciences). Serum was collected from each of the groups of mice treated with vehicle or simvastatin and examined according to the manufacturer’s specifications. IL-4 and IL-10 mRNA in submandibular lymph nodes were quantified by real-time PCR. Total RNA was prepared using a Qiagen kit, and cDNA was prepared using ThermoScript RT-PCR Systems (Invitrogen Life Technologies). Quantitative PCR was performed using the iCycler iQ Real-Time PCR Detection System (Bio-Rad) with SYBR Green 1 as a double-standard DNA-specific dye (Applied Biosystems). The following primers were used: GAT CATCGGCATTTTGAACGA (forward) and AGGACGTTTGGCACATCCAT (reverse) for IL-4; AGACAATAACTCCACCCACTTCC (forward) and GCTGGTCCTTTGTTTGAAAGAAAG (reverse) for IL-10; and CGTGAAAAGATGACCCAGATCA (forward) and TGGTACGACCAGAGGCATACAG (reverse) for β-actin.
Immunoprecipitation/Western blot
Submandibular lymph nodes from vehicle- and simvastatin-treated mice were harvested and homogenized. After determination of concentration with a protein assay kit (Pierce), 1 mg of protein was immunoprecipitated for STAT4 or STAT6 overnight with anti-STAT4 Ab or anti-STAT6 Ab (BD Pharmingen). The next day, after incubation with agarose beads, the protein was separated by SDS-PAGE and transferred to a polyvinylidene difluoride membrane (Millipore). The membrane was blocked with TBS containing 5% BSA (TBS-BSA) and incubated with the primary Ab overnight at 4°C in TBS-BSA. The membrane was washed three times with TBS containing 0.1% Tween 20 and incubated with secondary Ab for 1 h at room temperature. ECL Plus (Amersham Biosciences) was used for detection. The primary Abs used were anti-phospho-STAT6 Ab (Cell Signaling Technology) and anti-phospho-STAT4 Ab (Zymed Laboratories) at 1/1000 dilutions. The secondary Abs were anti-mouse or anti-rabbit IgG/HRP conjugate (Cell Signaling Technology) diluted 1/5000 in TBS containing 5% skim milk.
Statistical analysis
Results are shown as the mean ± SEM or SD. Differences between groups were determined by ANOVA and Student’s t test using SigmaPlot, and were considered statistically significant for p < 0.05.
Results
Simvastatin treatment does not alter cholesterol levels, but decreases atherosclerotic lesion area in apoE−/− and gld.apoE−/− mice
At 7 wk of age, wt, apoE−/−, gld, and gld.apoE−/− mice were put on Western diet and administered simvastatin (0.125 mg/kg/day) or saline by i.p. injection for 12 wk. Consistent with previous studies in rodents (14, 18, 19), simvastatin did not affect plasma cholesterol levels in the four groups of mice (Table I). Regardless of treatment, LDL levels were elevated in both apoE−/− and gld.apoE−/− mice. Consistent with previously published data (22), LDL levels were lower in the gld.apoE−/− mice than apoE−/− mice, which may result from a suppression of hepatic cholesterol synthesis by inflammatory cytokines (25). In addition, the ratio of LDL to high-density lipoprotein was markedly higher in apoE−/− or gld.apoE−/− than in wt or gld mice, and statin treatment had little or no effect on this ratio (data not shown).
Table I.
Cholesterol levels in mice treated with vehicle or simvastatin
| Total Cholesterol (mg/dl) |
||
|---|---|---|
| Vehicle treated | Statin treated | |
| wt | 158.1 ± 14.3 | 147.5 ± 8.4 |
| apoE−/− | 1450.3 ± 64.6 | 1290.3 ± 57.2 |
| gld | 149.5 ± 15.9 | 124.2 ± 12.5 |
| gld.apoE−/− | 757.7 ± 76.5* | 743.1 ± 75.6* |
, p < 0.001 vs wt, gld, and apoE−/−.
Oil Red O staining of aortae revealed that the atherosclerotic lesion area was lower in both apoE−/− and gld.apoE−/− mice that received simvastatin (Fig. 1). The lesion area in the gld.apoE−/− mice treated with statin was decreased ~25% compared with vehicle-treated mice: from 28.4 ± 2.1 to 21.6 ± 1.9 mm2, respectively. Similarly, statin treatment led to a 21% reduction in atherosclerotic lesion area in apoE−/− mice, from 9.1 ± 0.9 to 5.9 ± 0.8 mm2 (Fig. 1B), consistent with findings from previous studies (14, 26). No atherosclerotic lesions were observed in wt or gld mice (data not shown). Thus, statin treatment reduces atherosclerosis independent of detectable effects on serum cholesterol.
FIGURE 1.

Total atherosclerotic lesion area in vehicle- and simvastatin-treated mice. A, Aortae from mice maintained on Western diet for 12 wk and treated with either simvastatin or vehicle were dissected from the aortic root to the iliac bifurcation, and stained en face with Oil Red O. B, Atherosclerotic lesion area of Oil Red O-stained aortae was quantified in apoE−/− (n = 7) and gld.apoE−/− (n = 11) (*, p < 0.01).
Simvastatin treatment decreases lymphoproliferation and autoantibody production in gld.apoE−/− mice
Enlargement of spleen and lymph nodes is a feature of gld mice, and is more pronounced in gld.apoE−/− mice (22). After 12 wk of simvastatin treatment, submandibular lymph node sizes in the gld.apoE−/− mice decreased from 1.05 ± 0.09 to 0.41 ± 0.05 g (Fig. 2A). In addition, splenomegaly was significantly decreased in simvastatin-treated gld.apoE−/− mice compared with vehicle-treated controls (Fig. 2B). In contrast, gld mice did not show any response to statin treatment with regard to lymphadenopathy and splenomegaly. Simvastatin also had no effect on spleen or lymph node weight in wt or apoE−/− mice. Treatment with simvastatin significantly decreased ANA titer in gld.apoE−/− mice, from 822.9 ± 118 to 352 ± 78.4 (Fig. 2C). In contrast, simvastatin had no effect on ANA titers in gld mice.
FIGURE 2.

Statin treatment decreases the autoimmune phenotype in gld.apoE−/− mice. A, Submandibular lymph nodes were harvested from all groups of mice maintained on Western diet for 12 wk, and treated with either simvastatin or vehicle, and weighed (*, p < 0.001). B, The spleen was also harvested from mice in each group and weighed (*, p < 0.001). C, ANA titer was determined from serum samples using HEp-2-coated slides and is reported as the inverse value of the last positive dilution (*, p < 0.01). Data are expressed as means ± SEM.
Renal disease in gld.apoE−/− mice is improved with simvastatin treatment
In earlier studies, we observed that the gld.apoE−/− mice exhibited proteinuria, enlarged glomeruli, and tubular vacuolization (22), whereas these features are not seen in the gld mice on the C57BL/6 genetic background (27). Simvastatin treatment in gld.apoE−/− mice reduced proteinuria (Fig. 3A), and histological analyses of kidney revealed that glomerular tuft volume was significantly smaller when gld.apoE−/− mice were treated with simvastatin (Fig. 3, B and C). In contrast, simvastatin had no effects on normal urine protein levels and kidney glomeruli size in wt, apoE−/−, and gld mice. Thus, simvastatin treatment ameliorates several components of the pathological renal phenotype observed in gld.apoE−/− mice.
FIGURE 3.

Renal disease is ameliorated in gld.apoE−/− mice after simvastatin treatment. The wt (n = 5), apoE−/− (n = 7), gld (n = 9), and gld.apoE−/− (n = 11) mice were treated with simvastatin (+) or vehicle control (−) for 12 wk. A, Proteinuria was measured in a 24-h urine collection. B, Representative H&E-stained sections of kidney. Glomerulus size in the gld.apoE−/− mice is shown with arrows. C, Glomerular tuft size was measured by computer-assisted pixel counting (*, p < 0.001). Values shown are the mean ± SD.
Simvastatin decreases apoptotic debris in lymph nodes of gld.apoE−/− mice
Failure to clear apoptotic cells has been implicated in the development and progression of autoimmune diseases in several mouse models (28–31), and we previously observed impaired apoptotic cell clearance in the gld.apoE−/− mice (22). Thus, we examined whether simvastatin treatment would affect the levels of apoptotic cells in lymph nodes. Simvastatin treatment significantly decreased the amount of apoptotic cells within the submandibular lymph nodes of the gld.apoE−/− mice, whereas this treatment had little or no effect on the accumulation of apoptotic cells in gld mice (Fig. 4). Very few apoptotic cells could be detected in the lymph nodes of wt or apoE−/− mice, and statin treatment had no detectable effect on these levels (data not shown).
FIGURE 4.
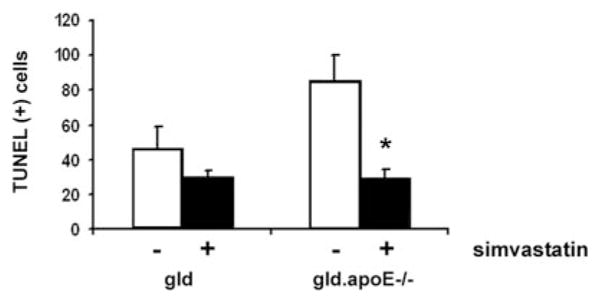
Apoptotic cell accumulation in lymph nodes is decreased in gld.apoE−/− mice following simvastatin treatment. Gld (n = 9) and gld.apoE−/− (n = 11) mice were maintained on a high cholesterol Western diet and treated with either simvastatin (+) or vehicle control (−) for 12 wk. The numbers of apoptotic cells per high-power field (×20) in the submandibular lymph nodes were quantitated by TUNEL staining (*, p < 0.001). Values shown are the mean ± SD.
Simvastatin up-regulates the Th2 immune response in gld.apoE−/− mice
The STAT6 transcription factor promotes Th2 immune responses and anti-inflammatory cytokine production (32, 33), whereas the STAT4 transcription factor promotes Th1 immune responses and the production of proinflammatory cytokines (34–36). Because murine SLE is associated with Th1 proinflammatory cytokine production, we examined whether simvastatin differentially regulates STAT4 and STAT6 phosphorylation in this model. Western blot analysis of STAT4 and STAT6 immunoprecipitated from submandibular lymph nodes of gld.apoE−/− mice revealed that simvastatin treatment increased STAT6 phosphorylation levels, whereas phosphorylated STAT4 was down-regulated (Fig. 5A), indicating a shift toward a Th2 response. Consistent with a modulation of the Th1/Th2 response in gld.apoE−/− mice, serum levels of proinflammatory cytokines TNF-α and IFN-β were lower in the gld.apoE−/− mice treated with simvastatin (Fig. 5B). In addition, treatment with simvastatin led to an increase in mRNA levels of anti-inflammatory cytokines IL-4 and IL-10 in the submandibular lymph nodes of gld.apoE−/− mice (Fig. 5C). Therefore, simvastatin modulates the immune response in gld.apoE−/− mice, at least in part, by altering the Th1/Th2 cytokine response.
FIGURE 5.

Simvastatin promotes Th2 responses, and reduces Th1 responses in gld.apoE−/− mice. Gld.apoE−/− mice were treated with simvastatin (+) or vehicle control (−) for 12 wk. A, Submandibular lymph nodes from individual mice in each group were pooled, and the level of phosphorylated STAT6 (p-STAT6) and STAT4 (p-STAT4) was measured by immunoprecipitation followed by Western blotting. B, Proinflammatory cytokine TNF-α and IFN-γ levels in the sera of gld.apoE−/− mice were measured by cytokine bead analysis (*, p < 0.05). C, mRNA extracted from submandibular lymph nodes of gld.apoE−/− mice was evaluated by quantitative real-time PCR for anti-inflammatory cytokines IL-4 and IL-10. Values shown are the mean − SEM from gld (n = 9) and gld.apoE−/− (n = 11) mice (*, p < 0.01).
Discussion
Premature atherosclerosis associated with SLE is a major cause of morbidity and mortality (37, 38). However, the pathogenesis of atherosclerosis is poorly understood in this patient population. Recently, we described the gld.apoE−/− mouse model of premature atherosclerosis in SLE (22). In the current study, we show that statin treatment substantially reduces both atherosclerosis and autoimmune disease in this model independent of effects on serum cholesterol levels. Simvastatin has a modest (21%) inhibitory effect on atherosclerosis, consistent with the findings of others in hyperlipidemic mice (14, 26). However, the effects of statins on autoimmunity and lymphoproliferation were more striking in the gld.apoE−/− model. Simvastatin treatment reduced ANA titers by 57%, lymphadenopathy by 66%, and splenomegaly by 48%. Furthermore, simvastatin treatment normalized kidney glomeruli size and protein levels in urine.
Although there may be several mechanisms by which simvastatin exerts its anti-inflammatory and immunomodulatory properties, we provide evidence that simvastatin influences the expression of proinflammatory cytokines in gld.apoE−/− mice. Statins have been shown to ameliorate signs of experimental autoimmune encephalomyelitis, a mouse model of multiple sclerosis, via effects on the Th1/Th2 balance of cytokines (20, 39). Several studies have implicated Th1 cytokines and their upstream transcription factors in the pathogenesis of lupus in mouse models (39–42). It has been shown that retinoic acid treatment to New Zealand Black/New Zealand White F1 mice resulted in decrease in proinflammatory cytokines IFN-γ and IL-2 as well as reduced renal dysfunction, contributing to increased survival (43). Furthermore, treatment with a peptide that inhibits SLE progression was associated with a down-regulation of IFN-γ and IL-2 and up-regulation of anti-inflammatory cytokine TGF-β (44). Proinflammatory cytokines have also been implicated in the pathology of lupus nephritis in human patients (45, 46). These studies provide evidence that proinflammatory cytokine signaling contributes to the progression of autoimmune diseases. In our study, simvastatin therapy promoted a decrease in the serum levels of the Th1 cytokines TNF-α and IFN-γ and an increase in the lymph node mRNA levels of the Th2 cytokines of IL-4 and IL-10 in gld.apoE−/− mice, indicating a shift toward a more anti-inflammatory immune response. Consistent with this interpretation, our immunoprecipitation experiments detected a statin-mediated increase in phosphorylated STAT6 and a decrease in STAT4 phosphorylation in submandibular lymph nodes. Collectively, these findings suggest that statin therapies down-regulate Th1 cytokine signaling, and promote a shift toward a Th2 response.
Although atherosclerosis is associated with a Th1 response, it is paradoxical that severe hypercholesterolemic conditions in mice will modulate the T cell response toward a Th2 type (47, 48). This Th2 shift is seen with diets containing 1.25% cholesterol and more so with diets containing 1.25% cholesterol and 0.5% cholic acid, but little or no Th2 shift is observed with the 0.15% cholesterol diet that is typically used to assess the anti-atherosclerotic effects of statins in apoE−/− mice (49). The diet used in the current study contains 0.15% cholesterol and no cholic acid. Thus, it is unlikely that the Th2 shift seen with severe hypercholesterolemia and dietary supplementation of cholic acid would confound our observations of a statin-induced Th2 shift in gld.apoE−/− mice under the conditions of our assays. Of interest, simvastatin treatment did not affect inflammation in gld mice. We speculate that there is no detectable effect of statins because the Th1 inflammatory response is minimal in gld compared with gld.apoE−/− mice. Consistent with this hypothesis, TNF-α levels are 58% lower in gld than gld.apoE−/− mice (p < 0.05) and simvastatin treatment had little or no effect on the level of this cytokine in gld mice or on the relative ratio of STAT4 to STAT6 phosphorylation levels in lymph nodes (data not shown).
The utility of statin therapy for the treatment of autoimmune disease is currently being investigated in the clinic (16, 17) and animal models (20, 39). Recently, Lawman et al. (50) reported that atorvastatin at a dose of 30 mg/kg/day diminished autoimmune disease in New Zealand Black/New Zealand White F1 mice. This level of statin approaches the lethal dose for this class of drugs (51), and it is much higher than the clinical dose (typically in the range of 0.1–0.5 mg/kg/day). High doses of statins can be toxic through their ability to interfere with synthesis of nonsterol products that are involved in cellular upkeep (52). Whereas it is well established that statins typically do not affect serum lipid levels in mice (14, 18, 19), Lawman et al. (50) reported a 50% decrease in serum cholesterol levels, indicative of high dosing in this model. Thus, an aim of our study was to evaluate a physiological level of simvastatin (0.125 mg/kg/day). Despite no detectable effect on serum lipid levels, statin treatment led to a marked reduction in the autoimmune and lymphoproliferative phenotypes in the gld.apoE−/− mice. These data suggest that statins can act to limit both atherosclerotic and autoimmune phenotypes via anti-inflammatory and immunomodulatory actions that are independent of serum cholesterol levels.
It is becoming increasingly evident that chronic inflammation is a significant component of atherosclerotic disease progression, and there are several possibilities by which the statins may be impacting the development of atherosclerosis in the setting of autoimmunity. First, it is reasonable to consider that statins decrease the severity of autoimmunity, and, consequently, the severity of atherosclerosis. However, statins are widely recognized to reduce atherosclerosis in murine models that display no autoimmune phenotype. Thus, a more likely possibility is that statins directly decrease the inflammatory components that are common to atherosclerosis and autoimmunity. For example, TNF-α is a Th1 proinflammatory cytokine that promotes SLE (53), and it has been shown that apoE−/− mice lacking TNF-α do not develop extensive atherosclerosis (54, 55). Therefore, the statin-mediated reduction in TNF-α and other Th1 cytokines could account for the atheroprotective and immunomodulatory properties of this drug.
Another issue of interest is that simvastatin treatment decreased the amount of apoptotic cells within lymph nodes of gld.apoE−/− compared with vehicle treatment. We have shown previously that accelerated atherosclerosis increases the number of apoptotic bodies within the lymph nodes of gld.apoE−/− mice, and this apoptotic material does not colocalize with macrophages, indicating an impaired recognition and clearance (22). Dysregulated apoptosis has been associated both with SLE pathogenesis (56) and atherosclerosis pathogenesis (57). Thus, statin therapy may decrease the accumulation of apoptotic debris by reducing inflammation in atherosclerotic lesions; however, further studies will be required to elucidate how simvastatin reduces the level of TUNEL-positive apoptotic material in the gld.apoE−/− mice.
The model presented in this study is the first to look at the effects of simvastatin on the inflammatory feedback mechanisms that are observed in a mouse model of accelerated atherosclerosis and autoimmune disease. Our results indicate that simvastatin promotes a shift from Th1 cytokine production toward a more anti-inflammatory phenotype. We propose that these immunomodulatory effects of statins, rather than their lipid-lowering actions, mediate their beneficial effects on accelerated atherosclerosis and autoimmunity in this model.
Footnotes
Abbreviations used in this paper: SLE, systemic lupus erythematosus; LDL, low-density lipoprotein; ANA, anti-nuclear Ab; HMG-CoA, 3-hydroxy-3-methyl-glutaryl coenzyme A; wt, wild type.
Disclosures
The authors have no financial conflict of interest.
This work was supported by National Institutes of Health Grants AG15052, HL77774, HL81587, and AR40197 (to K.W.). T.A. was supported by a National Heart, Lung, and Blood Institute postdoctoral research training fellowship.
References
- 1.Kotzin BL. Systemic lupus erythematosus. Cell. 1996;85:303–306. doi: 10.1016/s0092-8674(00)81108-3. [DOI] [PubMed] [Google Scholar]
- 2.Manzi S. Systemic lupus erythematosus: a model for atherogenesis? Rheumatology. 2000;39:353–359. doi: 10.1093/rheumatology/39.4.353. [DOI] [PubMed] [Google Scholar]
- 3.Lockshin MD, Salmon JE, Roman MJ. Atherosclerosis and lupus: a work in progress. Arthritis Rheum. 2001;44:2215–2217. doi: 10.1002/1529-0131(200110)44:10<2215::aid-art381>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 4.Esdaile JM, Abrahamowicz M, Grodzicky T, Li Y, Panaritis C, duBerger R, Côté R, Grover SA, Fortin PR, Clarke AE, Senécal JL. Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheum. 2001;44:2331–2337. doi: 10.1002/1529-0131(200110)44:10<2331::aid-art395>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 5.Nikpour M, Urowitz MB, Gladman DD. Premature atherosclerosis in systemic lupus erythematosus. Rheum Dis Clin N Am. 2005;31:329–354. doi: 10.1016/j.rdc.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 6.Riboldi P, Gerosa M, Luzzana C, Catelli L. Cardiac involvement in systemic autoimmune diseases. Clin Rev Allergy Immunol. 2002;23:247–261. doi: 10.1385/CRIAI:23:3:247. [DOI] [PubMed] [Google Scholar]
- 7.Van Doornum S, McColl G, Wicks IP. Accelerated atherosclerosis: an extraarticular feature of rheumatoid arthritis? Arthritis Rheum. 2002;46:862–873. doi: 10.1002/art.10089. [DOI] [PubMed] [Google Scholar]
- 8.Lusis AJ. Atherosclerosis. Nature. 2000;407:233–234. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ilowite NT, Samuel P, Ginzler E, Jacobson MS. Dyslipoproteinemia in pediatric systemic lupus erythematosus. Arthritis Rheum. 1988;31:859–863. doi: 10.1002/art.1780310706. [DOI] [PubMed] [Google Scholar]
- 10.Ettinger WH, Goldberg AP, Applebaum-Bowden D, Hazzard WR. Dyslipoproteinemia in systemic lupus erythematosus: effect of corticosteroids. Am J Med. 1987;83:503–508. doi: 10.1016/0002-9343(87)90762-5. [DOI] [PubMed] [Google Scholar]
- 11.Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med. 1998;339:1349–1357. doi: 10.1056/NEJM199811053391902. [DOI] [PubMed] [Google Scholar]
- 12.Shepherd J, Cobbe SM, Ford I, Isles CG, Lorimer AR, MacFarlane PW, McKillop JH, Packard CJ. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia: West of Scotland Coronary Prevention Study Group. N Engl J Med. 1995;333:1301–1307. doi: 10.1056/NEJM199511163332001. [DOI] [PubMed] [Google Scholar]
- 13.Scandinavian Simvastatin Survival Study Group. Randomized trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian simvastatin survival study (4S) Lancet. 1994;344:1383–1389. [PubMed] [Google Scholar]
- 14.Sparrow CP, Burton CA, Hernandez M, Mundt S, Hassing H, Patel S, Rosa R, Hermanowski-Vosatka A, Wang PR, Zhang D, et al. Simvastatin has anti-inflammatory and antiatherosclerotic activities independent of plasma cholesterol lowering. Arterioscler Thromb Vasc Biol. 2001;21:115–121. doi: 10.1161/01.atv.21.1.115. [DOI] [PubMed] [Google Scholar]
- 15.Abud-Mendoza C, de la Fuente H, Cuevas-Orta E, Baranda L, Cruz-Rizo J, Gonzalez-Amaro R. Therapy with statins in patients with refractory rheumatic diseases: a preliminary study. Lupus. 2003;12:607–611. doi: 10.1191/0961203303lu429oa. [DOI] [PubMed] [Google Scholar]
- 16.McCarey DW, I, McInnes B, Madhok R, Hampson R, Scherbakov O, Ford I, Capell HA, Sattar N. Trial of atorvastatin in rheumatoid arthritis (TARA): double-blind, randomized placebo-controlled trial. Lancet. 2004;363:2015–2021. doi: 10.1016/S0140-6736(04)16449-0. [DOI] [PubMed] [Google Scholar]
- 17.Vollmer T, Key L, Durkalski V, Tyor W, Corboy J, Markovic-Plese S, Preiningerova J, Rizzo M, Singh I. Oral simvastatin treatment in relapsing-remitting multiple sclerosis. Lancet. 2004;363:1607–1608. doi: 10.1016/S0140-6736(04)16205-3. [DOI] [PubMed] [Google Scholar]
- 18.Endo A, Tsujita Y, Kuroda M, Tanzawa K. Effects of ML-236B on cholesterol metabolism in mice and rats: lack of hypocholesterolemic activity in normal animals. Biochim Biophys Acta. 1979;575:266–276. [PubMed] [Google Scholar]
- 19.Kita T, Brown MS, Goldstein JL. Feedback regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase in livers of mice treated with mevinolin, a competitive inhibitor of the reductase. J Clin Invest. 1980;66:1094–1100. doi: 10.1172/JCI109938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nath N, Giri S, Prasad R, Singh AK, Singh I. Potential targets of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor for multiple sclerosis therapy. J Immunol. 2004;172:1273–1286. doi: 10.4049/jimmunol.172.2.1273. [DOI] [PubMed] [Google Scholar]
- 21.Fischetti F, Carretta R, Borotto G, Durigutto P, Bulla R, Meroni PL, Tedesco F. Fluvastatin treatment inhibits leukocyte adhesion and extravasation in models of complement-mediated acute inflammation. Clin Exp Immunol. 2004;135:186–193. doi: 10.1111/j.1365-2249.2003.02358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aprahamian T, Rifkin I, Bonegio R, Hugel B, Freyssinet JM, Sato K, Castellot JJ, Jr, Walsh K. Impaired clearance of apoptotic cells promotes synergy between atherogenesis and autoimmune disease. J Exp Med. 2004;199:1121–1131. doi: 10.1084/jem.20031557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, Sessa WC, Walsh K. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med. 2000;6:1004–1010. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meyer TW, Rennke HG. Increased single-nephron protein excretion after renal ablation in nephrotic rats. Am J Physiol. 1988;255:F1243–F1248. doi: 10.1152/ajprenal.1988.255.6.F1243. [DOI] [PubMed] [Google Scholar]
- 25.Ettinger WH, V, Varma K, Sorci-Thomas M, Parks JS, Sigmon RC, Smith TK, Verdery RB. Cytokines decrease apolipoprotein accumulation in medium from Hep G2 cells. Arterioscler Thromb. 1994;14:8–13. doi: 10.1161/01.atv.14.1.8. [DOI] [PubMed] [Google Scholar]
- 26.Kleemann R, Princen HM, Emeis JJ, Jukema JW, Fontijn RD, Horrevoets AJ, Kooistra T, Havekes LM. Rosuvastatin reduces atherosclerosis development beyond and independent of its plasma cholesterol-lowering effect in APOE*3-Leiden transgenic mice: evidence for antiinflammatory effects of rosuvastatin. Circulation. 2003;108:1368–1374. doi: 10.1161/01.CIR.0000086460.55494.AF. [DOI] [PubMed] [Google Scholar]
- 27.Kelley VE, Roths JB. Interaction of mutant lpr gene with background strain influences renal disease. Clin Immunol Immunopathol. 1985;37:220–229. doi: 10.1016/0090-1229(85)90153-9. [DOI] [PubMed] [Google Scholar]
- 28.Taylor PR, Carugati A, Fadok VA, Cook HT, Andrews M, Carroll MC, Savill JS, Henson PM, Botto M, Walport MJ. A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J Exp Med. 2000;192:359–366. doi: 10.1084/jem.192.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Licht R, Dieker JW, Jacobs CW, Tax WJ, Berden JH. Decreased phagocytosis of apoptotic cells in diseased SLE mice. J Autoimmun. 2004;22:139–145. doi: 10.1016/j.jaut.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 30.Cohen PL, Caricchio R, Abraham V, Camenisch TD, Jennette JC, Roubey RA, Earp HS, Matsushima G, Reap EA. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J Exp Med. 2002;196:135–140. doi: 10.1084/jem.20012094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, Nagata S. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science. 2004;304:1147–1150. doi: 10.1126/science.1094359. [DOI] [PubMed] [Google Scholar]
- 32.Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. 1996;4:313–319. doi: 10.1016/s1074-7613(00)80439-2. [DOI] [PubMed] [Google Scholar]
- 33.Takeda K, Tanaka T, Shi W, Matsumoto M, Minami M, Kashiwamura S, Nakanishi K, Yoshida N, Kishimoto T, Akira S. Essential role of Stat6 in IL-4 signalling. Nature. 1996;380:627–630. doi: 10.1038/380627a0. [DOI] [PubMed] [Google Scholar]
- 34.Holz A, Bot A, Coon B, Wolfe T, Grusby MJ, von Herrath MG. Disruption of the STAT4 signaling pathway protects from autoimmune diabetes while retaining antiviral immune competence. J Immunol. 1999;163:5374–5382. [PubMed] [Google Scholar]
- 35.Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. 1996;382:174–177. doi: 10.1038/382174a0. [DOI] [PubMed] [Google Scholar]
- 36.Jacobson NG, Szabo SJ, Weber-Nordt RM, Zhong Z, Schreiber RD, Darnell JE, Jr, Murphy KM. Interleukin 12 signaling in T helper type 1 (Th1) cells involves tyrosine phosphorylation of signal transducer and activator of transcription (Stat)3 and Stat4. J Exp Med. 1995;181:1755–1762. doi: 10.1084/jem.181.5.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Urowitz MB, Bookman AA, Koehler BE, Gordon DA, Smythe HA, Ogryzlo MA. The bimodal mortality pattern of systemic lupus erythematosus. Am J Med. 1976;60:221–225. doi: 10.1016/0002-9343(76)90431-9. [DOI] [PubMed] [Google Scholar]
- 38.Manzi S, Meilahn EN, Rairie JE, Conte CG, Medsger TAJ, Jansen-McWilliams L, D’Agostino RB, Kuller LH. Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: comparison with the Framingham study. Am J Epidemiol. 1997;145:408–415. doi: 10.1093/oxfordjournals.aje.a009122. [DOI] [PubMed] [Google Scholar]
- 39.Youssef S, Stuve O, Patarroyo JC, Ruiz PJ, Radosevich JL, Hur EM, Bravo M, Mitchell DJ, Sobel RA, Steinman L, Zamvil SS. The HMG-CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature. 2002;420:78–84. doi: 10.1038/nature01158. [DOI] [PubMed] [Google Scholar]
- 40.Nakajima A, Hirose S, Yagita H, Okumura K. Roles of IL-4 and IL-12 in the development of lupus in NZB/W F1 mice. J Immunol. 1997;158:1466–1472. [PubMed] [Google Scholar]
- 41.Santiago ML, Fossati L, Jacquet C, Muller W, Izui S, Reininger L. Interleukin-4 protects against a genetically linked lupus-like autoimmune syndrome. J Exp Med. 1997;185:65–70. doi: 10.1084/jem.185.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takahashi S, Fossati L, Iwamoto M, Merino R, Motta R, Kobayakawa T, Izui S. Imbalance towards Th1 predominance is associated with acceleration of lupus-like autoimmune syndrome in MRL mice. J Clin Invest. 1996;97:1597–1604. doi: 10.1172/JCI118584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kinoshita K, Yoo BS, Nozaki Y, Sugiyama M, Ikoma S, Ohno M, Funauchi M, Kanamaru A. Retinoic acid reduces autoimmune renal injury and increases survival in NZB/W F1 mice. J Immunol. 2003;170:5793–5798. doi: 10.4049/jimmunol.170.11.5793. [DOI] [PubMed] [Google Scholar]
- 44.Brosh N, Zinger H, Mozes E. Treatment of induced murine SLE with a peptide based on the CDR3 of an anti-DNA antibody reverses the pattern of pathogenic cytokines. Autoimmunity. 2002;35:211–219. doi: 10.1080/08916930290024584. [DOI] [PubMed] [Google Scholar]
- 45.Masutani K, Akahoshi M, Tsuruya K, Tokumoto M, Ninomiya T, Kohsaka T, Fukuda K, Kanai H, Nakashima H, Otsuka T, Hirakata H. Predominance of Th1 immune response in diffuse proliferative lupus nephritis. Arthritis Rheum. 2001;44:2097–2106. doi: 10.1002/1529-0131(200109)44:9<2097::AID-ART360>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 46.Chan RW, Tam LS, Li EK, Lai FM, Chow KM, Lai KB, Li PK, Szeto CC. Inflammatory cytokine gene expression in the urinary sediment of patients with lupus nephritis. Arthritis Rheum. 2003;48:1326–1331. doi: 10.1002/art.11062. [DOI] [PubMed] [Google Scholar]
- 47.Zhou X, Paulsson G, Stemme S, Hansson GK. Hypercholesterolemia is associated with a T helper (Th)1/Th2 switch of the autoimmune response in atherosclerotic apo E-knockout mice. J Clin Invest. 1998;101:1717–1725. doi: 10.1172/JCI1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Robertson AK, Zhou X, Strandvik B, Hansson GK. Severe hypercholesterolemia leads to strong Th2 responses to an exogenous antigen. Scand J Immunol. 2004;59:285–293. doi: 10.1111/j.0300-9475.2004.01403.x. [DOI] [PubMed] [Google Scholar]
- 49.Plump AS, Smith JD, Hayek T, Aalto-Setälä K, Walsh A, Verstuyft JG, Rubin EM, Breslow JL. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992;71:343–353. doi: 10.1016/0092-8674(92)90362-g. [DOI] [PubMed] [Google Scholar]
- 50.Lawman S, Mauri C, Jury EC, Cook HT, Ehrenstein MR. Atorvastatin inhibits autoreactive B cell activation and delays lupus development in New Zealand Black/White F1 mice. J Immunol. 2004;173:7641–7646. doi: 10.4049/jimmunol.173.12.7641. [DOI] [PubMed] [Google Scholar]
- 51.Asenjo-Barron JC, Cardenas-Vasquez R, Martinez F, Juarez-Oropeza MA, Diaz-Zagoya JC. High lovastatin doses combined with hypercholesterolemic diet induce hepatic damage and are lethal to the CD-1 mouse. Life Sci. 1999;64:2155–2161. doi: 10.1016/s0024-3205(99)00162-9. [DOI] [PubMed] [Google Scholar]
- 52.Skaletz-Rorowski A, Walsh K. Statin therapy and angiogenesis. Curr Opin Lipidol. 2003;14:599–603. doi: 10.1097/00041433-200312000-00008. [DOI] [PubMed] [Google Scholar]
- 53.Svenungsson E, Gunnarsson I, Fei GZ, Lundberg IE, Klareskog L, Frostegard J. Elevated triglycerides and low levels of high-density lipoprotein as markers of disease activity in association with up-regulation of the tumor necrosis factor α/tumor necrosis factor receptor system in systemic lupus erythematosus. Arthritis Rheum. 2003;48:2533–2540. doi: 10.1002/art.11264. [DOI] [PubMed] [Google Scholar]
- 54.Ohta H, Wada H, Niwa T, Kirii H, Iwamoto N, Fujii H, Saito K, Sekikawa K, Seishima M. Disruption of tumor necrosis factor-α gene diminishes the development of atherosclerosis in ApoE-deficient mice. Atherosclerosis. 2005;180:11–17. doi: 10.1016/j.atherosclerosis.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 55.Branen L, Hovgaard L, Nitulescu M, Bengtsson E, Nilsson J, Jovinge S. Inhibition of tumor necrosis factor-α reduces atherosclerosis in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2004;24:2137–2142. doi: 10.1161/01.ATV.0000143933.20616.1b. [DOI] [PubMed] [Google Scholar]
- 56.Munoz LE, Gaipl US, Franz S, Sheriff A, Voll RE, Kalden JR, Herrmann M. SLE: a disease of clearance deficiency? Rheumatology. 2005;44:1101–1107. doi: 10.1093/rheumatology/keh693. [DOI] [PubMed] [Google Scholar]
- 57.Stoneman VE, Bennett MR. Role of apoptosis in atherosclerosis and its therapeutic implications. Clin Sci. 2004;107:343–354. doi: 10.1042/CS20040086. [DOI] [PubMed] [Google Scholar]