

Abstract
The high complexity and large dynamic range of blood plasma proteins currently prohibit the sensitive and high‐throughput profiling of disease and control plasma proteome sample sets large enough to reliably detect disease indicating differences. To circumvent these technological limitations we describe here a new two‐stage strategy for the mass spectrometry (MS) assisted discovery, verification and validation of disease biomarkers. In an initial discovery phase N‐linked glycoproteins with distinguishable expression patterns in primary normal and diseased tissue are detected and identified. In the second step the proteins identified in the initial phase are subjected to targeted MS analysis in plasma samples, using the highly sensitive and specific selected reaction monitoring (SRM) technology. Since glycosylated proteins, such as those secreted or shed from the cell surface are likely to reside and persist in blood, the two‐stage strategy is focused on the quantification of tissue derived glycoproteins in plasma. The focus on the N‐glycoproteome not only reduces the complexity of the analytes, but also targets an information‐rich subproteome which is relevant for remote sensing of diseases in the plasma. The N‐glycoprotein based biomarker discovery and validation workflow reviewed here allows for the robust identification of protein candidate panels that can finally be selectively monitored in the blood plasma at high sensitivity in a reliable, non‐invasive and quantitative fashion.
Keywords: Clinical proteomics, Biomarker discovery, Glycopeptide capturing, Targeted Mass Spectrometry, Selected Reaction Monitoring (SRM)
1. Protein biomarkers for preventive and predictive medicine
The greatest benefits for patients are likely to be realized from the monitoring and management of early stage disease rather than from treatment of late stage disease. This concept, often called preventive medicine, has been a vision for many years. Recent technological advancements along with the information generated by the human genome project offer great hope for making the early detection of diseases a reality within the next few years in many disease settings (Goncalves et al., 2004; Hood et al., 2004; Jain, 2004).
Among the strategies that have the highest potential to realize the promises of preventive medicine is the detection of prognostic and diagnostic protein signatures in blood plasma 1 and other body fluids (Christensen et al., 2008; Gravett et al., 2007; Theodorescu et al., 2006). It has been shown that personalized molecular gene expression signatures can be detected in tissues and that these signatures can aid clinicians to diagnose early stage disease, stratify similar pathologies, and to distinguish those diseases which respond to current therapy from those that do not (Chang et al., 2003; Staunton et al., 2001; van 't Veer et al., 2002; van de Vijver et al., 2002). As an example, in 2007, the FDA cleared the first multivariate molecular test that profiles genetic activity. It is a breast cancer specific molecular prognostic test which correlates the expression pattern of 21 genes in paraffin‐embedded tumor tissue probes with the likelihood of distant reoccurrence in patients with node‐negative, tamoxifen‐treated breast cancer (Paik et al., 2004). Although the test is of highly predictive value it requires the complicated and costly clinical extraction of selective breast tissue samples.
Unfortunately, most human tissues are difficult to access and it is unlikely that human tissue will be routinely analyzed in large populations for the presence of such predictive gene expression signatures. In contrast, human blood is easily accessible for sampling and contains informational cues from all organs which is contacting through a network of arteries, veins and capillaries. During its journey through the cardiovascular system blood has been shown to collect molecular cues consisting of proteins secreted, shed or otherwise released from tissues (Liotta and Petricoin, 2006; Zhang et al., 2007). Therefore, the quantitative protein composition of blood plasma contains information about the state of organs and the whole organism in health and disease – an informational network which needs to be deciphered to allow for remote sensing of specific diseases. The mapping of this informational network requires robust, reproducible and sensitive measurements of single protein markers or selected protein panels. Such protein panels can be thought to reflect the perturbed molecular networks in the disease microenvironment. The task for a successful blood biomarker strategy therefore involves the analysis of the disease perturbed cellular networks, the identification of cellular proteins that indicate the state of the perturbed networks and their detection and quantification in blood plasma.
2. Currently used protein biomarkers and their limitations
Initial attempts to use the information contained in the blood proteome for early diagnosis were focused on the detection and quantitative measurement of single protein markers via affinity reagents. This is exemplified by the best known plasma biomarker, prostate specific antigen (PSA). Despite its now well recognized limited specificity for the detection of prostate cancer, PSA continues to be the most widely used tumor marker in the world. The discovery of PSA is beset with controversy as different researchers discovered it independently using immunological techniques, resulting in different names for the same marker (Rao et al., 2008). Originally, PSA was of interest for immunological reasons. Tissue specific antigens were believed to be targets for specific antibodies in order to destroy cancer (Flocks et al., 1960). Later PSA was also suggested as forensic evidence in cases of rape (Hara et al., 1971). Only in 1987, some 27 years after the first publication, Stamey et al. (1987) suggested in a landmark study to use PSA as a marker for prostate cancer.
PSA was found to be specifically expressed in prostate tissue and to be secreted into the blood stream at elevated levels upon disease progression. A second reason for using PSA as a tumor marker was the availability of specific antibodies for standardized and affordable ELISA blood tests. A second well‐known example of a single protein biomarker is the Her2/neu proto‐oncogene (CD340). This membrane bound receptor tyrosine kinase exemplifies the way scientists in the 1980s attempted to uncover new cancer‐causing oncogenes. By overexpressing genes of interest, the effect of potential oncogenes on cancer induction or development was assayed. In one such study the Her2/neu gene was found to cause breast cancer in rats (Schechter et al., 1984). Later, it was found to be amplified in up to 30% of invasive breast cancers and its over‐expression to be associated with a poor prognosis (Slamon et al., 1987). Elevated plasma levels of CD340 are therefore used as an indicator for higher aggressiveness in breast cancer (Luftner et al., 2003). Her2/neu is not only used as a biomarker, but also as a target of trastuzumab (Herceptin) in anti‐cancer therapy (Baselga et al., 1998). Both examples showcase single proteins which can “leak” from diseased tissue into the blood stream and are indicative for a disease if detected at elevated plasma levels. Unfortunately, neither PSA nor Her2/neu, nor for that matter any other single protein biomarker in clinical use, have sufficiently high sensitivity and specificity to predict the development of a particular form of disease and to accurately detect it at an early stage. In the Prostate Cancer Prevention Trial (PCPT), among 5112 men in the placebo arm of this trial, a PSA level >4 ng/ml had specificity of 93% and a rather low sensitivity of 24% (Thompson et al., 2006), while a study by Cook et al. (2001) revealed that at an upper limit at 15 ng/ml of Her2/neu the specificity for normal breast was 98% and the sensitivity for breast cancer stage IV disease was only 40%. In Her2/neu positive breast cancer patients, a Her2/neu serum concentration cutoff of 37 ng/ml resulted in 95% specificity and 62% sensitivity (Kong et al., 2006).
Therefore, additional test parameters are needed in combination with current biomarker tests to increase their performance. A panel of disease‐specific protein biomarkers is thought to be necessary to narrow down diagnosis and treatment options, and reliable strategies for the discovery of such panels need to be developed. Furthermore, both examples cited above highlight another major limitation for current protein biomarker measurements. Without suitable antibodies or other affinity reagents to sensitively and unambiguously detect and quantify the respective proteins, their validation and use as protein biomarkers has been substantially limited.
3. Protein biomarker discovery strategies and their limitations
Most clinically relevant biomarkers have been discovered serendipitously, as already mentioned in the case of PSA and Her2/neu, or via a circuitous route of trial and error. Substances thought to be associated with a certain disease were further investigated in several directions (Pritzker, 2002). For example, in 1847 Bence Jones detected large quantities of a particular protein in the urine of a multiple myeloma patient (Jones, 2006). More than a hundred years later the protein was identified as a free antibody light chain produced by the tumor (Kyle, 1994) that was also present in blood plasma (Sinclair et al., 1986). It was a 152 years after its first discovery that in the year 1998 the FDA approved a routine immunodiagnostic test for the protein as a diagnostic marker for multiple myeloma. Clearly, such non‐directed biomarker discovery efforts, while occasionally successful, lack the efficiency to be of general utility for medicine.
The emerging field of proteomics with its objective to comprehensively identify and quantify proteomes immediately raised high expectations for plasma biomarker discovery (Srinivas et al., 2002). Most biomarker discovery studies based on proteomics to date attempted to detect proteins specifically associated with disease by the comparative profiling of plasma proteomes (or specific fractions thereof) of healthy control and disease affected donors. Several proteomic techniques have been applied for this purpose, including two‐dimensional gel electrophoresis (Lee et al., 2002), SELDI‐TOF MS (Petricoin et al., 2002), label free LC–MS pattern comparison (Zhang et al., 2005), LC–MS/MS shotgun analysis (Chen and Yates, 2007; Hong et al., 2004; Radulovic et al., 2004), and protein array methods (Janzi et al., 2005; Loch et al., 2007). However, these purely discovery‐driven studies have achieved only modest success. While these methods, either by themselves or in combination, have achieved substantial progress in the quantity and quality of data generated, every presently known plasma proteomic method today still only samples a relatively small fraction of the proteome that mostly consists of the relatively highly expressed proteins (States et al., 2006). Both the large dynamic concentration range of up to 12 orders of magnitude for plasma proteins but also the presence of very high abundance proteins such as serum albumin (35–50 mg/ml) and immunoglobulins (5–18 mg/ml) which mask the lower abundance plasma proteins present major challenges for comprehensive plasma proteome analysis, especially for proteins below the microgram per milliliter concentration limit (Anderson and Anderson, 2002). The discrepancy between the sensitivity of present proteomics methods and the requirements for biomarker discovery is illustrated in Figure 1. The figure indicates the concentration of proteins identified by the HUPO plasma proteome collaborative study (States et al., 2006) and that of currently used plasma biomarkers (Polanski and Anderson, 2006). It is apparent that the concentration ranges of the two populations barely overlap, suggesting that it is unlikely that the continued application of the same methods in further studies will discover new biomarkers. The presently used proteomics methods mainly sample so called classical plasma proteins in the range of μg/ml to mg/ml, i.e. those proteins that carry out their functions in the circulation, thus excluding messengers and leakage products (Putnam, 1976). In contrast, the PSA concentration in blood plasma of healthy individuals is around 2 ng/ml (Herrmann et al., 2004; Ng et al., 2005), and the blood plasma concentration of Her2/neu is in the range of 10 ng/mL, about one order of magnitude higher than for PSA (Wu, 2002). Both plasma biomarkers are thus situated in the lower region of currently known plasma protein abundance levels and the same applies to other plasma biomarkers known today. Thus, future biomarker discovery technologies have to be able to reliably detect plasma proteins in the low ng/ml concentration range, or even below the ng/ml range.
Figure 1.
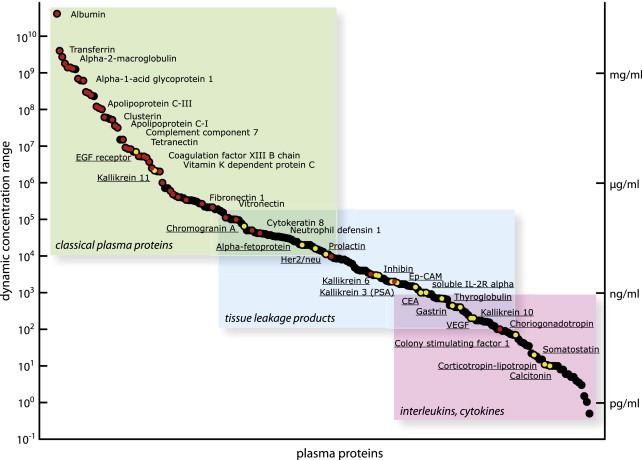
Depicted are the plasma protein concentration as described by Anderson and Anderson (2002). The proteins can be grouped in three main categories (classical plasma proteins, tissue leakage products, interleukins/cytokines). Red dots indicate proteins that were identified by the HUPO plasma proteome initiative (States et al., 2006) and yellow dots represent currently utilized biomarkers (Polanski and Anderson, 2006).
Because currently used LC–MS‐based proteomic methods have great difficulty to analyze low abundance proteins, numerous protein and/or peptide separation methods have been used to reduce plasma sample complexity and thus to increase sensitivity of detection in the thus generated fractions (Issaq et al., 2002). The most common protein fractionation methods use size fractionation by gel electrophoresis or chromatography. While protein fractionation methods performed upstream of a mass‐spectrometric shotgun analysis have to be performed offline, peptide separation based on hydrophobicity can be achieved online and is therefore easier to automate. Although sample fractionation is the key to a higher number and quality for protein identifications, in the context of a biomarker discovery effort such techniques can be problematic for mainly two reasons. First, sample fractionation increases the number of samples to be analyzed, which is time and labor intensive to a degree that routine measurements of larger patient groups become prohibitive. Second, variations along a multi‐step protein separation workflow, e.g. the slightly different distribution of specific proteins in collected fractions, will add another level of bioinformatic complexity toward the detection of disease related patterns. Another popular strategy to achieve higher sensitivity in MS assisted plasma proteome analyses has been the selective removal of high‐abundance proteins such as albumin and the various forms of immunoglobulins by selective immunodepletion. A study published by Echan et al. (2005) showed that effective depletion of six abundant proteins resulted in the ability to load larger equivalent amounts of plasma into downstream separation workflows including 2‐D gels, leading to a more in depth plasma proteome characterization. The removal of the six most abundant plasma proteins depletes 85% of the total plasma protein resulting in an estimated five‐fold enrichment of a potential biomarker (Brand et al., 2006). Another immunodepletion LC column removes 99% of the 20 most high abundance plasma proteins representing 97% of the proteomes giving rise to an up to 20‐fold enrichment as stated by the manufacturer (Sigma–Aldrich). Despite these improvements, the currently used MS assisted biomarker discovery efforts still lack the needed sensitivity and the throughput required to identify biomarker candidates in the low ng/ml concentration range by the comparative analysis of multiple samples.
Based on these observations we conclude that the next generation proteomics‐based protein biomarker discovery strategies need improved analytical sensitivity, robustness and sample throughput in order to reliably detect tissue‐specific protein patterns in plasma with high specificity. Moreover, special attention has to be paid to the time consuming and labor intensive validation of the large datasets produced by these methods in a relatively short time, an issue that so far has been neglected in most studies as pointed out in recent reviews (Domon and Aebersold, 2006; Zolg, 2006).
4. Performance boundaries for proteomic technologies in biomarker discovery
In the segment above we have discussed the limitations of current proteomic methods for protein biomarker discovery. Here we discuss the performance boundaries that have likely to be matched for a method to be successful.
The accessible human body fluids such as plasma, urine, ascites, semen, saliva, seminal plasma and cerebrospinal fluid are thought to contain tens of thousands of different proteins spanning more than 10 orders of magnitude in abundance (Anderson and Anderson, 2002). To comprehensively analyze such samples at the required sample throughput a proteomic technology has to meet a number of so far unmet requirements. First, the technology has to have the sensitivity to identify and quantify minute amounts of proteins in plasma to a concentration of at least low ng/ml, i.e. seven orders of magnitude in concentration below albumin. In that regard good signal to noise ratios are critical to exclude artifactual results. This is particularly challenging if the concentration range assayed exceeds the dynamic range of the MS platform used (Rifai et al., 2006). Second, any proteomic platform for the discovery of candidate disease protein biomarkers must have the capacity for the automated, repetitive and reproducible analysis of hundreds of patient samples in a relatively short time period and in a cost‐effective manner to achieve sufficient statistical power to be clinically useful. Third, clinically relevant diagnostic markers require both high sensitivity and specificity (Gutman and Kessler, 2006). To achieve this goal, any proteomic platform in a biomarker discovery workflow requires robustness in the sense that repetitive measurements achieve coefficients of variance in the low single digit range, and fourth, the assay that measures the protein or sets of proteins in question needs to be portable between laboratories in a way that guarantees comparable results obtained in different studies, given that the sample taking, handling and storage thereof is well controlled and standardized.
Apart from the technological challenges, the quest for standardized protein biomarkers as measurable disease predicting indicators is further complicated by the genetic variation among individuals (Altmüller et al., 2001). This genetic variation causes measurable protein abundance changes within the plasma of individuals that are independent of any disease state, making it difficult to define “normal” protein levels. Furthermore, one has to consider that the plasma proteome is dynamic over time and a function of a multitude of factors (daytime, age, sex, etc.). Therefore, disease related protein abundance changes especially in the onset of a disease can be buried within normal plasma protein fluctuations within the individuals tested (Coombes et al., 2005). On top of this, even at the single protein level an array of protein modifications such as posttranslational modifications as well as point mutations frequently occur expanding the potential variation among patients boundlessly (Nedelkov et al., 2005). In order to circumvent some of the above mentioned challenges at least in the biomarker discovery phase genetically stable mouse models of disease could simplify the initial protein biomarker candidate selection (Kuick et al., 2007).
5. A role for proteomic technology in all phases of biomarker discovery
The process of identifying new protein biomarkers can be mainly divided into four major phases as suggested by Rifai et al. (2006). In the initial discovery phase, proteins of differential abundance in plasma are identified and classified. Subsequently, promising candidates are qualified in a second phase and a subset of these verified in a third phase. In the last phase, the surviving candidates are validated as potential biomarkers by using a specifically developed high‐throughput assay, usually ELISA. During this four‐step biomarker discovery process, the number of samples that need to be analyzed increases while the number of potential biomarker candidates decreases from initially hundreds to a few candidates. These remaining candidates must be verified and validated by showing discriminative power in clinical studies among large cohorts of cancer positive and negative patients. The rationale for choosing this path of progressive attrition of biomarker candidates is rooted in the practical challenge to quantify large numbers of proteins in large numbers of samples, rather than in fundamental considerations. In fact, in an ideal scenario all the putative biomarkers would be subjected to rigorous validation in large sample sets, thus avoiding the application of arbitrary rules to reduce the candidate pool at each step.
Until very recently mass spectrometry has been used almost exclusively for the identification of potential biomarker candidates, whereas their verification and validation have traditionally been carried out by higher throughput affinity methods. Emerging new MS‐based analytical platforms with increased selectivity and sensitivity have now the capacity to be instrumental not only in the initial phase of biomarker discovery but also for the follow‐up studies in clinical settings. Multiplexed measurements of biomarker candidates via targeted MS methods such as selected ion monitoring (SRM; also referred to as multiple reaction monitoring, MRM) have the potential to speed‐up the expensive and time‐consuming biomarker verification and validation phases (Lange et al., 2008b). Therefore, the application of emerging targeted mass spectrometry‐based proteomics methods with their proven ability to reliably and sensitively detect and quantify pre‐determined sets of proteins in complex samples will be instrumental for protein biomarker discovery as well as qualification, verification and validation, respectively (Whiteaker et al., 2007).
6. Generation of biomarker candidate sets by quantitative cell and tissue proteomics
Above we discussed the challenges in identifying protein biomarker candidates by comparative plasma proteomics. Compared to detecting meaningful disease related protein differences in plasma the challenges of identifying proteins that differentiate cancerous and normal cells and tissues are significantly reduced. This is due to the fact that the protein concentration range in tissue is expected to be lower than in blood which facilitates the measurement of a higher percentage of the proteome in a single analysis (Eriksson and Fenyö, 2007; Tyers and Mann, 2003). Cell lines have the additional advantage that in most cases the amount of sample needed for MS analysis is not limited which makes cell lysates compatible with extensive fractionation schema, further increasing the likelihood of discovering proteins of lower abundance. A key benefit of tissue samples is the fact that the differential proteome profiles can be directly investigated at the origin of the disease. Therefore, disease indicating protein concentration differences are expected to be more pronounced in suitable tissue samples compared to the blood stream where the relevant tissue derived proteins are expected to be detected after significant dilution. Assuming that a protein gets secreted from the prostate into the blood, the protein would be a thousand times more concentrated in the tissue by simply comparing the volume of prostate and plasma. In turn, protein abundance ratio changes in the tissue compared to blood are also expected to be higher and thus easier to detect with the MS‐based quantification strategies currently available. In addition, the biological material assayed for discovery can also originate from proximal fluids, i.e. biofluids in close or direct contact with the site of disease. In contrast to blood, it is highly plausible that the protein concentration of potential biomarkers is enriched in such a “sink” and that therefore such liquids are valuable resources for initial biomarker discovery (Celis et al., 2004; Soltermann et al., 2008).
For the detection of biomarker candidates from tissue or proximal fluids, it is critical to start out with a well‐defined group of samples, i.e. the disease samples must be classified clearly and differentiated from the control group (Rifai et al., 2006). For statistical analysis, at least three independent samples of each condition need to be available to account for biological variations (Molloy et al., 2003). In order to follow the progression of the disease, it is also beneficial to have defined samples at different stages of disease development, as pointed out earlier. Conditional gene knock‐in/out models leading to a specific disease phenotype are excellent systems as starting points for biomarker discovery efforts, provided that they closely recapitulate the known human disease stages (Pitteri et al., 2008). Such systems offer the opportunity for sampling at the very early stage of the disease where the genetic preposition for the disease is present but no disease‐specific phenotype is detectable. Upon the possible sampling at the onset of the disease, genetic model systems also allow for consistent sampling at different disease stages.
Cancerous diseases are categorized according to the following stages as defined by the National Cancer Instiute (NCI, 2004): Stage 0 – the amount of cancerous cells is relatively small and constrained to the organ within which it developed. Stage I–III – from stage I to III, the cancer gets more extensive and the tumor size increases. Sometimes nearby lymph nodes contain cancer cells and the cancer spreads to organs adjacent to the primary tumor. Stage IV – the cancer has spread from where it started to another body organ, such as the liver, bones or lungs (see Table 1). Thus, a valid animal model should recapitulate the different stages so that valuable conclusions can be drawn from the discovery phase. Nowadays, cancer progression is described using TNM staging and various disease‐specific grading such as the Gleason score for prostate cancer (Gleason, 1992). In the TNM staging system the disease is assessed using a combination of tumor size or depth (T), lymph node spread (N), and presence or absence of metastases (M) (Ludwig and Weinstein, 2005).
Table 1.
Description of the different stages used TNM classification as defined by NCI (2004).
| Stage | Definition |
|---|---|
| Stage 0 | Carcinoma in situ (early cancer that is present only in the layer of cells in which it began). |
| Stage I, Stage II, and Stage III | Higher numbers indicate more extensive disease: greater tumor size, and/or spread of the cancer to nearby lymph nodes and/or organs adjacent to the primary tumor. |
| Stage IV | The cancer has spread to another organ. |
Valid disease and benign control tissue samples, or cell lines representing different disease states are crucial resources for MS‐assisted biomarker discovery, especially if used with new mass spectrometry‐based workflows of increased throughput and sensitivity (Rifai et al., 2006).
In the following, we describe a new biomarker discovery strategy based upon the directed analysis of glycoproteins in plasma. The approach presented circumvents most of the above mentioned limitations and supports the multiplexed measurement of protein targets with increased sensitivity and high quantitative accuracy, and the throughput required for discovering and evaluating new biomarker candidates.
7. The glyco‐proteome enrichment strategy
In searching for a method having the potential to detect tissue‐specific protein signatures in blood plasma, we have developed methods for the selective analysis of de‐glycosylated peptides that are N‐glycosylated in the intact protein, termed solid‐phase extraction of N‐glycopeptides (SPEG) (Zhang et al., 2003). We refer to these peptides as N‐glycosites. The focus on the subproteome of N‐glycosites is based on the fact that most proteins that are localized on the cell surface or secreted from cells are glycosylated (Gahmberg and Tolvanen, 1996). Our working assumption was that disease‐associated glycoproteins secreted or shed from cell surfaces, or otherwise released from tissue, might be detectable by remote detection in the blood stream. The potential of such a strategy focusing onto the N‐glycosite subproteome was further supported by a re‐evaluation of a list of current plasma biomarkers published by Polanski and Anderson (2006). Thirty out of the 38 proteins within the list of protein biomarkers currently used in the clinic and thus a vast majority is known to be glycosylated (Table 2).
Table 2.
List of markers in clinical use including their status of glycosylation.
| Protein Names | Plasma concentration in controls pg/ml | Clinical Markers | SwissProt # (human) | Glycosylation | FDA approved |
|---|---|---|---|---|---|
| Alkaline phosphatase, placental type | √ | P05187 | yes | ||
| Alpha‐fetoprotein | 2.00E+04 | √ | P02771 | yes | √ |
| CA 125 | √ | Q8WXI7 | yes | √ | |
| CA 15.3 | √ | P15941 | yes | √ | |
| CA 19.9 | √ | x | yes | √ | |
| CA 27.29 | √ | x | yes | ||
| CA 72‐4 | √ | x | yes | ||
| Carcinoembryonic antigen | 1.00E+03 | √ | P06731 | yes | √ |
| Choriogonadotropin beta chain | 1.00E+02 | √ | P01233 | yes | |
| Chromogranin A (parathyroid secretory protein 1) | 6.50E+04 | √ | P10645 | yes | |
| Colony stimulating factor 1 (macrophage) | 7.00E+01 | √ | P09603 | yes | |
| Complement factor H related protein | √ | Q03591 | yes | √ | |
| Corticotropin‐lipotropin contains ACTH | 1.10E+01 | √ | P01189 | yes | |
| Epidermal growth factor receptor | 6.94E+06 | √ | P00533 | yes | |
| Follicle‐stimulating hormone | √ | P01225 | yes | ||
| Hepatocyte growth factor | 2.00E+02 | √ | P14210 | yes | |
| Inhibin | 3.00E+03 | √ | P05111 | yes | |
| Kallikrein 10 | 4.39E+02 | √ | O43240 | yes | |
| Kallikrein 11 | 2.15E+06 | √ | Q9UBX7 | yes | |
| Kallikrein 3 (prostate specific antigen) | 1.86E+03 | √ | P07288 | yes | √ |
| Kallikrein 5 | √ | Q9Y337 | yes | ||
| Kallikrein 6 | 2.90E+03 | √ | Q92876 | yes | |
| Kallikrein 7 | √ | P49862 | yes | ||
| Kallikrein 8 | √ | O60259 | yes | ||
| Luteinizing hormone‐releasing hormone receptor | √ | P22888 | yes | ||
| Mesothelin | √ | Q13421 | yes | ||
| MK‐1 protein, Ep‐CAM | 2.00E+03 | √ | P16422 | yes | |
| OVX1 | √ | x | yes | ||
| Prolactin | 1.60E+04 | √ | P01236 | yes | √ |
| soluble IL‐2R alpha | 1.42E+03 | √ | P01589 | yes | |
| Somatotropin growth factor, growth hormone | 4.00E+02 | √ | P01241 | yes | |
| Thyroglobulin | 1.00E+03 | √ | P01266 | yes | √ |
| V‐erb‐b2, Her2/neu | 1.12E+04 | √ | P04626 | yes | √ |
| Vascular endothelial growth factor A, VEGF | 2.01E+02 | √ | P15692 | yes | |
| Calcitonin | 1.00E+01 | √ | P01258 | no | |
| Estrogen receptor 1 | √ | P03372 | no | ||
| Gastrin | 6.90E+02 | √ | P01350 | no | |
| Insulin | √ | P01308 | no | ||
| Parathyroid hormone‐related protein | √ | P12272 | no | ||
| Progesterone receptor | √ | P06401 | no | ||
| Somatostatin | 2.00E+01 | √ | P61278 | no | |
| Vasoactive intestinal peptide | √ | P01282 | no |
8. Identification of N‐glycosites from plasma
Protein glycosylation has long been recognized as a common co‐translational modification. Typically, carbohydrates are linked to serine or threonine residues (O‐linked glycosylation) or to asparagine residues (N‐linked glycosylation). N‐linked glycosylation sites generally fall into the NxS/T sequence motif in which × denotes any amino acid except proline. In contrast, a consensus primary amino acid sequence for O‐glycosylation sites has not been identified. Glycoproteins can be enriched either via lectins (Yang and Hancock, 2004) leaving the carbohydrate structure intact or via coupling to a hydrazide support (Bayer et al., 1988). In this case, the carbohydrates are oxidized with sodium periodate and the aldehydes formed for affinity purification can be covalently coupled to a hydrazide containing support as described by Bayer et al. (1988) and Zhang et al. (2003). For subsequent mass‐spectrometric identification, N‐glycosites can be specifically released from the solid support by PNGase F. The catalytic action of the enzyme also converts the formerly glycosylated asparagine residue via deamidation into aspartic acid. This enzymatic conversion leads to a mass shift of 0.98 Da which can readily be detected by high mass accuracy mass spectrometers. The MS detectable mass shift improves the confidence of the peptide identification and unambiguously identifies the asparagine residue(s) to which the carbohydrate was linked in the intact protein. We have investigated the rate of miss assignment of the monoisotopic peak on an LTQ‐FTICR from Thermo Finnigan using a monoisotopic toggle and found that in 99.5% of the time the right monoisotopic signal was indeed identified, which is a requisite for the unambiguous assignment of N‐glycosites (unpublished result). Optionally, to gain increased confidence in the N‐glycosite identification, the hydrolysis of the glycan–asparagine bond catalyzed by PNGase F can be performed in heavy water (D2O), which leads to an increased mass shift of 1.98 Da.
9. Selective isolation of N‐glycosites from cells and tissues
While soluble glycoproteins in plasma can be readily isolated and analyzed, glycoproteins embedded in cellular membranes within tissues are more difficult to isolate and identify. Typically, the tissue/cell samples have to be homogenized prior to glycoprotein isolation, or a cell free supernatant of collagenase digested tissues has to be used for N‐glycosites extraction as described by Tian et al. (2007).
To isolate the N‐glycosites specifically from cell surface glycoproteins we have developed a variant of the SPEG method, the cell surface capturing (CSC) method, where glycoproteins can be selectively enriched from the plasma membrane of intact, living cells (unpublished data/manuscript under revision, Wollscheid et al., Nature Biotechnology, 2008). CSC allows for the selective isolation, identification and quantification of cell surface glycoproteins, and the MS data reveals a snapshot of the cell surface protein landscape at the time of labeling. The selective and multiplexed identification of cell surface glycoproteins of a specific cell type is especially interesting for targeted therapeutic approaches. Almost two‐thirds of the currently used therapeutic targets are among these plasma membrane proteins (Yildirim et al., 2007).
In contrast to the identification of N‐glycosites, O‐glycosites are more difficult to study mainly due to a lack of a consensus sequence around the carbohydrate attachment site and the lack of an enzyme analogous to PNGase F that generally removes O‐linked carbohydrate from the glycoprotein. Thus, chemical approaches for the efficient de glycosylation of O‐glycosites in complex samples, such as beta‐elimination are currently being explored albeit with limited success so far.
To date we have developed a suite of methods for the specific MS identification of N‐glycosites from the cell surface, complete cells, tissue and plasma. All these methods have been extensively applied toward the identification of N‐glycosites from human and murine cells, tissue and plasma. In particular, plasma samples were extensively fractionated in several dimensions on the protein and peptide level to reach an extensive coverage of the human plasma glycoproteome (unpublished data). The identified peptides/proteins were consistently annotated and imported into the established database UniPep (http://www.unipep.org), a publicly accessible repository for N‐glycosites (Zhang et al., 2006). UniPep protein entries are annotated by the number of times a particular N‐glycosite was observed including relevant meta information about the source of origin and associated parameters which are critical for biomarker discovery efforts. UniPep is part of the PeptideAtlas project (http://www.peptideatlas.org) which comprises a growing publicly accessible database of peptides not restricted to N‐glycosites that were detected in many MS‐based proteomic studies (Deutsch et al., 2005) and an instance of the PeptideAtlas database (Desiere et al., 2005). Such databases are cornerstones for the future target selection of peptides in emerging MS‐assisted biomarker studies relying on directed proteomic workflows as reviewed by Deutsch et al. (2008).
10. Detection of cell/tissue N‐glycosites in plasma
As pointed out earlier, we initially assumed that proteins released by tissue (secreted, shed or otherwise released) into the blood stream could be detected in plasma for remote sensing of the state of specific cells/tissues in health and disease. Our approach, the comprehensive analysis of N‐glycosites seemed to be ideally suited for the remote sensing of such signatures, due to the fact that glycoproteins and therefore N‐glycosites are an information‐rich subproteome with the benefit of a reduced proteome complexity. In initial studies we therefore set out to determine whether N‐glycosites identified in various cell and tissue samples were represented in the plasma by comparing the MS identified N‐glycosites within the relational database UniPep, being the perfect tool for such a comparison. As shown in Figure 2a, a large number of N‐glycosites and proteins that were identified from lymphocytes, bladder, prostate, breast and liver were also detected in the respective plasma samples (Zhang et al., 2007). The data provided proof that it is possible to identify cellular N‐glycosites in the plasma. However, the data represent only an indirect proof of our concept, since the N‐glycosites identified within the plasma cannot be attributed at this point directly to the cell or tissue of origin. For example, we identified 202 unique N‐glycosites in both prostate tissue and plasma. Of these, 96 likely to originate from classic plasma proteins, 94 are likely to have originated from prostate tissue and cells, and the remaining 12 originated from hypothetical proteins with no protein information to determine their source.
Figure 2.
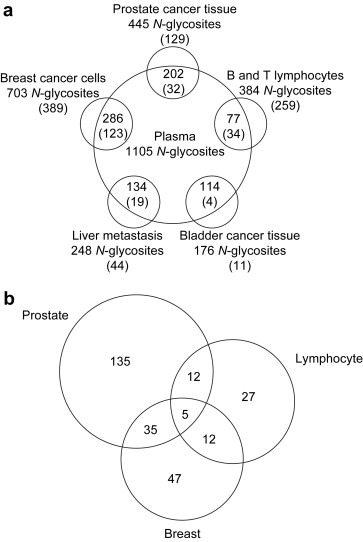
Schematic diagram of analysis of N‐glycosites from tissues/cells and plasma. Cell surface proteins and secreted proteins from tissues/cells and plasma are processed using glycopeptide capture method, glyco‐peptides are analyzed by mass spectrometry and identified by SEQUEST search. The identified peptides and proteins from tissues/cells and plasma are compared and the tissue/cell specific proteins are identified. Figure was adapted from Zhang et al. (2007).
Nevertheless, a subsequent comparison of N‐glycosites identified from SK‐BR‐3 breast cancer cells and Jurkat T lymphocytes by using the CSC technology with prostate N‐glycosites and plasma identified N‐glycosites provided further evidence for our concept. As shown in Figure 2a, 77 peptides identified from lymphocytes and 286 peptides from breast cancer cells were also detected in plasma. When we compared the peptides identified from lymphocytes and breast cancer cells with the peptides identified from prostate tissue, only 5 peptides were found to be common to all three samples (Figure 2b). This indicates that N‐glycosites derived from cells and tissues can be detected in plasma and might be linked to their specific cell/tissue of origin. Collectively these data indicate that we are able to detect cell/tissue‐derived N‐glycosites specifically in plasma via newly developed N‐glycosite capturing workflows in combination with tandem mass spectrometry.
11. Quantification of N‐glycosites
Peptides extracted from either cells, tissue or plasma can be MS quantified by using a number of stable isotope labeling technologies as reviewed by Bantscheff et al. (2007) and Mueller et al. (2008). The introduction of stable isotopes through stable isotope labeling into protein samples has the advantage that after the labeling process the samples can be processed in parallel and thus the variability among the samples can be limited (Gygi et al., 1999). However, isotopic labeling increases sample complexity due the differential labeling and combining the samples. Furthermore, the number of samples that can be compared directly is limited by the number of isotopic labels (ICAT/2; iTRAQ/4‐8; SILAC/3). In contrast, recently emerging label‐free quantification workflows using peptide elution ion trace profiles (Listgarten and Emili, 2005; Mueller et al., 2007) or semi‐quantitative methods using spectral counting (Ishihama et al., 2005; Liu et al., 2004) have the advantage that they are not limited in the number of samples analyzed, rather by the sample amount itself. Although label‐free quantification of peptides reduces the individual sample manipulation steps, which is beneficial for pattern detection, the workflow requires the independent MS analysis of the samples. This in turn requires sophisticated computational tools for the alignment of the MS runs and the subsequent quantification of peptide ratios. Because this approach seemed suitable for the bioinformatic MS analysis of clinical samples in a high‐throughput manner we developed the software SuperHirn (Mueller et al., 2007) and applied label‐free quantitative proteomics to the detection of N‐glycosites. The experiments revealed that the combination of label‐free quantification and SuperHirn are a robust quantitative technology platform to profile N‐glycosites (Schiess et al., 2008).
12. Selective quantification of tissue derived N‐glycosites in plasma by targeted mass spectrometry
To identify N‐glycosites with diagnostic information in plasma it is necessary to screen multiple plasma samples for the presence of the respective proteins in a selective, parallel and absolute quantitative fashion. This can be accomplished by combining the previously generated knowledge about N‐glycosites with targeted MS assisted via SRM. The requirements for targeted MS are two‐fold. First, one needs to know which proteins/peptides are to be targeted and secondly, a selective SRM assay has to be established for the absolute quantification of the protein of interest. The necessary information about individual N‐glycosites required for establishing the SRM assay can be retrieved either by searching the UniPep (or PeptideAtlas) databases, or bioinformatically estimated using a suite of software tools (Kuster et al., 2005; Mallick et al., 2007; Tang et al., 2006). Recently, we have also made an effort to setup a database of targeted proteomics assays to detect and quantify proteins (http://www.mrmatlas.org) (Picotti et al., 2008). For each protein to be measured at least one peptide which is unique for the selected protein, a so‐called proteotypic peptide, has to be chosen (Kuster et al., 2005). By measuring only selected proteotypic peptides, the presence or absence of a protein and its abundance, respectively, can be definitively established. Sets of proteotypic peptides can be detected and quantified very precisely by using triple quadrupole MS or triple quadrupole/linear ion trap hybrid MS instruments by applying SRM. Highly sensitive and selective SRM analyses are performed by monitoring fragmentation channels specific to each peptide of interest (Kuhn et al., 2004). From a technical point of view, a precursor ion is selected by the first quadrupole, fragmented in the second quadrupole and characteristic fragment ions of the precursor are detected and counted upon selection in the third quadrupole by a sensitive detector. The SRM technology ensures higher selectivity by eliminating co‐eluting interferences and thus allows for the detection of low abundance components. This gained increase in sensitivity compared to shotgun MS workflows is critical for the success of MS assisted biomarker discovery efforts. Absolute quantification of the selected peptides can be performed by concomitantly monitoring the fragmentations of the corresponding isotopically labeled peptides that are added to the plasma samples prior to their analysis. A linear response over a wide concentration range of at least five orders of magnitude was observed by Stahl‐Zeng et al. (2007). The detection limit for peptides present in the mixture was around 30atmol (i.e. amount actually injected into the LC/MS system), which translates to a protein concentration in the original plasma sample of some 100 pg/ml. However, it is important to note that this sensitivity could only be achieved by reducing the sample complexity through N‐glycosite capturing. Similar results cannot be achieved to date by measuring whole plasma samples due to sample complexity in combination with signal to noise issues in available MS instrumentation. Apart from the increased sensitivity of N‐glycosite SRM assays compared to shotgun proteomic workflows, such a strategy enables the quantitative measurement of currently up to 500 peptides per MS run in a selective, repetitive and automated manner (Lange et al., 2008a).
13. Conclusions
Currently used MS assisted biomarker discovery platforms are not sensitive enough and lack throughput. Sensitivity is mainly hampered by the huge complexity of the protein samples obtained from human body fluids. Here we suggest that MS can play a role in all phases of biomarker discovery. To circumvent current limitations, we suggest enriching for a subproteome, the glycoproteome. The selective focus on this particular subproteome allows for the discovery‐driven identification of glycoproteins in tissue and cell culture followed by the directed analysis of these secreted or otherwise released proteins in blood plasma. Therefore SRM assays have to be established for N‐glycosites originating from those tissue‐derived glycoproteins.
Importantly, new biomarkers must outperform currently available markers. To do so, proteins need to be reliably and routinely detected at the low ng/ml range. Current MS techniques can still be improved in terms of sample throughput and reproducibility as well as software tools for automated SRM scheduling need to be developed and improved. Furthermore, resources for the community such as an SRM atlas need to be built up.
We believe that the proposed strategy by choosing directed MS could speed‐up biomarker discovery (Figure 3). Furthermore we have demonstrated that directed MS in combination with solid phase enrichment of N‐glycosites reaches desired sensitivity and due to the fact that up to 500 candidates can be monitored in parallel, this approach finally has the potential to compete current ELISA techniques in preclinical biomarker evaluation studies.
Figure 3.
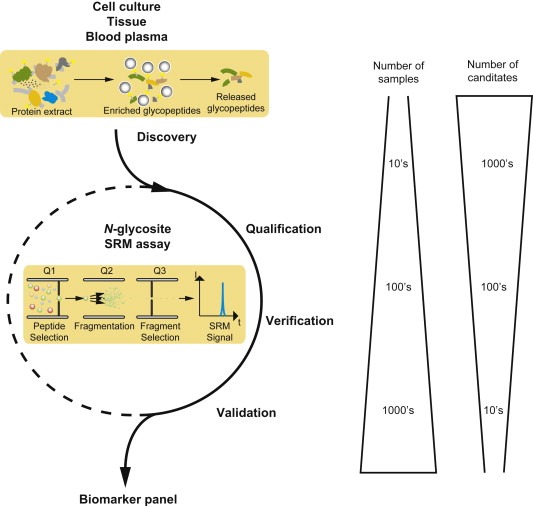
Scheme for biomarker discovery, qualification, verification and validation. Solid phase enrichment of N‐glycopeptides (SPEG) can be performed to discover in vivo disease‐specific signatures using cells, tissue and finally blood plasma and MS‐based label‐free quantification. The selected reaction monitoring (SRM) assays of these protein panel are then qualified and later verified in human patients by SRM and eventually validated again by SRM or ELISA.
Glossary
Selected Reaction Monitoring (SRM): A mass spectrometry based method for the targeted quantification of peptides at high selectivity and sensitivity.
Solid‐Phase Extraction of N‐Glycopeptides (SPEG): Isolation procedure for glycoproteins from biological samples based on hydrazide chemistry.
N‐glycosites: A peptide that is N‐glycosylated in the intact protein in its de‐glycosylated form.
Cell Surface Capturing (CSC): Selective isolation of glycoproteins from the cell surface of living cells.
Schiess Ralph, Wollscheid Bernd, Aebersold Ruedi, (2009), Targeted proteomic strategy for clinical biomarker discovery, Molecular Oncology, 3, doi: 10.1016/j.molonc.2008.12.001.
Footnotes
In this paper, the term plasma is used to indicate serum or plasma.
References
- Altmüller, J. , Palmer, L.J. , Fischer, G. , Scherb, H. , Wjst, M. , 2001. Genomewide scans of complex human diseases: true linkage is hard to find. Am. J. Hum. Genet.. 69, 936–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, N.L. , Anderson, N.G. , 2002. The human plasma proteome: history, character, and diagnostic prospects. Mol. Cell. Proteomics. 1, 845–867. [DOI] [PubMed] [Google Scholar]
- Bantscheff, M. , Schirle, M. , Sweetman, G. , Rick, J. , Kuster, B. , 2007. Quantitative mass spectrometry in proteomics: a critical review. Anal. Bioanal. Chem.. 389, 1017–1031. [DOI] [PubMed] [Google Scholar]
- Baselga, J. , Norton, L. , Albanell, J. , Kim, Y.M. , Mendelsohn, J. , 1998. Recombinant humanized anti-HER2 antibody (Herceptin) enhances the antitumor activity of paclitaxel and doxorubicin against HER2/neu overexpressing human breast cancer xenografts. Cancer Res.. 58, 2825–2831. [PubMed] [Google Scholar]
- Bayer, E.A. , Ben-Hur, H. , Wilchek, M. , 1988. Biocytin hydrazide–a selective label for sialic acids, galactose, and other sugars in glycoconjugates using avidin-biotin technology. Anal. Biochem.. 170, 271–281. [DOI] [PubMed] [Google Scholar]
- Brand, J. , Haslberger, T. , Zolg, W. , Pestlin, G. , Palme, S. , 2006. Depletion efficiency and recovery of trace markers from a multiparameter immunodepletion column. Proteomics. 6, 3236–3242. [DOI] [PubMed] [Google Scholar]
- Celis, J.E. , Gromov, P. , Cabezón, T. , Moreira, J.M. , Ambartsumian, N. , Sandelin, K. , Rank, F. , Gromova, I. , 2004. Proteomic characterization of the interstitial fluid perfusing the breast tumor microenvironment: a novel resource for biomarker and therapeutic target discovery. Mol. Cell Proteomics. 3, 327–344. [DOI] [PubMed] [Google Scholar]
- Chang, J.C. , Wooten, E.C. , Tsimelzon, A. , Hilsenbeck, S.G. , Gutierrez, M.C. , Elledge, R. , Mohsin, S. , Osborne, C.K. , Chamness, G.C. , Allred, D.C. , O’Connell, P. , 2003. Gene expression profiling for the prediction of therapeutic response to docetaxel in patients with breast cancer. Lancet. 362, 362–369. [DOI] [PubMed] [Google Scholar]
- Chen, E.I. , Yates, J.R. , 2007. Cancer proteomics by quantitative shotgun proteomics. Mol. Oncol.. 1, 144–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen, E. , Evans, K.R. , Ménard, C. , Pintilie, M. , Bristow, R.G. , 2008. Practical approaches to proteomic biomarkers within prostate cancer radiotherapy trials. Cancer Metastasis Rev.. 375–385. [DOI] [PubMed] [Google Scholar]
- Cook, G.B. , Neaman, I.E. , Goldblatt, J.L. , Cambetas, D.R. , Hussain, M. , Luftner, D. , Yeung, K.K. , Chan, D.W. , Schwartz, M.K. , Allard, W.J. , 2001. Clinical utility of serum HER-2/neu testing on the Bayer Immuno 1 automated system in breast cancer. Anticancer Res.. 21, 1465–1470. [PubMed] [Google Scholar]
- Coombes, K.R. , Morris, J.S. , Hu, J. , Edmonson, S.R. , Baggerly, K.A. , 2005. Serum proteomics profiling – a young technology begins to mature. Nat. Biotechnol.. 23, 291–292. [DOI] [PubMed] [Google Scholar]
- Desiere, F. , Deutsch, E.W. , Nesvizhskii, A.I. , Mallick, P. , King, N.L. , Eng, J.K. , Aderem, A. , Boyle, R. , Brunner, E. , Donohoe, S. , 2005. Integration with the human genome of peptide sequences obtained by high-throughput mass spectrometry. Genome Biol.. 6, R9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch, E.W. , Eng, J.K. , Zhang, H. , King, N.L. , Nesvizhskii, A.I. , Lin, B. , Lee, H.K. , Yi, E. , Ossola, R. , Aebersold, H.R. , 2005. Human plasma PeptideAtlas. Proteomics. 5, 3497–3500. [DOI] [PubMed] [Google Scholar]
- Deutsch, E.W. , Lam, H. , Aebersold, R.H. , 2008. PeptideAtlas: a resource for target selection for emerging targeted proteomics workflows. EMBO Rep.. 9, 429–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domon, B. , Aebersold, R.H. , 2006. Challenges and opportunities in proteomics data analysis. Mol. Cell Proteomics. 5, 1921–1926. [DOI] [PubMed] [Google Scholar]
- Echan, L.A. , Tang, H.Y. , Ali-Khan, N. , Lee, K. , Speicher, D.W. , 2005. Depletion of multiple high-abundance proteins improves protein profiling capacities of human serum and plasma. Proteomics. 5, 3292–3303. [DOI] [PubMed] [Google Scholar]
- Eriksson, J. , Fenyö, D. , 2007. Improving the success rate of proteome analysis by modeling protein-abundance distributions and experimental designs. Nat. Biotechnol.. 25, 651–655. [DOI] [PubMed] [Google Scholar]
- Flocks, R.H. , Urich, V.C. , Patel, C.A. , Opitz, J.M. , 1960. Studies on the antigenic properties of prostatic tissue. I. J. Urol.. 84, 134–143. [DOI] [PubMed] [Google Scholar]
- Gahmberg, C.G. , Tolvanen, M. , 1996. Why mammalian cell surface proteins are glycoproteins. Trends Biochem. Sci.. 21, 308–311. [PubMed] [Google Scholar]
- Gleason, D.F. , 1992. Histologic grading of prostate cancer: a perspective. Hum. Pathol.. 23, 273–279. [DOI] [PubMed] [Google Scholar]
- Goncalves, A. , Borg, J. , Pouyssegur, J. , 2004. Biomarkers in cancer management: a crucial bridge toward personalized medicine. Drug Discov. Today: Ther. Strategies. 1, 305–311. [Google Scholar]
- Gravett, M.G. , Thomas, A. , Schneider, K.A. , Reddy, A.P. , Dasari, S. , Jacob, T. , Lu, X. , Rodland, M. , Pereira, L. , Sadowsky, D.W. , 2007. Proteomic analysis of cervical-vaginal fluid: identification of novel biomarkers for detection of intra-amniotic infection. J. Proteome Res.. 6, 89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutman, S. , Kessler, L.G. , 2006. The US Food and Drug Administration perspective on cancer biomarker development. Nat. Rev. Cancer. 6, 565–571. [DOI] [PubMed] [Google Scholar]
- Gygi, S.P. , Rist, B. , Gerber, S.A. , Turecek, F. , Gelb, M.H. , Aebersold, R.H. , 1999. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol.. 17, 994–999. [DOI] [PubMed] [Google Scholar]
- Hara, M. , Koyanagi, Y. , Inoue, T. , Fukuyama, T. , 1971. Some physico-chemical characteristics of “-seminoprotein”, an antigenic component specific for human seminal plasma. Forensic immunological study of body fluids and secretion. VII. Nihon Hoigaku Zasshi. 25, 322–324. [PubMed] [Google Scholar]
- Herrmann, W. , Stöckle, M. , Sand-Hill, M. , Hübner, U. , Herrmann, M. , Obeid, R. , Wullich, B. , Loch, T. , Geisel, J. , 2004. The measurement of complexed prostate-specific antigen has a better performance than total prostate-specific antigen. Clin. Chem. Lab. Med.. 42, 1051–1057. [DOI] [PubMed] [Google Scholar]
- Hong, S.H. , Misek, D.E. , Wang, H. , Puravs, E. , Giordano, T.J. , Greenson, J.K. , Brenner, D.E. , Simeone, D.M. , Logsdon, C.D. , Hanash, S.M. , 2004. An autoantibody-mediated immune response to calreticulin isoforms in pancreatic cancer. Cancer Res.. 64, 5504–5510. [DOI] [PubMed] [Google Scholar]
- Hood, L. , Heath, J.R. , Phelps, M.E. , Lin, B. , 2004. Systems biology and new technologies enable predictive and preventative medicine. Science. 306, 640–643. [DOI] [PubMed] [Google Scholar]
- Ishihama, Y. , Oda, Y. , Tabata, T. , Sato, T. , Nagasu, T. , Rappsilber, J. , Mann, M. , 2005. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell Proteomics. 4, 1265–1272. [DOI] [PubMed] [Google Scholar]
- Issaq, H.J. , Conrads, T.P. , Janini, G.M. , Veenstra, T.D. , 2002. Methods for fractionation, separation and profiling of proteins and peptides. Electrophoresis. 23, 3048–3061. [DOI] [PubMed] [Google Scholar]
- Jain, K.K. , 2004. Role of oncoproteomics in the personalized management of cancer. Expert Rev. Proteomics. 1, 49–55. [DOI] [PubMed] [Google Scholar]
- Janzi, M. , Odling, J. , Pan-Hammarstrom, Q. , Sundberg, M. , Lundeberg, J. , Hammarstrom, L. , Nilsson, P. , 2005. Serum microarrays for large scale screening of protein levels. Mol. Cell. Proteomics. 4, 1942–1947. [DOI] [PubMed] [Google Scholar]
- Jones, H. , 2006. On a new substance occurring in the urine of a patient with mollities ossium. Philosophical Transactions of the Royal Society of London (1776–1886). 138, 55–62. [Google Scholar]
- Kong, S.Y. , Nam, B.H. , Lee, K.S. , Kwon, Y. , Lee, E.S. , Seong, M.W. , Lee, D.H. , Ro, J. , 2006. Predicting tissue HER2 status using serum HER2 levels in patients with metastatic breast cancer. Clin. Chem.. 52, 1510–1515. [DOI] [PubMed] [Google Scholar]
- Kuhn, E. , Wu, J. , Karl, J. , Liao, H. , Zolg, W. , Guild, B.C. , 2004. Quantification of C-reactive protein in the serum of patients with rheumatoid arthritis using multiple reaction monitoring mass spectrometry and 13C-labeled peptide standards. Proteomics. 4, 1175–1186. [DOI] [PubMed] [Google Scholar]
- Kuick, R. , Misek, D.E. , Monsma, D.J. , Webb, C.P. , Wang, H. , Peterson, K.J. , Pisano, M. , Omenn, G.S. , Hanash, S.M. , 2007. Discovery of cancer biomarkers through the use of mouse models. Cancer Lett.. 249, 40–48. [DOI] [PubMed] [Google Scholar]
- Kuster, B. , Schirle, M. , Mallick, P. , Aebersold, R.H. , 2005. Scoring proteomes with proteotypic peptide probes. Nat. Rev. Mol. Cell Biol.. 6, 577–583. [DOI] [PubMed] [Google Scholar]
- Kyle, R.A. , 1994. Multiple myeloma: how did it begin?. Mayo Clin. Proc.. 69, 680–683. [DOI] [PubMed] [Google Scholar]
- Lange, V. , Malmstrom, J.A. , Didion, J. , King, N.L. , Johansson, B.P. , Schäfer, J. , Rameseder, J. , Wong, C.H. , Deutsch, E.W. , Brusniak, M.Y. , 2008. Targeted quantitative analysis of Streptococcus pyogenes virulence factors by multiple reaction monitoring. Mol. Cell Proteomics. 7, 1489–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange, V. , Picotti, P. , Domon, B. , Aebersold, R.H. , 2008. Selected reaction monitoring for quantitative proteomics: a tutorial. Mol. Syst. Biol.. 4, 222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S.J. , Evers, S. , Roeder, D. , Parlow, A.F. , Risteli, J. , Risteli, L. , Lee, Y.C. , Feizi, T. , Langen, H. , Nussenzweig, M.C. , 2002. Mannose receptor-mediated regulation of serum glycoprotein homeostasis. Science. 295, 1898–1901. [DOI] [PubMed] [Google Scholar]
- Liotta, L.A. , Petricoin, E.F. , 2006. Serum peptidome for cancer detection: spinning biologic trash into diagnostic gold. J. Clin. Invest.. 116, 26–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Listgarten, J. , Emili, A. , 2005. Statistical and computational methods for comparative proteomic profiling using liquid chromatography-tandem mass spectrometry. Mol. Cell Proteomics. 4, 419–434. [DOI] [PubMed] [Google Scholar]
- Liu, H. , Sadygov, R.G. , Yates, J.R. , 2004. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem.. 76, 4193–4201. [DOI] [PubMed] [Google Scholar]
- Loch, C. , Ramirez, A. , Liu, Y. , Sather, C. , Delrow, J. , Scholler, N. , Garvik, B. , Urban, N. , Mcintosh, M. , Lampe, P. , 2007. Use of high density antibody arrays to validate and discover cancer serum biomarkers. Mol. Oncol.. 1, 313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig, J.A. , Weinstein, J.N. , 2005. Biomarkers in cancer staging, prognosis and treatment selection. Nat. Rev. Cancer. 5, 845–856. [DOI] [PubMed] [Google Scholar]
- Luftner, D. , Lüke, C. , Possinger, K. , 2003. Serum HER-2/neu in the management of breast cancer patients. Clin. Biochem.. 36, 233–240. [DOI] [PubMed] [Google Scholar]
- Mallick, P. , Schirle, M. , Chen, S.S. , Flory, M.R. , Lee, H. , Martin, D.B. , 2007. Computational prediction of proteotypic peptides for quantitative proteomics. Nat. Biotechnol.. 25, 125–131. [DOI] [PubMed] [Google Scholar]
- Molloy, M.P. , Brzezinski, E.E. , Hang, J. , McDowell, M.T. , VanBogelen, R.A. , 2003. Overcoming technical variation and biological variation in quantitative proteomics. Proteomics. 3, 1912–1919. [DOI] [PubMed] [Google Scholar]
- Mueller, L.N. , Brusniak, M.Y. , Mani, D.R. , Aebersold, R.H. , 2008. An assessment of software solutions for the analysis of mass spectrometry based quantitative proteomics data. J. Proteome Res.. 7, 51–61. [DOI] [PubMed] [Google Scholar]
- Mueller, L.N. , Rinner, O. , Schmidt, A. , Letarte, S. , Bodenmiller, B. , Brusniak, M.Y. , Vitek, O. , Aebersold, H.R. , Müller, M. , 2007. SuperHirn – a novel tool for high resolution LC–MS-based peptide/protein profiling. Proteomics. 7, 3470–3480. [DOI] [PubMed] [Google Scholar]
- Staging: questions and answers, 2004. National Cancer Institute Fact Sheet 5.32.
- Nedelkov, D. , Kiernan, U.A. , Niederkofler, E.E. , Tubbs, K.A. , Nelson, R.W. , 2005. Investigating diversity in human plasma proteins. Proc. Natl. Acad. Sci. U S A. 102, 10852–10857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, T.K. , Vasilareas, D. , Mitterdorfer, A.J. , Maher, P.O. , Lalak, A. , 2005. Prostate cancer detection with digital rectal examination, prostate-specific antigen, transrectal ultrasonography and biopsy in clinical urological practice. BJU Int.. 95, 545–548. [DOI] [PubMed] [Google Scholar]
- Paik, S. , Shak, S. , Tang, G. , Kim, C. , Baker, J.O. , Cronin, M. , Baehner, F.L. , Walker, M.G. , Watson, D. , Park, T. , 2004. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N. Engl. J. Med.. 351, 2817–2826. [DOI] [PubMed] [Google Scholar]
- Petricoin, E.F. , Ardekani, A.M. , Hitt, B.A. , Levine, P.J. , Fusaro, V.A. , Steinberg, S.M. , Mills, G.B. , Simone, C. , Fishman, D.A. , Kohn, E.C. , Liotta, L.A. , 2002. Use of proteomic patterns in serum to identify ovarian cancer. Lancet. 359, 572–577. [DOI] [PubMed] [Google Scholar]
- Picotti, P. , Lam, H. , Campbell, D. , Deutsch, E.W. , Mirzaei, H. , Ranish, J. , Domon, B. , Aebersold, H.R. , 2008. A database of mass spectrometric assays for the yeast proteome. Nat. Methods. 5, 913–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitteri, S.J. , Faca, V.M. , Kelly-Spratt, K.S. , Kasarda, A.E. , Wang, H. , Zhang, Q. , Newcomb, L. , Krasnoselsky, A. , Paczesny, S. , Choi, G. , 2008. Plasma proteome profiling of a mouse model of breast cancer identifies a set of up-regulated proteins in common with human breast cancer cells. J. Proteome Res.. 7, 1481–1489. [DOI] [PubMed] [Google Scholar]
- Polanski, M. , Anderson, N.L. , 2006. Candidate cancer markers. Biomarker Insights. 2, 1–48. [PMC free article] [PubMed] [Google Scholar]
- Pritzker, K.P. , 2002. Cancer biomarkers: easier said than done. Clin. Chem.. 48, 1147–1150. [PubMed] [Google Scholar]
- Putnam, F.W. , 1976. The trace components of plasma: an overview. Prog. Clin. Biol. Res.. 5, 1–24. [PubMed] [Google Scholar]
- Radulovic, D. , Jelveh, S. , Ryu, S. , Hamilton, T.G. , Foss, E. , Mao, Y. , Emili, A. , 2004. Informatics platform for global proteomic profiling and biomarker discovery using liquid chromatography-tandem mass spectrometry. Mol. Cell. Proteomics. 3, 984–997. [DOI] [PubMed] [Google Scholar]
- Rao, A.R. , Motiwala, H.G. , Karim, O.M. , 2008. The discovery of prostate-specific antigen. BJU Int.. 101, 5–10. [DOI] [PubMed] [Google Scholar]
- Rifai, N. , Gillette, M.A. , Carr, S.A. , 2006. Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat. Biotechnol.. 24, 971–983. [DOI] [PubMed] [Google Scholar]
- Schechter, A.L. , Stern, D.F. , Vaidyanathan, L. , Decker, S.J. , Drebin, J.A. , Greene, M.I. , Weinberg, R.A. , 1984. The neu oncogene: an erb-B-related gene encoding a 185,000-Mr tumour antigen. Nature. 312, 513–516. [DOI] [PubMed] [Google Scholar]
- Schiess, R., Mueller, L.N., Schmidt, A., Mueller, M., Wollscheid, B., Aebersold, R.H., 2008. Analysis of cell surface proteome changes via label-free, quantitative mass spectrometry. Mol. Cell Proteomics, in press, doi: 10.1074/mcp.M800172-MCP200. [DOI] [PMC free article] [PubMed]
- Sinclair, D. , Dagg, J.H. , Smith, J.G. , Stott, D.I. , 1986. The incidence and possible relevance of Bence-Jones protein in the sera of patients with multiple myeloma. Br. J. Haematol.. 62, 689–694. [DOI] [PubMed] [Google Scholar]
- Slamon, D.J. , Clark, G.M. , Wong, S.G. , Levin, W.J. , Ullrich, A. , McGuire, W.L. , 1987. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 235, 177–182. [DOI] [PubMed] [Google Scholar]
- Soltermann, A. , Ossola, R. , Kilgus-Hawelski, S. , Von Eckardstein, A. , Suter, T. , Aebersold, R.H. , Moch, H. , 2008. N-glycoprotein profiling of lung adenocarcinoma pleural effusions by shotgun proteomics. Cancer. 114, 124–133. [DOI] [PubMed] [Google Scholar]
- Srinivas, P.R. , Verma, M. , Zhao, Y. , Srivastava, S. , 2002. Proteomics for cancer biomarker discovery. Clin Chem. 48, 1160–1169. [PubMed] [Google Scholar]
- Stahl-Zeng, J. , Lange, V. , Ossola, R. , Eckhardt, K. , Krek, W. , Aebersold, R.H. , Domon, B. , 2007. High sensitivity detection of plasma proteins by multiple reaction monitoring of N-glycosites. Mol. Cell Proteomics. 6, 1809–1817. [DOI] [PubMed] [Google Scholar]
- Stamey, T.A. , Yang, N. , Hay, A.R. , McNeal, J.E. , Freiha, F.S. , Redwine, E. , 1987. Prostate-specific antigen as a serum marker for adenocarcinoma of the prostate. N. Engl. J. Med.. 317, 909–916. [DOI] [PubMed] [Google Scholar]
- States, D.J. , Omenn, G.S. , Blackwell, T.W. , Fermin, D. , Eng, J. , Speicher, D.W. , Hanash, S.M. , 2006. Challenges in deriving high-confidence protein identifications from data gathered by a HUPO plasma proteome collaborative study. Nat. Biotechnol.. 24, 333–338. [DOI] [PubMed] [Google Scholar]
- Staunton, J.E. , Slonim, D.K. , Coller, H.A. , Tamayo, P. , Angelo, M.J. , Park, J.H. , Scherf, U. , Lee, J.K. , Reinhold, W.O. , Weinstein, J.N. , 2001. Chemosensitivity prediction by transcriptional profiling. Proc. Natl. Acad. Sci. U S A. 98, 10787–10792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, H. , Arnold, R.J. , Alves, P. , Xun, Z. , Clemmer, D.E. , Novotny, M.V. , Reilly, J.P. , Radivojac, P. , 2006. A computational approach toward label-free protein quantification using predicted peptide detectability. Bioinformatics. 22, e481–e488. [DOI] [PubMed] [Google Scholar]
- Theodorescu, D. , Wittke, S. , Ross, M.M. , Walden, M. , Conaway, M. , Just, I. , Mischak, H. , Frierson, H.F. , 2006. Discovery and validation of new protein biomarkers for urothelial cancer: a prospective analysis. Lancet Oncol. 7, 230–240. [DOI] [PubMed] [Google Scholar]
- Thompson, I.M. , Chi, C. , Ankerst, D.P. , Goodman, P.J. , Tangen, C.M. , Lippman, S.M. , Lucia, M.S. , Parnes, H.L. , Coltman, C.A. , 2006. Effect of finasteride on the sensitivity of PSA for detecting prostate cancer. J. Natl. Cancer Inst.. 98, 1128–1133. [DOI] [PubMed] [Google Scholar]
- Tian, Y. , Zhou, Y. , Elliott, S. , Aebersold, R.H. , Zhang, H. , 2007. Solid-phase extraction of N-linked glycopeptides. Nat. Protoc.. 2, 334–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyers, M. , Mann, M. , 2003. From genomics to proteomics. Nature. 422, 193–197. [DOI] [PubMed] [Google Scholar]
- van 't Veer, L.J. , Dai, H. , van de Vijver, M.J. , He, Y.D. , Hart, A.A. , Mao, M. , Peterse, H.L. , van der Kooy, K. , Marton, M.J. , Witteveen, A.T. , 2002. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 415, 530–536. [DOI] [PubMed] [Google Scholar]
- van de Vijver, M.J. , He, Y.D. , van't Veer, L.J. , Dai, H. , Hart, A.A. , Voskuil, D.W. , Schreiber, G.J. , Peterse, J.L. , Roberts, C. , 2002. A gene-expression signature as a predictor of survival in breast cancer. N. Engl. J. Med.. 347, 1999–2009. [DOI] [PubMed] [Google Scholar]
- Whiteaker, J.R. , Zhang, H. , Zhao, L. , Wang, P. , Kelly-Spratt, K.S. , Ivey, R.G. , Piening, B.D. , Feng, L.C. , Kasarda, E. , Gurley, K.E. , 2007. Integrated pipeline for mass spectrometry-based discovery and confirmation of biomarkers demonstrated in a mouse model of breast cancer. J. Proteome Res.. 6, 3962–3975. [DOI] [PubMed] [Google Scholar]
- Wu, J. 2002 Circulating Tumor Markers of the New Millennium: Target Therapy, Early Detection, and Prognosis American Association for Clinical Chemistry.
- Yang, Z. , Hancock, W.S. , 2004. Approach to the comprehensive analysis of glycoproteins isolated from human serum using a multi-lectin affinity column. J. Chromatogr. A. 1053, 79–88. [PubMed] [Google Scholar]
- Yildirim, M.A. , Goh, K.I. , Cusick, M.E. , Barabási, A.L. , Vidal, M. , 2007. Drug-target network. Nat. Biotechnol.. 25, 1119–1126. [DOI] [PubMed] [Google Scholar]
- Zhang, H. , Li, X.J. , Martin, D.B. , Aebersold, R.H. , 2003. Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotechnol.. 21, 660–666. [DOI] [PubMed] [Google Scholar]
- Zhang, H. , Liu, A.Y. , Loriaux, P. , Wollscheid, B. , Zhou, Y. , Watts, J.D. , Aebersold, H.R. , 2007. Mass spectrometric detection of tissue proteins in plasma. Mol. Cell Proteomics. 6, 64–71. [DOI] [PubMed] [Google Scholar]
- Zhang, H. , Loriaux, P. , Eng, J.K. , Campbell, D.S. , Keller, A. , Moss, P. , Bonneau, R. , Zhang, N. , Zhou, Y. , Wollscheid, B. , 2006. UniPep–a database for human N-linked glycosites: a resource for biomarker discovery. Genome Biol.. 7, R73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H. , Yi, E.C. , Li, X.J. , Mallick, P. , Kelly-Spratt, K.S. , Masselon, C.D. , Camp, D.G. , Smith, R.D. , Kemp, C.J. , Aebersold, H.R. , 2005. High throughput quantitative analysis of serum proteins using glycopeptide capture and liquid chromatography mass spectrometry. Mol. Cell Proteomics. 4, 144–155. [DOI] [PubMed] [Google Scholar]
- Zolg, W. , 2006. The proteomic search for diagnostic biomarkers: lost in translation?. Mol. Cell Proteomics. 5, 1720–1726. [DOI] [PubMed] [Google Scholar]