

Abstract
Our aim was to identify an insulin response element (IRE) in the lipoprotein lipase (LPL) gene. We identified a 19 bp sequence as a putative IRE in LPL non-coding exon 10 using bioinformatics. Upon sequencing the IRE region, a novel 5 bp deletion was identified in Hispanics (N=406) with a carrier frequency of 4.2% but not in non-Hispanic whites (N=604) or Africans. Electrophoretic mobility shift assay revealed binding sites for regulatory factor(s) in muscle cell nuclear extracts with putative IRE sequence. Antibody supershift assay using human aorta smooth muscle cell nuclear extract revealed that Elk-1 specifically binds to putative IRE. TaqMan real-time RT-PCR of the 5 bp deletion, the mutant and wild type cDNA expressed in COS-1 and human muscle cells revealed that the 5 bp deletion was associated with modest reduction in LPL expression. There was also a slight reduction in LPL translation in the deletion mutant. Our data suggest the presence of an IRE in the 3′UTR of the LPL gene.
Keywords: Lipoprotein lipase, Exon 10 mutation, Electrophoretic mobility shift assay
1. Introduction
Lipoprotein lipase (LPL) plays a central role in lipoprotein metabolism, primarily by regulating the catabolism of triglyceride (TG)-rich lipoprotein particles. The regulation of gene expression in LPL occurs at multiple levels and sensitive to several nutritional and hormonal factors. The 3′-UTR region of LPL is important for both mRNA stability and translational efficiency due to inhibitory actions of hormones and catecholamines [1] and absence of the proximal 3′-UTR is associated with translational changes [2]. Numerous studies have shown that LPL gene is sensitive to hormonal regulation of insulin [3,4,5], but the insulin sensitive area in LPL is unknown. Furthermore, LPL activity is reduced in the state of insulin resistance, which is associated with hypertriglyceridaemia. This is because the ability of insulin to up-regulate LPL gene is impaired in insulin resistant individuals, and this contributes to impaired clearance of TG-rich particles in these individuals [6].
Insulin impacts LPL in both adipose and skeletal muscle tissues. Smooth muscle LPL is modestly down-regulated by insulin in normal weight insulin responsive individuals, but is unaffected in insulin resistant obese or type 2 diabetic individuals [7,8]. Insulin influences regulation of expression of many genes involved in energy homeostasis at the levels of transcription, mRNA stability, or translation [9]. The effect of insulin on gene expression is mediated through specific regulatory sequence elements called insulin response elements (IREs). Several distinct classes of IREs (so far eight classes) have been defined that can up/down regulate the expression of a gene [10]. Although most IREs have been found mainly in the promoter region of relevant genes, existence of such elements in intronic regions has also been reported [11]. A gene can be regulated through multiple, yet functionally distinct IREs [12]. Although no trans-acting factor common to all IREs has been defined, a novel transcriptional factor, named insulin-response element-binding protein 1 (IRE-BP1) has been shown to mediate insulin action on multiple target genes involved in PI(3)K/Akt pathway [13]. A consensus sequence of negative IRE in the promoter of several genes, including apolipoprotein C3 (apoC3), which is an inhibitor of LPL, has been identified to contain [T(G/A)TTT(T/G)(G/T)] [14]. The 5′-UTR region of the apoC3 gene (-482 to -455) contains a negative insulin-response element and a mutation in this element results in a defective IRE [14].
The IRE in the LPL gene has not been identified. The purpose of this study was to identify a putative IRE in the LPL gene using a combination of bioinformatics, genetics and functional studies.
2. Materials and Methods
2.1. Insulin response element (IRE) identification
The core sequence of IRE from apoC3 promoter was used as a probe to search for IRE in the LPL gene of ten species using Multiple Sequence Alignment in GCG Software (Accelrys).
2.2. Subjects
The population samples used in this study consisted of 1,011 normoglycemic subjects, comprising 608 non-Hispanic whites (NHWs) and 403 Hispanics, and 193 diabetic Hispanics. Randomly selected 100 Africans were also screened for the LPL exon 10 mutation. The age range of study participants was 20 to 74 years. For the initial mutation screening in exon 10 of LPL, 86 normoglycemic individuals (39 NHWs, 47 Hispanics) falling in the upper tertile of TG and fasting insulin and in the lower tertile of HDL-C were chosen as described earlier by us [15]. Post-heparin blood was collected from subsets of 76 Hispanic normoglycemic individuals in order to analyze the impact of the exon 10 mutation on LPL mass. Blood samples were collected from the participants in the morning after a 12-14 hour fast in 0.1% EDTA. An intravenous heparin bolus of 60 IU/Kg was then administered, and after 10 min, blood was collected in iced lithium-heparin tubes for measurement of LPL mass. Randomly selected 100 Africans were also screened for the LPL exon 10 mutation. The study was approved by the University of Pittsburgh Institutional Review Board.
2.3. Determination of LPL mass
LPL mass was determined by an enzyme immunoassay (ALPCO Diagnostics, Salem, NH). Briefly, 10 μl of post-heparin plasma diluted in a provided phosphate buffer was bound to anti-LPL monoclonal antibody coating a 96-well plate. Anti-LPL polyclonal antibody was added, followed by an enzyme-labeled polyclonal antibody. Finally o-phenylenediamine was added and allowed to develop. The colorimetric reaction was terminated by 7.7% H2SO4. The plates were read on a Tecan Infinate F200 microplate reader (Männedorf, Switzerland). The absorbance at 494 nm is proportional to the amount of LPL in each sample. A provided calibrator was used as a standard.
2.4. Identification of deletion mutation in IRE
A 189 bp fragment in exon 10 containing the putative IRE was amplified using forward (5′-CAGAGTAAAATAAGGCTCCTTC-3′) and reverse (3′-GTAGATGTGTCGTCATACTTA-5′) primers followed by vertical gradient temperature-single strand conformation polymorphism (VGT-SSCP) method as described elsewhere [16]. The 5 bp deletion was confirmed by DNA sequencing and restriction digestion with DdeI.
2.5. LPL cDNA constructs and site-directed mutagenesis
Expression vector of pGEM-4Z containing LPL-full cDNA of 3.6 kb was kindly provided by Dr. Gouri Ranganathan, University of Arkansas. We reconstructed the vector by adding 1.5 kb full length functional human LPL promoter (Supplementary Fig. 1a-c) and confirmed the 5′-3′ orientation by sequencing. The majority of the regulatory elements for LPL expression lies within the first 900 bp of its initiation site and includes TATA Box (-27), OCT1 (-46), CαRM (-54), CAAT Box (-65), LSEs (-81 to -225), cAMP-RE (-306), FSE2 (-362), LP (-440), LP-β (-468), OCT1 (-589), GRE (-664), LP (-669), LP-α (-702), and GRE (-881) [17]. The 5.1 kb promoter-cDNA construct was removed from the vector by double digestion with KpnI and PstI and ligated into pALTER-1 vector using T4 ligase. In this vector, we added a ‘tag’ containing three copies of the termination codon (tga tga tga) by site-directed mutagenesis just before the actual stop codon, TGA (Supplementary Fig. 1d). The incorporation of the above ‘tag’ in the vector enabled us to design a specific primer, which amplified a unique fragment only from the exogenous LPL mRNA. This way, we were able to distinguish exogenous LPL mRNA from endogenous ones. This wild type (WT) vector was used for site-directed mutagenesis to create two mutant constructs: 1) LPL-Mut1 containing the exon 10 mutation with 5 bp deletion (nt. 2015 to 2019 deleted), and 2) LPL-Mut2 containing the deleted 19 bp of putative IRE (nt. 2003 to 2022 deleted) (supplementary Fig. 1e). Each construct was double digested with KpnI and PstI and ligated back to two separate pGEM-4Z vectors. The three LPL constructs (WT, Mut1, Mut2; Supplementary Fig. 1e) were transfected into human aorta smooth muscle or COS-1 cell lines using Optifect™ Transfection Reagent (Invitrogen). β-Gal plasmid DNA was co-transfected with the above LPL plasmids, respectively for the downstream internal control and transfection efficiency assay.
2.6. Cell culture and gene transfection of LPL in human aorta smooth muscle cell Line and COS-1 cells
Human aorta smooth muscle cells (HA-VSMC) and COS-1 cells were obtained from American Type Culture Collection (Rockville, MD). COS-1 cells were cultured with Dulbecco's modified Eagle's medium with 4 mM L-glutamine adjusted to contain 1.5 g/L sodium bicarbonate, 4.5 g/L glucose and 10% fetal bovine serum. Human aorta smooth muscle cells were grown in 60-mm culture dishes in F12K medium with 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin, at 37°C in 5% CO2. Transfection was performed when COS-1 cells approached 70% confluence and HA-VSMC cells approached 60% and one day before transfection, it changed to growth medium 2 ml for 6-well dish without antibiotics. For each 60-mm culture dish, we first diluted 4μg constructed plasmid DNA in 250 μl of Opti-MEM® I Medium (Invitrogen), then diluted 20μl Optifect Transfection Reagent (Invitrogen) in 250μl Opti-MEM and allowed to sit at room temperature for 5 min and finally, combined and mixed the 250 μl of diluted DNA with 250 μl diluted Optifect and allowed to form a complex at room temperature for 20 min. The COS-1 or HA-VSMC cultures were removed from the incubator and the medium from each 60-mm culture dish was aspirated, cells were rinsed twice with 1 ml Opti-MEM® I Medium and replaced with 2 ml Opti-MEM® I Medium. The cells were incubated at 37°C until the time of addition of the DNA-Optifect complexes. After a 20 min incubation period, the 500 μl DNA-Optifect complexes were added to each 60-mm culture dish. The cells were incubated at 37°C in 5% CO2. The medium was replaced with 3 ml of F12K medium (containing 10% FBS) 12 hr after transfection. Transfection efficiency was confirmed by β-galactocidase assay for each transfection experiment.
2.7. Reverse transcription and quantitative real-time PCR for LPL mRNA expression and stability
48 hr after transfection, insulin (10μg/ml, Sigma) was added to the transfected HA-VSMC cells in newly changed medium and incubated at 37°C for 2 hr. The control cells were treated with a diluted carrier (25 μM HEPES) in culture for the same length of time. After treatment the cells were collected and total RNA was extracted using RNAqueous-4PCR Kit with the treatment of TURBO DNase (Ambion Inc., Austin, TX). The absence of genomic contamination of the isolated RNA was confirmed by performing β-actin PCR reactions on the purified RNA samples. The amount of total RNA for each sample was quantitated with the UV spectrophotometer. For each reverse transcription-PCR reaction, 2μg of total RNA was converted into cDNA using M-MLV reverse transcriptase (Ambion Inc., Austin, TX). Five micro liter of reverse transcription reaction was used for quantitative real-time PCR with the 7900 Real-time PCR system and ABI TaqMan® PCR Master Mix (Applied Biosystems). All samples were amplified in triplicate. No-template and no-reverse transcription controls were included in each 96-well plate for amplification. Primers and probes were synthesized at Qiagen and Applied Biosystems. Primers used for LPL were: forward primer, containing triple tga as a ‘tag’ for exogenous LPL amplification: 5′- AAGTCAGGCtgatgatgaTGAAACT-3′ and reverse primer, 5′- CCCCAAACACTGGGTATGTTTT-3′. The TaqMan probe used for LPL was 5′-FAM-CGAATCTACAGAACAAAGAACGGCATGTGAAT-3′TAMRA designed with Primer Express software (Applied Biosystems). The probe contained 6-carboxyfluorescein (FAM) at the 5′ end and 6-carboxytetramethylrhodamine (TAMRA) at the 3′ end and was designed to hybridize to the sequence located between the PCR primers. TaqMan primers and probe for eukaryotic 18S rRNA as endogenous control was from Applied Biosystems. The TaqMan PCR products were verified by Melting Curve analysis and gel electrophoresis and visualized by ethidium bromide staining. The identity of each expected LPL fragment was confirmed by cloning into pCR 4-TOPO vector (Invitrogen) and nucleotide sequence analysis.
2.8. Western blot analysis for LPL translation level and phosphorylations of Elk-1
The cultured HA-VSMC and COS-1 cells were changed to serum-free media 4 hr prior to incubation at 37°C with insulin for 30 min and immediately lysed with M-PER Mammalian Protein Extraction Reagent supplemented with 0.5% SDS and a mixture of Protease Inhibitors and Phosphatase Inhibitors (as above). Cleared total cell lysates were denatured with NuPAGE LDS Sample Buffer plus Reducing agent (Invitrogen) at and resolved by SDS-PAGE on 4-20% NuPAGE gradient Bis-Tris polyacrylamide electrophoresis gel and transferred to PVDF membranes, as previously described [18]. Western blot analysis of LPL expression in HA-VSMC and COS-1 cells were performed using the total cell lysates and probed with anti-LPL monoclonal antibody (Abcam Inc, Cambridge, MA). After stripping with 0.2 M glycine-HCl, pH 2.4 for 1h at room temperature, the blots were re-probed with anti-GAPDH antibody (Abcam Inc, Cambridge, MA) to demonstrate comparable amounts of LPL protein in all the lanes. For Elk-1 phosphorylation analysis the blots were first probed with phospho-Elk-1 antibodies (Cell Signaling Technology Inc., Beverly, MA) and re-probed with anti-Elk-1 antibody (Sigma).
2.9. Competitive electrophoretic mobility shift assay and antibody gel supershift analysis
Electrophoretic mobility shift assay (EMSA) was performed using two double-stranded 29-mer oligonucleotides corresponding to the wild type and 5 bp deletion mutant encompassing the putative IRE in LPL exon 10. For cold EMSA, we developed a simple EMSA technique without use of any radioactive labeling. Briefly, 1.75 pmol of the IRE wild type and mutant oligos were incubated with 2 μ1 (∼20 ng) of the nuclear extracts from human aorta smooth muscle cells (Geneka, Toronto, Canada), and 6 μl of ddH2O at room temperature for 15 minutes. 1 μl of 10× loading buffer [250 mM Tris-HCl, pH 7.5, 40% glycerol, 0.2% bromophenol blue] was added to the reaction and the mixture was loaded on 6% polyacrylamide retardation gel (Novax). The gel was run at 25 °C constant temperature in 0.5× TBE buffer with constant voltage of 250V for 15 minutes. The gel was then stained with a 1:10,000 diluted SYBR Gold (BioProbes) for 10 minutes and photographed. For the competitive EMSA, the oligonucleotides were 5′-end–labeled with α-32P ATP and purified by using the QIAquick Purification Kit (Qiagen). Equally concentrated, nonradioactive competitor DNA (wild type and mutant oligonucleotides) was added at 3×, 10×, 30×, and 100× excess volumes of the labeled probe. The mixture of unlabeled and labeled oligos were incubated with 1.5 μg of aorta smooth muscle cell nuclear extracts (Geneka, Toronto, Canada) for 20 minutes at 25°C in binding buffer (1 mM MgCl2, 0.5 mM EDTA, 0.5 mM dithiothreitol, 50 mM NaCl, 10 mM Tris-HCl pH 7.5, 50μg/ml poly (dI:dC) and 20% glycerol). In antibody gel supershift analysis, aorta smooth muscle cell nuclear extracts were incubated at 4 °C for 15 min with anti-Elk-1 antibody (Sigma) before binding reactions. The DNA-protein complexes were then separated on 5% nondenaturing polyacrylamide gel at 120 V for 2 hours, and the gel was dried and autoradiographed overnight at room temperature.
2.10. Statistical analyses
For genetic studies, allele frequency was calculated by the allele-counting method. Genotype frequency was determined by dividing the number in each genotype by the total number of individuals genotyped. For LPL expression studies statistical significance was determined by ANOVA and Student's t-test, and the levels of probability were noted. The results are expressed as mean ±SD for at least three separate (replicate) experiments for each treatment. For quantitative real time PCR, data analyses were performed with SDS, version 2.1, software (Applied Biosystems, CA). A comparative threshold cycle (Ct) method was applied for the quantification of each LPL construct treated with or without insulin. For each sample, ΔCt is the value obtained by subtracting the Ct value of the endogenous control 18S rRNA from the Ct value of the LPL mRNA. All samples were examined in triplicate and the mean value of ΔCt was used for further comparative analysis. The quantitative data of mRNA expression for LPL was calculated by 2−ΔCt and representative of experimental results obtained from three independent experiments.
3. Results
3.1. Putative insulin response element (IRE) in LPL exon 10
The sequence of the insulin response element (IRE) from the promoter region of the apoC3 gene [14] was used as a probe to search for similar elements in the LPL gene of ten species: human, mouse, cow, pig, sheep, cat, guinea pig, rat, baboon and chicken. The results were aligned and scored for a consensus sequence. The putative IRE was found to be evolutionarily conserved in all LPL sequences examined from various species. In human, mouse, cow and pig, where the LPL gene organization is known, the putative IRE was assigned to exon 10. In other species, where the complete LPL gene organization is unknown, the putative IRE was also assigned to the 3′UTR (Supplementary Fig. 2). Three clusters of consensus sequence were found: (i) TTCT corresponding to positions 9-12 in apoC3 IRE, (ii) TTT corresponding to positions 18-20 in apoC3 IRE, and (iii) TCCAA corresponding to positions 23-27 in apoC3 IRE. Out of the 10 possible scores (10 species examined) at each IRE position in apoC3, a minimum score of 6 was chosen for the consensus sequence that ranged 6-9 for the three clusters. The probability (and computed p-value) for a nucleotide match at a specific position was: 0.9965 (p=0.0035) for 6 matches, 0.9996 (p=0.0004) for 7 matches, 0.9999 (p=2.96×10-5) for 8 matches and 0.9999 (p=9.5×10-7) for 9 matches. Based on this bioinformatics data, we propose the following 19 nucleotide sequence to be a putative IRE in exon 10 of the human LPL gene: TTCTCAAACTTTACTCCAA that corresponds to the three clusters noted above and the intervening sequence from position 9 to 27 in the apoC3 IRE (Supplementary Fig. 2).
3.2. Identification of a 5 bp deletion in the putative IRE of human LPL
The putative IRE is located about 400 bp downstream from the beginning of exon 10, which is 1,947-bp long in the human LPL gene. Thus we targeted this region for mutation detection, rather than screening the whole exon. Initially the 86 normoglycemic individuals (47 Hispanics, 39 NHWs) having the phenotype of high TG/low HDL-C were screened for mutations. Two of the 86 individuals showed a variant pattern on SSCP (Fig. 1A), which upon sequencing revealed a 5 bp deletion within the putative IRE (Fig. 1B, C). The deletion also abolished the restriction site for DdeI (Fig.1D). The 5 bp deletion is flanked by TTT in the 5′ end and AA in the 3′ end in the core sequence of TTTACTCTAA and thus it could comprise TACTC, ACTCT or CTCTA. Following the identification of the 5 bp deletion in 86 individuals, we screened additional 356 normoglycemic Hispanics (total=403) and 569 normoglycemic NHWs (total=608) and 100 Africans to ascertain its frequency in the general population. While 17 of the 403 Hispanics (4.2%) were carriers of the deletion (heterozygous), none of the 608 NHWs and 100 Africans had this mutation. Since the African sample was small we cannot rule out the possibility that it occurs as a rare variant in African or African-derived populations. We also screened 193 diabetic Hispanics for the deletion mutation that found to have a carrier frequency of 3.1% (6 out of 193). The genotype distribution in normoglycemic and diabetic Hispanics were in Hardy-Weinberg equilibrium. The genotype distribution between normoglycemic and diabetic Hispanics was not statistically significant (p=0.511). We invited all Hispanic carriers with the 5 bp to give post-heparin blood for LPL measurement, however, only 6 agreed to do. Additionally, we collected post-heparin blood for 70 age- and sex-matched Hispanics. LPL mass was non-significantly lower in the deletion carriers than non-carriers (448.6 ± 203.0 ng/ml vs. 502.2 ± 105.7 ng/ml; p = 0.55).
Fig. 1.
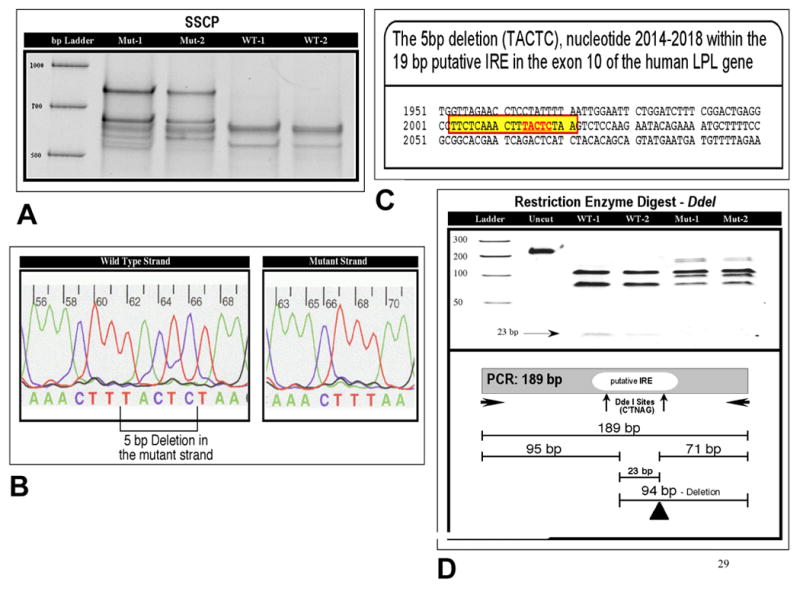
(A) SSCP. The results of SSCP showing mutant (Mut-1, Mut-2) and wild type (WT-1, WT-2) patterns in 4 different samples. (B) 5 bp Deletion in the Mutant Strand. DNA sequence of the wild type and mutant type with 5 bp deletion. The location of this deletion is marked in the wild type strand. (C) Insulin Response Element (IRE). The 19 bp sequence of the putative IRE in exon 10 of the human LPL gene is highlighted with the surrounding sequence. (D) Restriction Enzyme Digest. DdeI restriction banding patters of wild type (WT-1, WT-2) and 5 bp deletion (Mut-1, Mut-2) along with schematic diagram are shown.
3.3. Effect of insulin on LPL Regulation
Two mutant constructs (LPL-Mut1 containing 5 bp deletion in exon 10; LPL-Mut2 containing the deleted 19 bp sequence of putative IRE in exon 10) were created from the expression vector of pGEM-4Z containing full-length LPL cDNA and LPL promoter (see Supplementary Fig. 1). We monitored gene expression of these three LPL constructs regulated by insulin treatment. The wild type and two mutant constructs were expressed in HA-VSMC (Fig. 2A) and COS-1 cells (Fig. 2B) followed by real-time RT-PCR on isolated total RNA. Mock transfection of the empty vector (without LPL gene) showed no amplification of LPL mRNA (data not shown). As compared to the untreated wild type the expression was significantly lower in construct with the untreated 19 bp deletion in HA-VSMC (0.698 ± 0.064 vs. 1.313 ± 0.09; p<0.001). While the insulin treatment increased LPL mRNA expression of wild type (1.313 ± 0.09 vs. 2.695 ± 0.11; p<0.001) and 5 bp deletion (1.083 ± 0.076 vs. 1.402 ± 0.091; p< 0.05), no effect was seen on mRNA from 19 bp deletion construct, indicating that the putative 19 bp IRE is required for insulin action. Similar results were obtained from repeated experiments in COS-1 cells (Fig. 2B).
Fig. 2.

Real-time RT-PCR results of plasmid constructs expressing wild type 5 bp deletion and 19 bp deletion in exon 10 of human LPL in human aorta smooth muscle cells (HA-VSMC) (A) and COS-1 cells (B). Insulin treatment is indicated with (+) and lack of insulin treatment with (–). Mock transfection of the empty vector showed no amplification (not included in the figure). Equivalent amounts of total RNA were reversed transcribed and then the cDNAaliquots used for the RT-qPCR. Samples were amplified in triplicate and normalized internally using the 18S rRNA. Data are representative of experimental results obtained from three independent experiments. *p < 0.05 difference between 5 bp insulin treated and untreated constructs. **p<0.001 difference between wild type insulin treated and untreated constructs, and between wild type untreated and 19 bp untreated constructs (ANOVA).
Immunoblotting analysis was performed to see if deletion mutants influence the LPL gene expression via post-transcriptional or translational events. There was a slight reduction in LPL translation level in the deletion mutants compared to the LPL wild type and insulin treatment increased the LPL translation in wild type as shown in a representative of three repeated experiments in COS-1 cells (Fig. 3A & B). Similar results were also seen from three repeated experiments on HA-VSMC (Fig. 3C & D).
Fig. 3.
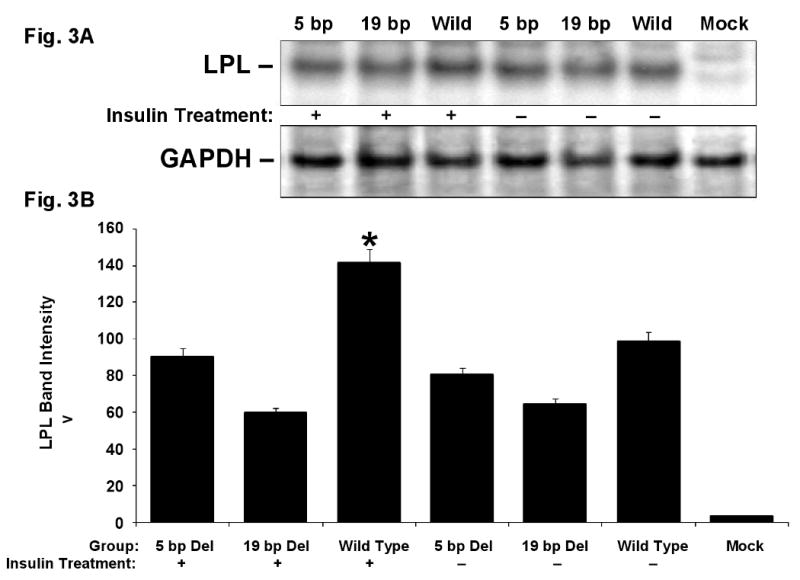
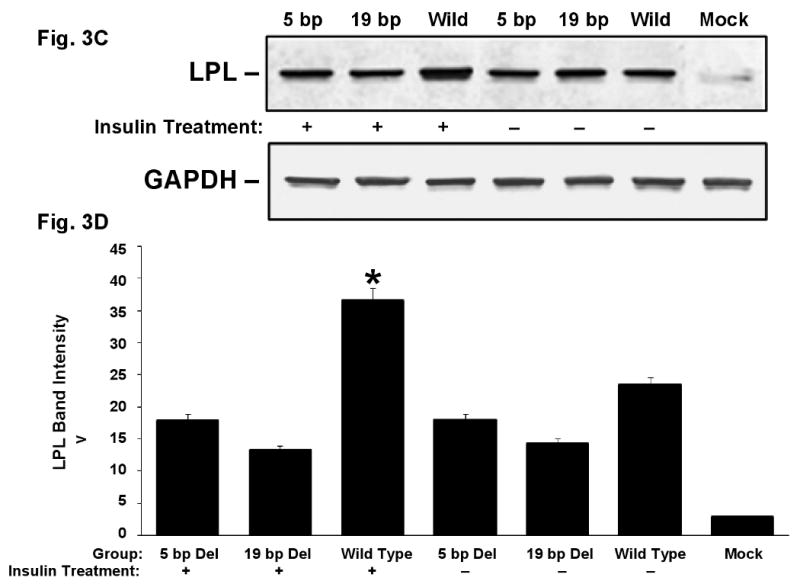
LPL change in translation. COS-1 cells (Fig. A and B) and HA-VSMCs (Fig. C and D) were transfected with the expression vectors for exon 10 wild type, 5 bp deletion mutant, 19 bp deletion mutant and empty vector (mock) and treated with insulin (indicated with “+”) or without insulin (indicated with “–”). A. Representative Western blot of the total COS-1 cell lysates using anti-LPL monoclonal antibody (upper panel). The lower panel shows a re-probing of the same filter with anti-GAPDH monoclonal antibody that demonstrates comparable amounts of protein in all the lanes. B. Quantitative results from three experiments for LPL translation in COS-1 cells after normalized to GAPDH. C. Representative Western blot of LPL (upper panel) and GAPDH (lower panel) from HA-VSMC lysates. D. Quantitative results from three experiments for LPL translation in HA-VSMCs after normalized to GAPDH. *p<0.05 between the wild type with insulin treatment and the rest of the constructs.
3.4. EMSA and supershift analysis
To assess if the putative IRE binds to a transcription factor and if the 5 bp deletion in the putative IRE affects the binding of this factor, we performed both cold and hot EMSA using nuclear extracts from human smooth muscle cell and 32P-labeled oligonucleotide probes corresponding to the wild type and 5 bp deletion mutant. A DNA-protein complex was detected on EMSA, indicating that a transcription factor binds to the putative IRE and that the 5 bp deletion was associated with less affinity with the transcription factor than the wild type (Fig. 4A). To confirm the specific binding we performed competitive EMSA to examine if this mutation alters the affinity of a transcription factor (Fig. 4B). When increasing amounts of unlabeled competitor oligos (wild type or 5 bp deletion mutant) competed with the 32P-labeled wild type oligo, it completely abolished the radiolabeled DNA-protein complex at the 100 fold molar excess amount of wild type competitor oligos. Comparison of lane 3-6 versus lane 7-10 in Fig. 4B shows that the wild type oligo had an average ∼3-fold higher binding affinity than the 5 bp deletion oligos. In contrast, excess amounts of unrelated oligonucleotides did not affect the formation of the DNA-protein complex (data not shown). Identical allele-dependent differences in binding were observed on repeating EMSA at least 5 times. These data indicate that the 5 bp deletion in 3′UTR affects the binding of a putative transcription factor in a sequence-specific manner.
Fig. 4.
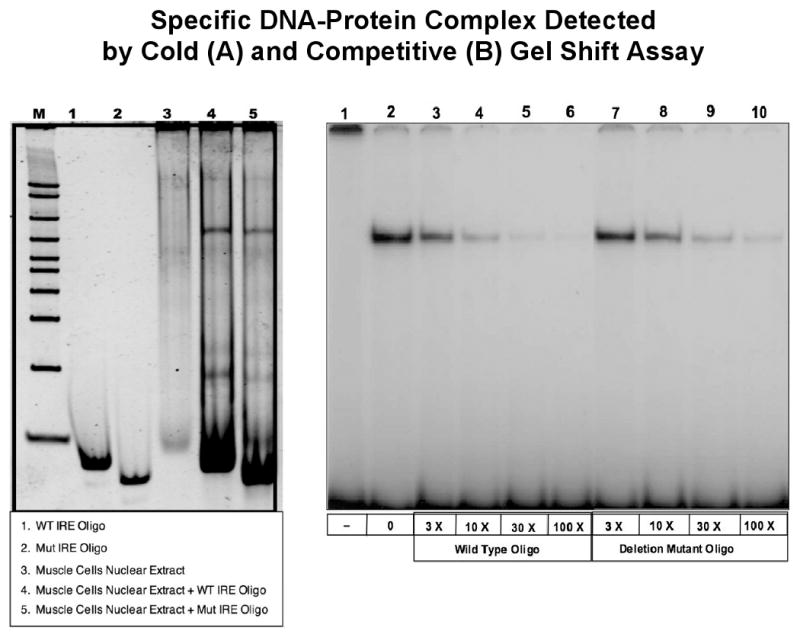
Specific DNA-Protein Complex Detected by Cold (A) and Competitive (B) Gel Shift Assay. (A) Non-radioactive EMSA was initially used to see if a factor from nuclear extract from human smooth muscle cell line would bind to the IRE wild type and mutant sequences. A single band was observed for both sequences. However, there was no significant difference in the intensity of the binding between the wild type and mutant probes. (B) Wild type 29 bp DNA fragment from the putative IRE region in exon 10 of LPL gene was used as 32P-labeled probe. No nuclear extract was incubated with the probe (lane 1). Nuclear extracts prepared from human aorta smooth muscle cells were incubated in the absence (lane 2) and the presence of molar excess amount of unlabeled wild type (lane 3-6) and 5 bp deletion mutant (lane 7-10) oligo competitors.
Next we performed antibody supershift assay to identify the potential nuclear protein that bind to the putative IRE. After a small scale screening that utilized antibodies against ADD1, Sap-1, SRF and Elk-1, we found that incubation of the aorta smooth muscle cell nuclear extracts with Elk-1 antibody resulted in an antibody dose-dependent supershifted band, indicating that Elk-1 is involved in the DNA-protein complex (Fig. 5A). Protein phosphorylation experiments revealed phosphorylated Elk-1 in response to insulin stimulation in human aorta smooth muscle cells expressing LPL and significantly reduced Elk-1 phosphorylations in the deletion mutants (Fig. 5B).
Fig. 5.
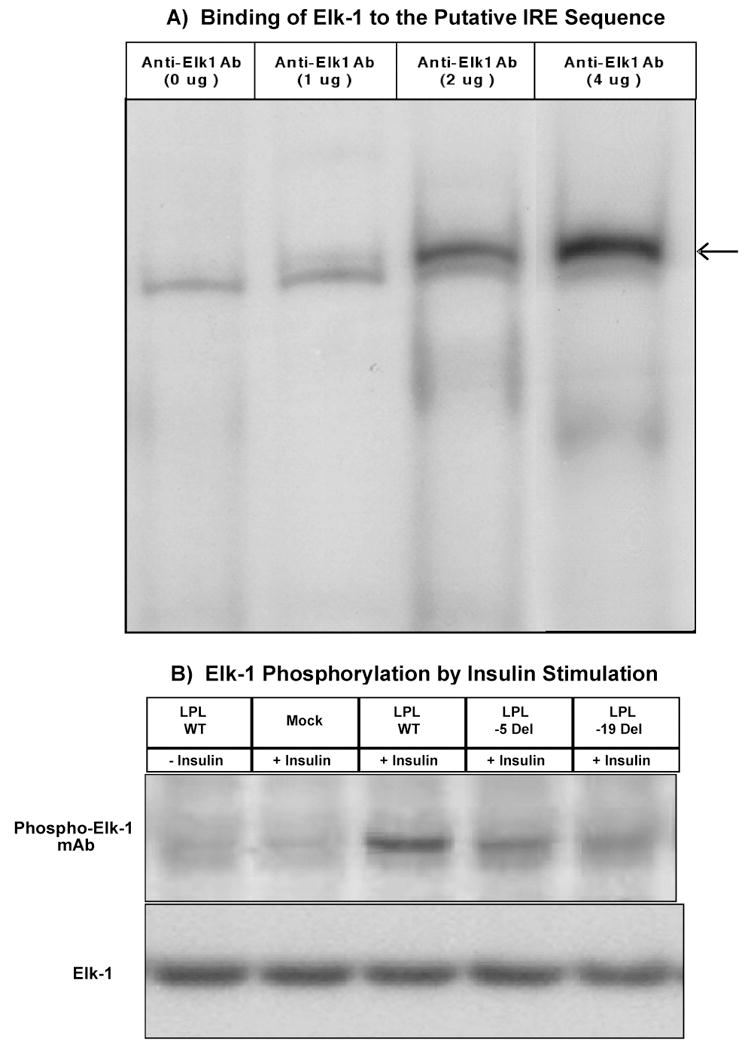
(A) Binding of Elk-1 to the Putative IRE Sequence. Super Gel Shift analysis. 29-bp DNA fragment from the putative IRE region in exon 10 of the LPL gene was used as 32P-labeled probe. Nuclear extracts prepared from human aorta smooth muscle cells were incubated in the absence (0 μg) or presence (1μg, 2 ug and 4 μg) of anti-Elk-1 antibody added prior to gel mobility shift analysis. Please note that in the absence of anti-Elk1 antibody (0 μg) there is only one band. However, in the presence of 1 ug of anti-Elk1 antibody a small supershift band appeared whose intensity increased proportionately in the presence of 2 μg and 4 μg of anti-Elk1 antibody, respectively (super shift band is indicated by an arrow). (B) Elk-1 Phosphorylation by Insulin Stimulation. Nuclear protein from human aorta smooth muscle cells transfected with expression plasmids for exon 10 wild type, 5 bp deletion and 19 bp deletion mutant, treated with carrier control (25 μM HEPES, − insulin) or insulin (+ insulin) for 30 min. and then subjected to Western blot analysis using Phospho-Elk-1 (Ser383) monoclonal antibody (upper panel).
4. Discussion
Insulin regulates the expression of at least 150 genes [8] and plays a central role in the postprandial response of adipose and muscle LPL [7,8]. Insulin regulates LPL gene expression at the mRNA level [3,5,19] and, accordingly, LPL mRNA levels are shown to be inversely correlated with the degree of insulin resistance [6]. However, the molecular mechanism by which insulin affects the LPL expression is not known. Conceivably, the LPL gene harbors an insulin response element (IRE), similar to that reported in the apoC3 gene [14], which mediates the effect of insulin on the LPL expression. In search of a putative IRE in the LPL gene, we used an ‘evolutionary trace’ approach and found a putative IRE in the human LPL gene about 400 bp downstream from the beginning of the non-coding exon 10 in 3′UTR. We have identified a 19-nucleotide sequence comprising TTCTCAAACTTTACTCCAA as a putative IRE in the human LPL gene. The putative IRE was also assigned to exon 10 in mouse, cow and pig where the LPL gene organization is known. Although IRE is found mainly in the promoter region of some genes, existence of such element in intronic regions has been reported [11], indicating that it is plausible that the 3′UTR of LPL may also harbor a functional IRE. The identification of a single consensus sequence for an insulin response element has been intangible due to the existence of multiple mechanisms by which insulin affects transcription. It is possible that insulin may have different impacts on the regulation of gene expression depending on its interaction with different transcriptional factors. These factors may bind to specific yet related IRE elements.
Adipose tissue LPL activity is decreased in diabetic patients and the mechanism by which this takes place is inhibition of translation, which involves the 3′UTR of the LPL mRNA and a RNA-binding protein [2]. Accumulating evidence strongly implicates the 3′UTR of mRNA in the regulation of gene expression [20]. Furthermore, the 3′UTR of mRNA plays a critical role in its function and mutations in this region can disrupt gene regulation [21]. MicroRNAs (miRNAs) regulate gene expression through base pairing to their targets within the 3′UTR of genes. If SNPs occur at this target sites, they can affect mRNA regulation and the resulting phenotype [22,23]. We targeted the sequence surrounding the putative IRE in LPL 3′UTR and identified a 5 bp deletion within the putative IRE that was restricted to individuals with Hispanic ancestry at a carrier frequency of about 4%. Interestingly, all heterozygous individuals for the 5 bp deletion were homozygous for the LPL/HindIII wild type H+ allele, indicating that the deletion arose on the HindIII/H+ background. Vohl et al. [24] have suggested that the HindIII/H+ allele may be associated with a functional LPL enzyme that is less sensitive to insulin and may result in a reduced response of LPL to insulin in visceral obesity. The 5 bp deletion that is associated with the HindIII/H+ allele may be one of the factors responsible for such observation in the Hispanic population, although its low frequency suggests the involvement of other functional elements in the LPL gene. In our study we did not find a significant association of the 5 bp deletion with plasma LPL mass although the carriers of the deletion had modestly lower LPL mass. This lack of association could be due to the availability of post-heparin samples from only 6 individuals who were heterozygotes for the deletion. Previously, Vohl et al. [24] have shown non-significantly lower LPL activity in HindIII/H+ homozygotes. It is noteworthy that post-heparin plasma LPL activity studies may not fully show the effect of LPL polymorphisms on LPL activity as exemplified by the inconsistent findings with the LPL S447X polymorphism [25].
We hypothesized that the putative IRE in exon 10 is a regulatory sequence through which insulin influences the expression of LPL and consequently, the 5 bp deletion in the IRE may make the sequence less sensitive to insulin action. To test this hypothesis, we created the 5 bp and 19 bp deletion mutations in LPL cDNA and expressed them in muscle and COS-1 cells in the presence or absence of insulin followed by measurement of its mRNA. Depending on whether the sequence harbors a negative or positive IRE, we predicted the 5 bp deletion would be associated with altered LPL expression. While the insulin treatment increased LPL expression of wild type and 5 bp deletion, this did not show effect on 19 bp deletion suggesting that the 19 bp sequence in 3′UTR is required for insulin action and that this sequence harbors a positive IRE (Fig. 2). However, the positive effect of insulin treatment was modest on the wild type and 5 bp deletion suggesting the possible presence of additional IREs in LPL. One possible explanation for the modest change in our LPL expression that we observed is that there may be several IRE mechanisms involved in regulating LPL gene and ours is one of them. Similar mechanisms have been observed in other genes. For example, multiple androgen response elements are involved in regulating neutral endopeptidase (NEP) gene [26].
Our experiments on LPL translation (Fig. 3) show that insulin treatment increased LPL translation of the wild type construct, but not those constructs containing 5 bp or 19 bp deletions, suggesting that insulin regulation via IRE is at the level of protein translation as well as LPL mRNA expression.
Next we reasoned that if the 5 bp deletion mutation is associated with altered insulin regulation then a possible explanation is that the mutation alters the affinity of trans-acting transcription factor(s) that mediates the insulin response. To test this possibility, we performed EMSA and gel supershift analysis. Indeed our EMSA data showed that the putative IRE sequence containing the 5 bp deletion is involved in binding with a transcription factor and that the transcriptional factor could be Elk-1. Protein phosphorylation revealed phosphorylated Elk-1 in response to insulin stimulation. It has been shown that Elk1 transcriptional factor plays a major role regulating human liver X receptor B (LXRβ), which is important for proper insulin production and glucose metabolism. Mutations in the binding site of Elk1 (5′-CGGACCGGAAGTTCGT-3′) and serum responsive factor (SRF) have been found to be associated with significantly reduced promoter activity of LXRβ and impaired glucose response [27]. However, the IRE we identified in LPL to which Elk1 binds is different from the Elk1 binding site reported for LXRβ. This may be due to complexity of the Elk1 binding sites. Further studies on Elk1 should elucidate its role in different genes.
In summary, using a combination of bioinformatics, genetic and functional studies, we have identified a putative IRE and a naturally occurring 5 bp deletion in the putative IRE in the LPL gene. We have shown that the putative IRE is a regulatory sequence in the LPL noncoding exon 10, which may play a role in the stability of the LPL mRNA and the 5 bp deletion in the IRE may make the LPL gene less sensitive to insulin action.
Supplementary Material
Acknowledgments
We are grateful to Ms. Noel Harrie for her secretarial help with the preparation of the manuscript and Mr. Ryan Minster for technical assistance. We thank Dr. Richard F. Hamman, University of Colorado Denver, for kindly providing us with the SLVDS population samples. This study was supported by the National Heart, Lung, and Blood Institute (NHLBI) Grant HL 070169.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ranganathan G, Ong JM, Yukht A, Saghizadeh M, Simsolo RB, Pauer A, Kern PA. Tissue-specific expression of human lipoprotein lipase. Effect of the 3′-untranslated region on translation. J Biol Chem. 1995;270:7149–7155. doi: 10.1074/jbc.270.13.7149. [DOI] [PubMed] [Google Scholar]
- 2.Hensley LL, Ranganathan G, Wagner EM, Saghizadeh M, Simsolo RB, Pauer A, Kern PA. Transgenic mice expressing lipoprotein lipase in adipose tissue. Absence of the proximal 3′-untranslated region causes translational upregulation. J Biol Chem. 2003;278:32702–32709. doi: 10.1074/jbc.M304200200. [DOI] [PubMed] [Google Scholar]
- 3.Ong JM, Kirchgessner TG, Schotz MC, Kern PA. Insulin increases the synthetic rate and messenger RNA level of lipoprotein lipase in isolated rat adipocytes. J Biol Chem. 1988;263:12933–12938. [PubMed] [Google Scholar]
- 4.Semenkovich CF, Wims M, Noe L, Etienne J, Chan L. Insulin regulation of lipoprotein lipase activity in 3T3-L1 adipocytes is mediated at posttranscriptional and posttranslational levels. J Biol Chem. 1989;264:9030–9038. [PubMed] [Google Scholar]
- 5.Raynolds MV, Awald PD, Gordon DF, Gutierrez-Hartmann A, Rule DC, Wood WM, Eckel RH. Lipoprotein lipase gene expression in rat adipocytes is regulated by isoproterenol and insulin through different mechanisms. Mol Endocrinol. 1990;4:1416–1422. doi: 10.1210/mend-4-9-1416. [DOI] [PubMed] [Google Scholar]
- 6.Maheux P, Azhar S, Kern PA, Chen YD, Reuven GM. Relationship between insulin-mediated glucose disposal and regulation of plasma and adipose tissue lipoprotein lipase. Diabetologia. 1997;40:850–858. doi: 10.1007/s001250050759. [DOI] [PubMed] [Google Scholar]
- 7.Pulawa LK, Eckel RH. Overexpression of muscle lipoprotein lipase and insulin sensitivity. Curr Opin Clin Nutr Metab Care. 2002;5:569–574. doi: 10.1097/00075197-200209000-00017. [DOI] [PubMed] [Google Scholar]
- 8.Preiss-Landl K, Zimmermann R, Hämmerle G, Zechner R. Lipoprotein lipase: the regulation of tissue specific expression and its role in lipid and energy metabolism. Curr Opin Lipidol. 2002;13:471–481. doi: 10.1097/00041433-200210000-00002. [DOI] [PubMed] [Google Scholar]
- 9.O'Brien RM, Granner DK. Regulation of gene expression by insulin. Physiol Rev. 1996;76:1109–1161. doi: 10.1152/physrev.1996.76.4.1109. [DOI] [PubMed] [Google Scholar]
- 10.O'Brien RM, Streeper RS, Ayala JE, Stadelmaier BT, Hornbuckle LA. Insulin-regulated gene expression. Biochem Soc Trans. 2001;29:552–558. doi: 10.1042/bst0290552. [DOI] [PubMed] [Google Scholar]
- 11.Lo HW, Ali-Osman F. Structure of the human allelic glutathione S-transferase-π gene variant, hGSTP1*C, cloned from a glioblastoma multiforme cell line. Chem-Biol Interact. 1998;111-112:91–102. doi: 10.1016/s0009-2797(97)00153-1. [DOI] [PubMed] [Google Scholar]
- 12.Vander Kooi BT, Streeper RS, Svitek CA, Oeser JK, Powell DR, O'Brien RM. The three insulin response sequences in the glucose-6-phosphatase catalytic subunit gene promoter are functionally distinct. J Biol Chem. 2003;278:11782–11793. doi: 10.1074/jbc.M212570200. [DOI] [PubMed] [Google Scholar]
- 13.Villafuerte BC, Phillips LS, Rane MJ, Zhao W. Insulin-response element-binding protein 1: a novel Akt substrate involved in transcriptional action of insulin. J Biol Chem. 2004;279:36650–36659. doi: 10.1074/jbc.M404349200. [DOI] [PubMed] [Google Scholar]
- 14.Li WW, Dammerman MM, Smith JD, Metzger S, Breslow JL, Leff T. Common genetic variation in the promoter of the human apo CIII gene abolishes regulation by insulin and may contribute to hypertriglyceridemia. J Clin Invest. 1995;96:2601–2615. doi: 10.1172/JCI118324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Razzaghi H, Aston CE, Hamman RF, Kamboh MI. Genetic screening of the lipoprotein lipase gene for mutations associated with high triglyceride/low HDL-cholesterol levels. Hum Genet. 2000;107:257–267. doi: 10.1007/s004390000367. [DOI] [PubMed] [Google Scholar]
- 16.Razzaghi H, Kamboh MI. A highly sensitive and nonradioactive mutation detection method based on vertical gradient temperature single-strand conformation polymorphism. Electrophoresis. 2001;22:2665–2669. doi: 10.1002/1522-2683(200108)22:13<2665::AID-ELPS2665>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 17.Bey L, Etienne J, Tse C, Brault D, Noé L, Raisonnier A, Arnault F, Hamilton MT, Galibert F. Cloning, sequencing and structural analysis of 976 base pairs of the promoter sequence for the rat lipoprotein lipase gene. Comparison with the mouse and human sequences. Gene. 1998;209:31–38. doi: 10.1016/s0378-1119(98)00003-1. [DOI] [PubMed] [Google Scholar]
- 18.Yang LX, Nelson PG. Glia cell line-derived neurotrophic factor regulates the distribution of acetylcholine receptors in mouse primary skeletal muscle cells. Neuroscience. 2004;128:497–509. doi: 10.1016/j.neuroscience.2004.06.067. [DOI] [PubMed] [Google Scholar]
- 19.Semb H, Olivecrona T. Two different mechanisms are involved in nutritional regulation of lipoprotein lipase in guinea-pig adipose tissue. Biochem J. 1989;262:505–511. doi: 10.1042/bj2620505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Conne B, Stutz A, Vassalli JD. The 3′ untranslated region of messenger RNA: A molecular ‘hotspot’ for pathology? Nat Med. 2000;6:637–641. doi: 10.1038/76211. [DOI] [PubMed] [Google Scholar]
- 21.Pesole G, Mignone F, Gissi C, Grillo G, Licciulli F, Liuni S. Structural and functional features of eukaryotic mRNA untranslated regions. Gene. 2001;276:73–81. doi: 10.1016/s0378-1119(01)00674-6. [DOI] [PubMed] [Google Scholar]
- 22.Sethupathy P, Borel C, Gagnebin M, Grant GR, Deutsch S, Elton TS, Hatzigeorgiou AG, Antonarakis SE. Human microRNA-155 on chromosome 21 differentially interacts with its polymorphic target in the AGTR1 3′ untranslated region: a mechanism for functional single-nucleotide polymorphisms related to phenotypes. Am J Hum Genet. 2007;81:405–413. doi: 10.1086/519979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gehring NH, Frede U, Neu-Yilik G, Hundsdoerfer P, Vetter B, Hentze MW, Kulozik AE. Increased efficiency of mRNA 3′ end formation: a new genetic mechanism contributing to hereditary thrombophilia. Nat Genet. 2001;28:389–392. doi: 10.1038/ng578. [DOI] [PubMed] [Google Scholar]
- 24.Vohl MC, Lamarche B, Moorjani S, Prud'homme D, Nadeau A, Bouchard C, Lupien PJ, Després JP. The lipoprotein lipase HindIII polymorphism modulates plasma triglyceride levels in visceral obesity. Arterioscler Thromb Vasc Biol. 1995;15:714–720. doi: 10.1161/01.atv.15.5.714. [DOI] [PubMed] [Google Scholar]
- 25.Rip J, Nierman MC, Ross CJ, Jukema JW, Hayden MR, Kastelein JJ, Stroes ES, Kuivenhoven JA. Lipoprotein lipase S447X: a naturally occurring gain-of-function mutation. Arterioscler Thromb Vasc Biol. 2006;16:1236–1245. doi: 10.1161/01.ATV.0000219283.10832.43. [DOI] [PubMed] [Google Scholar]
- 26.Zheng R, Shen R, Goodman OB, Jr, Nanus DM. Multiple androgen response elements cooperate in androgen regulated activity of the type 1 neutral endopeptidase promoter. Mol Cell Endocrinol. 2006;259:10–21. doi: 10.1016/j.mce.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 27.Nilsson M, Dahlman-Wright K, Karelmo C, Gustafsson JA, Steffensen KR. Elk1 and SRF transcription factors convey basal transcription and mediate glucose response via their binding sites in the human LXRB gene promoter. Nucleic Acids Res. 2007;35:4858–4868. doi: 10.1093/nar/gkm492. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.