

Abstract
The SKN-1 transcription factor specifies early embryonic cell fates in Caenorhabditis elegans. SKN-1 binds DNA at high affinity as a monomer, by means of a basic region like those of basic-leucine zipper (bZIP) proteins, which bind DNA only as dimers. We have investigated how the SKN-1 DNA-binding domain (the Skn domain) promotes stable binding of a basic region monomer to DNA. A flexible arm at the Skn domain amino terminus binds in the minor groove, but a support segment adjacent to the carboxy-terminal basic region can independently stabilize basic region–DNA binding. Off DNA, the basic region and arm are unfolded and, surprisingly, the support segment forms a molten globule of four α-helices. On binding DNA, the Skn domain adopts a tertiary structure in which the basic region helix extends directly from a support segment α-helix, which is required for binding. The remainder of the support segment anchors this uninterrupted helix on DNA, but leaves the basic region exposed in the major groove. This is similar to how the bZIP basic region extends from the leucine zipper, indicating that positioning and cooperative stability provided by helix extension are conserved mechanisms that promote binding of basic regions to DNA.
Keywords: basic region, SKN-1, DNA binding, bZIP, molten globule, α-helix
Members of two large and distinct families of transcription factors, the basic leucine zipper (bZIP) and basic helix–loop–helix (bHLH) proteins, bind to DNA through segments of 15–20 residues that are termed basic regions (BRs) (Ellenberger 1994). These BRs can mediate specific DNA recognition (Ellenberger 1994), but are incapable of binding to DNA stably as peptide monomers. bZIP and bHLH proteins promote stable BR–DNA binding by forming dimers. Their dimerization is mediated by α-helical ZIP or HLH segments, which are located immediately carboxyl-terminal to their respective BRs (Ellenberger 1994). The BR remains unstructured off DNA, but upon DNA binding it folds into an α-helix that recognizes a specific half-site in the major groove (O’Neil et al. 1990; Patel et al. 1990; Shuman et al. 1990; Weiss et al. 1990; Anthony-Cahill et al. 1992), and extends from its respective dimerization segment to form an uninterrupted α-helix (Ellenberger 1994). In part, dimerization promotes binding of bZIP and bHLH proteins to DNA simply by linking two BRs together, thus decreasing the entropy cost of binding an individual BR to DNA (Stanojevic and Verdine 1995). For example, bZIP BR peptides can bind to DNA sequence specifically when they are tethered together chemically as a dimer (Talanian et al. 1990; Park et al. 1992; Cuenoud and Schepartz 1993a,b; Pellegrini and Ebright 1996). Unlike bZIP proteins, these tethered BR dimers cannot dissociate into monomers. Nevertheless, the complexes that they form with DNA are generally less stable than bZIP–DNA complexes, indicating that the ZIP (and presumably HLH) segments contribute more to BR–DNA stability than simple tethering (Talanian et al. 1990).
The Caenorhabditis elegans SKN-1 protein provides a unique tool for examining how binding of an individual BR to DNA can be stabilized, because SKN-1 is an exception to the dimeric paradigm for BR–DNA binding. SKN-1 is a maternally expressed transcription factor that is required for proper cell fate specification during the earliest stages of embryogenesis (Bowerman et al. 1992, 1993; Blackwell et al. 1994). It binds to DNA as a monomer with high affinity, by means of a bZIP-like BR that lacks an adjacent ZIP segment (Blackwell et al. 1994). The preferred SKN-1-binding site is composed of an AT-rich region (A/T,A/T, T) located 5′ of a single AP-1-like bZIP half site (GTCAT), to which SKN-1 binds in the minor and major grooves, respectively (Blackwell et al. 1994). The carboxy-terminal 85 residues of SKN-1 mediate DNA-binding affinity and specificity, and are thus defined functionally as a novel DNA-binding motif referred to as the Skn domain (Fig. 1) (Blackwell et al. 1994). The BR lies at the Skn domain carboxyl terminus (Fig. 1). At the Skn domain amino terminus is a segment (the amino-terminal arm; Fig. 1) which is identical to the flexible arm that the homeodomain protein Antennapedia places in the minor groove (Blackwell et al. 1994). Similar minor-groove binding arms are present in other homeodomains and in various other helix–turn–helix proteins (Gehring et al. 1994). The residues located between the Skn domain amino-terminal arm and BR are required for DNA binding (Blackwell et al. 1994) and are designated here as the support segment. By understanding how the Skn domain promotes high-affinity DNA binding by a BR monomer, it should be possible to derive principles that are generally applicable to interactions between BRs and DNA.
Figure 1.

The Skn domain. This motif consists of the carboxyl-terminal 85 residues of SKN-1 and is compared with the corresponding portions of representative bZIP proteins of the cap ’n collar (CNC) subgroup (Blackwell et al. 1994). The CNC bZIP subgroup includes at least 12 known genes and numerous expressed cDNA sequences (not shown). The arm, support, and BR segments are indicated by bars above the sequences and the beginning of the ZIP segment by an arrow. Related residues are shaded, and the homeodomain turn motifs (Blackwell et al. 1994) are surrounded by boxes. Skn domain residues present in the mutants Δ1–9 (Blackwell et al. 1994) and SknT are indicated by lines. SknT also contains a Cys → Ser substitution at position 70. Sequences are referenced in Blackwell et al. (1994).
We have investigated how the Skn domain folds when it binds to DNA, and how the amino-terminal arm and support segment contribute to BR–DNA binding. The data show that the amino-terminal arm provides binding energy by interacting with the AT-rich region in the minor groove, but the support segment can independently stabilize specific binding. Off DNA, the amino-terminal arm and BR are unstructured, and the support segment consists of four α-helices that, surprisingly, lack a stable tertiary structure. These segments together adopt a cooperative fold when the Skn domain binds specifically to DNA. Unlike other monomeric domains that recognize DNA through α-helices, the support segment helices do not pack directly against the DNA-bound BR. Instead, BR–DNA binding is promoted entirely through formation of an uninterrupted α-helix consisting of the BR, and a helix within the support segment. This latter helix is essential for DNA binding, and is stabilized and positioned by the remainder of the support segment. Extension of the BR helix from the Skn domain support segment is reminiscent of how bZIP and bHLH BR helices extend directly from their respective dimerization segments, indicating that these monomeric and dimeric BR–DNA complexes derive stability from similar mechanisms.
Results
Stabilization of a Skn domain structure by DNA binding
The precedents set by bZIP and bHLH proteins (O’Neil et al. 1990; Patel et al. 1990; Shuman et al. 1990; Weiss et al. 1990; Anthony-Cahill et al. 1992) predict that the Skn domain BR is likely to be unstructured off DNA and to form an α-helix upon DNA binding. Circular dichroism (CD) spectroscopy is a useful method for examining Skn-domain folding, because it is a good indicator of α-helical content (Johnson 1988). At 25°C, the far-ultraviolet CD spectrum of the free Skn-domain displays the characteristic α-helix minimum at 222 nm and indicates a helical content of ∼26% (Fig. 2A, see Materials and Methods). When bound to cognate DNA, the helical content of the Skn domain is ∼46% (Fig. 2A), an increase consistent with formation of a BR α-helix. Addition of nonspecific DNA does not affect the Skn domain CD spectrum (not shown), indicating that this folding transition requires specific DNA binding.
Figure 2.
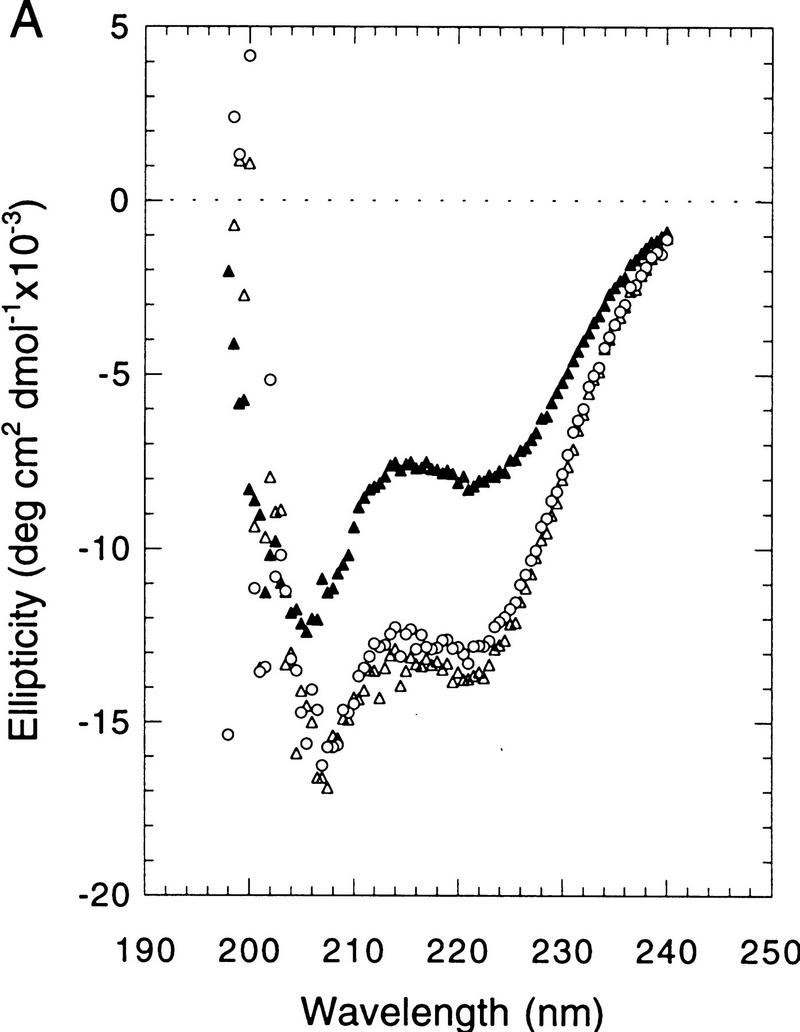

CD spectroscopy of Skn domain folding and DNA binding. (A) CD wavelength spectra of the Skn domain at 25°C. (▴) The spectrum of the Skn domain off DNA; (○ and ▵) the spectrum of the Skn domain–DNA complex before and after thermal unfolding, respectively (shown in B). (B) Thermal unfolding of the Skn domain. Ellipticity was monitored at 222 nm, as an indicator of α-helical content. (▴) Denaturation of the Skn domain off DNA; (○ and ▵) denaturation and renaturation of the Skn domain–DNA complex, respectively.
Monitoring of secondary structure during thermal denaturation can reveal whether a protein is folded cooperatively. When the Skn domain is denatured in the absence of specific DNA, its helical content decreases approximately linearly with temperature (as indicated by increasing ellipticity; Fig. 2B), showing that it has little stable tertiary structure. In contrast, the Skn-domain–DNA complex shows a broad cooperative unfolding transition that has a midpoint at 37°C (Fig. 2B), indicating that the Skn domain adopts a tertiary structure when it forms a complex with cognate DNA. This transition is reversible, as shown by the nearly superimposable denaturation and renaturation curves, and by the identical CD spectra at 25°C before and after denaturation (Figs. 2A,B). Even when the Skn domain is bound to specific DNA, its melting point is relatively low (Tm = 37°C, as compared with 55–65°C for a ZIP dimer) (O’Shea et al. 1989; Weiss 1990), and the percent helix increases monotonically as the temperature approaches 0°C (to ∼52%, Fig. 2B) suggesting that its structure is relatively labile. Consistent with this idea, the on- and off-rates of the Skn domain–DNA complex are too rapid to be measurable by electrophoretic mobility shift assay (EMSA) (not shown; see Materials and Methods).
Contribution of the Skn domain amino-terminal arm
Like the homeodomain (Gehring et al. 1994), the Skn domain has an arm segment at its amino terminus and a DNA recognition helix (the BR) at its carboxyl terminus (Fig. 1), suggesting that its amino-terminal arm is likely to engage the minor groove in the AT-rich element of its binding site (Blackwell et al. 1994). If this interaction were required to stabilize the Skn domain structure, or to position the BR on DNA, then deletion of the amino-terminal arm should eliminate specific DNA binding. EMSA titrations indicate that the Skn domain binds to an oligonucleotide containing its cognate site with a dissociation constant (Kd) of ∼1 (±0.5) × 10−9 m (not shown; see Materials and Methods). This binding affinity is comparable to that of full-length SKN-1 (Blackwell et al. 1994). Remarkably, a Skn domain derivative lacking the amino-terminal arm (Δ1–9, Fig. 1) binds to this site with a Kd of ∼5 (±3) × 10−9 m. The affinities of the Skn domain and Δ1–9 for nonspecific DNA are ∼200-fold lower than their respective specific binding affinities (not shown). The Δ1–9 mutant thus binds DNA with far greater affinity and specificity than individual BR peptides (Park et al. 1996), indicating that the support segment can independently stabilize specific DNA binding. The difference between the specific Kd values of the Skn domain and Δ1–9 is ∼10-fold less than that contributed by the amino-terminal arm of the fushi tarazu homeodomain (Percival-Smith et al. 1990), suggesting that the Skn domain arm may bind the minor groove less tightly.
To investigate how the amino-terminal arm contributes to SKN-1 DNA binding, we have compared the hydroxyl radical footprinting and interference patterns of the Skn domain and Δ1–9 bound to a cognate site. Hydroxyl radical protection footprinting detects close contact with the DNA backbone, and binding in the minor groove, because hydroxyl radicals cleave backbone sugar residues (Dixon et al. 1991). Both the Skn domain and Δ1–9 protect the DNA backbone from hydroxyl radicals on both sides of the major groove through the bZIP half-site (Fig. 3A,B,E), indicating contributions from the BR and support segment. However, the Δ1–9 footprint is relatively diminished on both sides of the minor groove in the AT-rich region, and its maximum on the bottom strand is changed from −4 to −3 (Fig. 3A,B,E), suggesting that removal of the amino-terminal arm results in a loss of minor groove binding in this region. A hydroxyl radical interference assay, in which the DNA is treated with hydroxyl radicals prior to protein binding, reveals the consequences of breaking the DNA backbone at a particular position, and of losing the corresponding base (Dixon et al. 1991). Removal of the amino-terminal arm decreases the hydroxyl radical interference at −1 on the top strand (Fig. 3C–E), suggesting a loss of binding. In addition, prior hydroxyl radical cleavage at positions −3 through −5 on the bottom strand, and at −3 on the top strand, enhances binding by the Skn domain, but not by Δ1–9 (Fig. 3C–E). This last observation suggests that prior hydroxyl radical cleavage relieves torsional stress that is placed on the DNA by the amino-terminal arm. Together, these findings indicate that the amino-terminal arm binds in the AT-rich region in the minor groove, but is not essential for stabilizing the fold of the Skn domain, or for positioning it on DNA.
Figure 3.

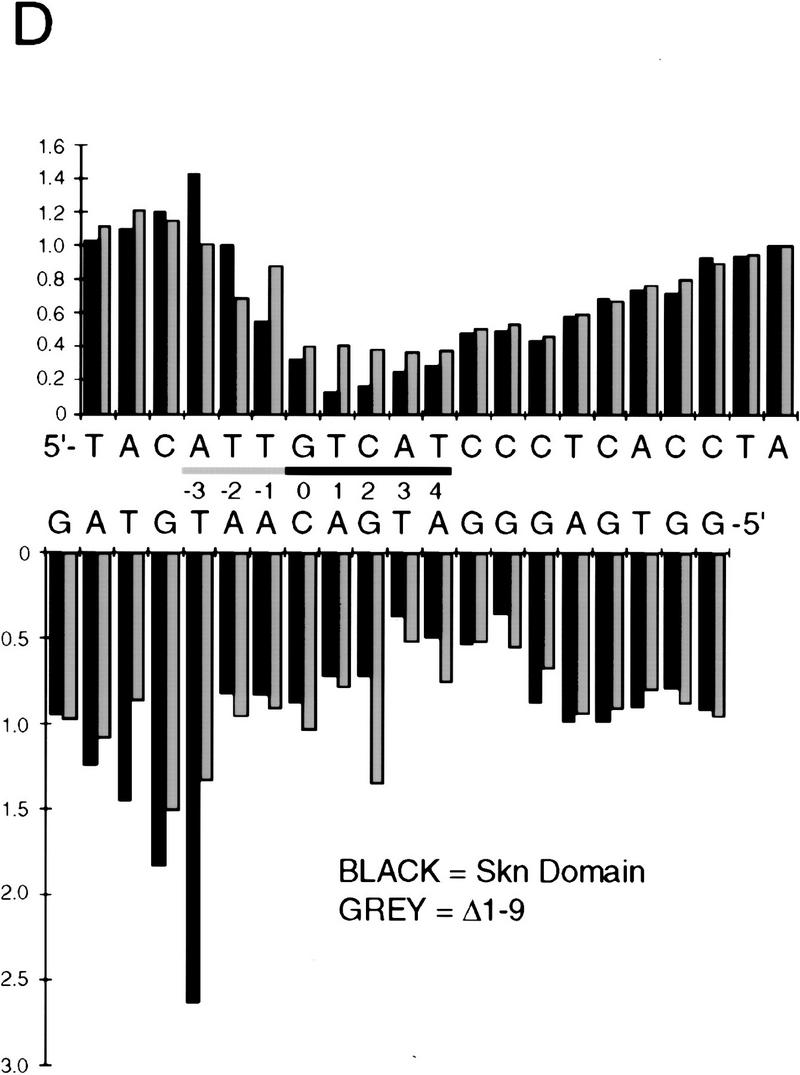

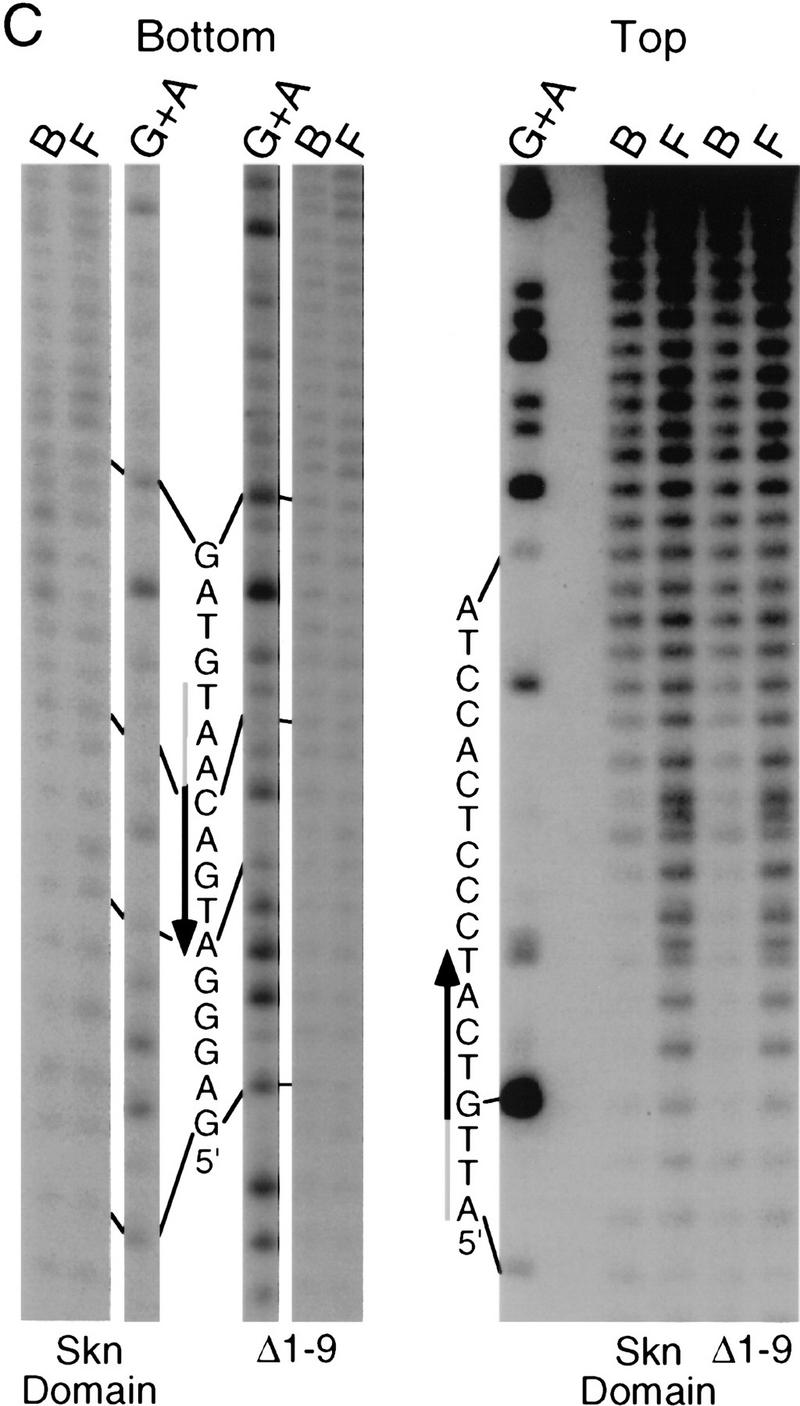
Hydroxyl radical protection footprints of the Skn domain and Δ1–9. (A) Hydroxyl radical protection analysis. An end-labeled SKN-1-binding site (Blackwell et al. 1994) is bound by the indicated protein (Skn domain or Δ1–9), or incubated without added protein (Free), then cleaved by hydroxyl radicals. Reaction products are then electrophoresed on a sequencing gel alongside a G + A track. (Top and bottom) Individual DNA strands. The AT-rich region is indicated by a shaded bar and the bZIP half-site by a black arrow that points away from the center of the complete bZIP site (indicated by 0 in B). (B) Graph of hydroxyl radical protection at individual site positions. The samples shown in A were analyzed by a PhosphorImager, and the data were converted to a ratio of bound-to-free at each position and normalized to 1 at positions at which no binding occurred. (C) Hydroxyl radical interference analysis. An end-labeled SKN-1-binding site (Blackwell et al. 1994) was cleaved by hydroxyl radicals, then bound by the indicated protein. Bound (B) and free (F) DNA were separated on an EMSA gel and analyzed as in A. (D) Graph of hydroxyl radical interference, calculated as in B. A bound-to-free ratio <1 indicates binding interference, and a ratio >1 indicates that prior cleavage enhances binding. (E) Skn domain footprinting along a DNA helix. Large ovals indicate a hydroxyl radical protection bound/free ratio of <0.5, and small ovals a ratio of <0.7, as depicted in B. Arrows indicate positions at which prior hydroxyl radical cleavage enhanced DNA binding (taken from D). A box indicates the approximate location of the BR α-helix.
Exposure of the SKN-1 BR in the major groove
The structure of the homeodomain also suggests how the support segment might promote BR–DNA binding. According to this model (model 1, Fig. 4), the support segment would stabilize the BR helix, and position it on DNA, by packing directly against the face of the BR that points away from the DNA (its back side), and by interacting with the DNA backbone (Fig. 4; Kissinger et al. 1990; Wolberger et al. 1991; Gehring et al. 1994). This model is in approximate agreement with footprinting and mutagenesis data (Blackwell et al. 1994), and is similar to other DNA-binding domains with arms that bind in the minor groove (Gehring et al. 1994). A critical prediction of this model is that residues on the BR back side, which do not contact DNA (Ellenberger et al. 1992; König and Richmond 1993; Glover and Harrison 1995; Keller et al. 1995), would be important for binding because they would be involved directly in critical packing interactions that stabilize the Skn domain fold.
Figure 4.

Models for Skn domain-DNA binding. Model 1 is based on the homeodomain, in which helices 1, 2, and 3 pack together to form a globular bundle, and helix 3 is inserted in the major groove as a recognition helix (Kissinger et al. 1990; Wolberger et al. 1991; Gehring et al. 1994). A homeodomain is depicted for simplicity, but the Skn domain would actually differ somewhat, because NMR data (Fig. 6A) indicate that its support segment forms four α-helices, and because interactions between the Skn domain BR and the support segment would occur only on DNA binding (Fig. 2). According to model 2, the BR does not interact directly with the support residues, but instead derives its stability entirely by extending from an adjacent α-helix (helix 4). The dashed region in model 2 outlines roughly how the other support segment helices could pack against helix 4 and the DNA backbone to stabilize and orient the BR, and place the amino-terminal arm in the minor groove. In both cartoons, the BR is depicted as a spiral, the amino-terminal arm by an arrow (of arbitrary direction in model 2), and helices by cylinders.
If model 1 were correct, nonconservative substitutions on the BR back side would disrupt DNA binding, either by eliminating important interactions or by interfering with domain folding. We have made such substitutions in SknT (Fig. 5A,B), a Skn domain truncation mutant that lacks seven residues at its carboxyl terminus (Fig. 1) and has comparable secondary structure and DNA-binding characteristics (Fig. 5C, lanes 1,2; not shown). Simultaneous alanine (Ala) substitution of back side residues G61, V65, R68, Q72, T75, and D76 (Ala BR, Fig. 5A,B) does not impair DNA binding (Fig. 5C, lane 4), indicating that their specific side chains are not required. Replacement of the Skn domain BR with that of GCN4 (GCN4 BR; Fig. 5A), which binds to the same half-site (Ellenberger et al. 1992), dramatically alters the charge distribution on the BR back side by swapping a glutamic acid for V65 and an arginine for T69, but increases the level of DNA binding (Fig. 5C, lane 5). In the GBR–ER mutant, which also binds well to DNA (Fig. 5C, lane 6), charges at GCN4 BR residues E65 and R68 have been switched, and an additional basic residue has been substituted for Q72 (Fig. 5A). Substitution of tryptophan for either V65 or R68 in GCN4 BR (Fig. 5A) also fails to prevent binding (Fig. 5C, lanes 7 and 8). Finally, substitution of the BR segment from the bZIP protein C/EBP (CCAAT/enhancer-binding protein) introduces multiple changes, including acidic substitutions of G61 and Q72 (C/EBP BR; Fig. 5A), but still allows binding to a SKN-1 site (Fig. 5C, lane 9). Remarkably, C/EBP BR binds even more efficiently to a substituted SKN-1 site that contains a C/EBP bZIP half-site (C/EBP half swap) and is not bound by the Skn domain or GCN4 BR (Fig, 5C, lanes 13–16). The Skn domain can accommodate radical substitutions on the BR back side, and can also promote binding by BR segments that specify different DNA targets. These findings demonstrate that the BR back side is exposed in the major groove as in bZIP proteins, and, therefore, they rule out model 1 (Fig. 4) as a possibility. By showing that the Skn domain support segment does not pack against or constrain the BR in the major groove, these experiments indicate that it stabilizes the Skn domain BR–DNA complex through the residues located immediately amino-terminal to the BR (model 2; Fig. 4).
Figure 5.
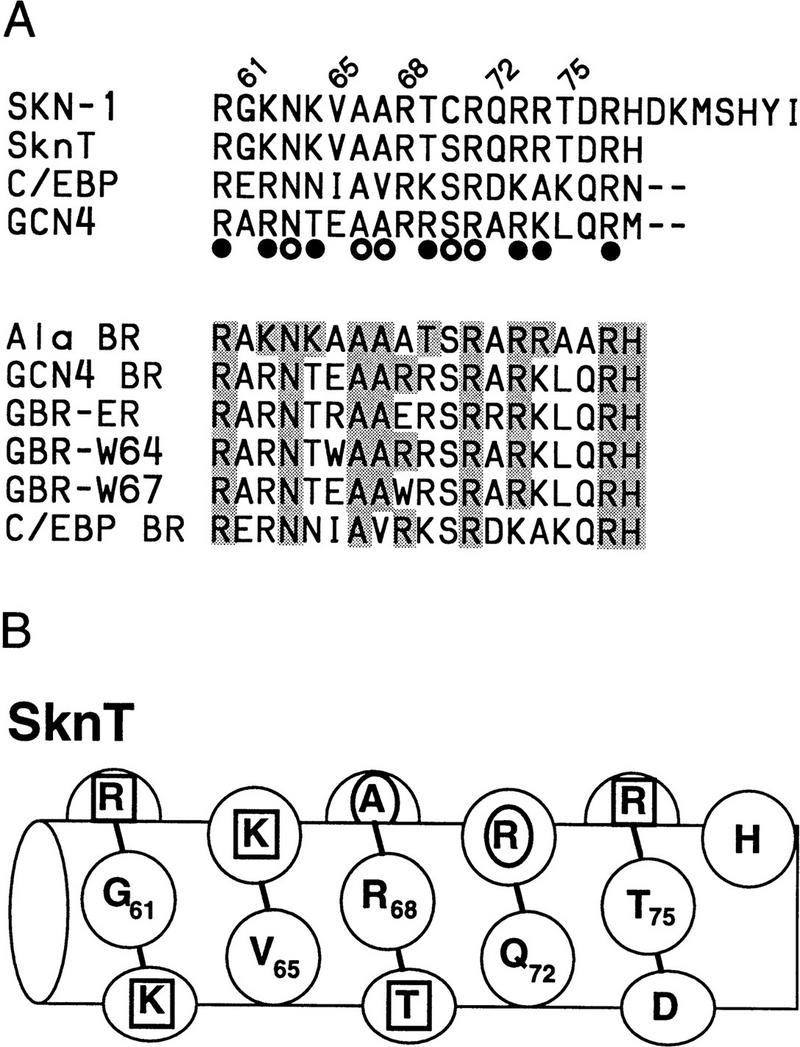

DNA binding by Skn domain BR mutants. (A) BRs of mutants constructed in SknT (Fig. 1). These constitute the carboxyl terminus of each protein, except where indicated by dashes. (○ and •) GCN4 residues that contact bases and backbone phosphates (Ellenberger et al. 1992), respectively. BR back side residues predicted not to contact DNA are numbered. Within the mutants, residues that are identical to SKN-1 are shaded. The C/EBP sequence is derived from Landschulz et al. (1988). (B) The SknT BR, depicted as a helix. Residues that correspond to GCN4 base contact residues are indicated by an oval; those that correspond to backbone contact residues are indicated by a square. For simplicity, this helix is depicted with 3.5 residues per turn. (C) An EMSA comparing binding of in vitro translated Skn domain, Δ1–9, SknT, and the indicated BR mutants to the SKN-1-binding site SK1, or the C/EBP half-swap site. Lysate indicates unprogrammed reticulocyte lysate.
α-Helical secondary structure of the support segment
We have investigated the structure of the Skn domain off DNA by nuclear magnetic resonance (NMR) spectroscopy. Analysis of two- and three-dimensional nuclear Overhauser effect spectroscopy (NOESY) spectra show that the amino-terminal arm and BR segments are in a random coil configuration, as expected, and that the support segment residues form four α-helices (Fig. 6A). Within these helices, the chemical shift indices of the alpha protons are predominantly negative (Fig. 6A), as is characteristic of a helical conformation (Wishart et al. 1992). However, although numerous short and medium range NOEs define the α-helical secondary structures, no long-range NOEs are observed. This makes it impossible to orient the helices relative to one another and supports the idea that they are not folded in a stable tertiary structure.
Figure 6.


NMR spectra of the Skn domain. (A) Summary of NOE data for the free Skn domain, numbered as in Fig. 1. The intensity of the sequential NOEs, αN, βN, and NN, is indicated by the thickness of the line. An overlap of resonances is indicated by +. The CSI is the chemical shift index for α protons (Wishart et al. 1992), in which 0 indicates a chemical shift close to random coil values, + indicates a chemical shift higher than random coil, and − indicates a value less than the random coil chemical shift. The approximate boundaries of helices 1–4 are indicated by cylinders. (B) HSQC spectra of free Skn domain (1) and 1:1 complex of Skn domain and DNA (2).
Helix 1 (residues 12–21) is defined by only a few NOEs (Fig. 6A), suggesting that it is in rapid exchange with an unfolded conformation, but helices 2–4 are defined by multiple αN (i, i + 3) and strong sequential NN NOEs (Fig. 6A). Helix 4 (residues 47–60) appears to be the best defined and most stable of these helices and, significantly, it includes the BR amino terminus (R60; Fig. 5 A and B, and 6A). The helical content derived from these NMR assignments (45%; Fig. 6A) is higher than indicated by CD (26%; Fig. 2A), as has been observed for some helical peptides (Bradley et al. 1990). Residues 33–38 and 45–50, which correspond to the homeodomain turn motif (Fig. 1) (Blackwell et al. 1994) overlap with spaces between helices (Fig. 6A). In homeodomains, related sequences form the turn between helices 2 and 3, and the amino terminus of helix 3 (Gehring et al. 1994). At the amino terminus of each helix is an SXXE or Q capping box (Harper and Rose 1993), and at the carboxyl terminus of helix 4 is an apparent G cap (Presta and Rose 1988; Richardson and Richardson 1988) that is flanked by residues favoring helix termination (Aurora et al. 1994).
Amide protons of the Skn domain exchange within 10 min at pH 5 in D2O (not shown), indicating that the hydrogen bonds within the helices are fluctuating. Comparison of heteronuclear single-quantum coherence (HSQC) spectra (Fig. 6B) of the free Skn domain, however, and of a 1:1 complex of the Skn domain bound specifically to DNA, reveals structural changes accompanying DNA binding. The free protein spectrum (Fig. 6B, panel 1) is highly overlapped and has a narrow range of amide proton chemical shifts, consistent with α-helical and random coil structure. In contrast, the spectrum of the complex (Fig. 6B, panel 2) shows much improved resolution of the cross peaks, and a somewhat broader range of amide proton chemical shifts, consistent with the BR becoming helical and the Skn domain adopting a tertiary structure. These changes are not observed in the presence of nonspecific DNA (not shown), suggesting that the Skn domain is not fully folded when binding DNA nonspecifically.
Direct extension of the SKN-1 BR from helix 4
The mutagenesis experiments described above, and the observation that Skn domain helix 4 (Fig. 6A) overlaps the BR segment, together suggest that both the position and helical fold of the BR are stabilized by formation of an uninterrupted helix together with helix 4 (Fig. 7A). This model (Fig. 4, model 2) predicts that the integrity and stability of helix 4 should be essential for DNA binding. Accordingly, DNA binding was prevented, even at 0°C, by insertion of residues that should form a flexible loop into the carboxy-terminal end of helix 4, and between residues 56 and 57 (link 1 and link 2, respectively; Fig. 7B; Fig. 7, C and D, respectively, lanes 6 and 7). Various proline substitutions (Fig. 7B) similarly inhibited binding (Fig. 7, E and F, respectively, lanes 15–18), as did insertion of two Ala residues [54(AA)55; Fig. 7B], or of two-residue duplications [55(KI)56 and 56(IR)57; Fig. 7, B, C, and D, respectively, lanes 8–10]. This latter group of insertions should preserve the helical character of helix 4, but shift the register of the carboxyl-terminal BR with respect to the rest of the Skn domain. By demonstrating that DNA binding depends upon the integrity of helix 4, and requires an appropriate configuration of its side chains relative to the BR, these data support the idea that the BR helix extends directly from helix 4 (Fig. 4, model 2).
Figure 7.
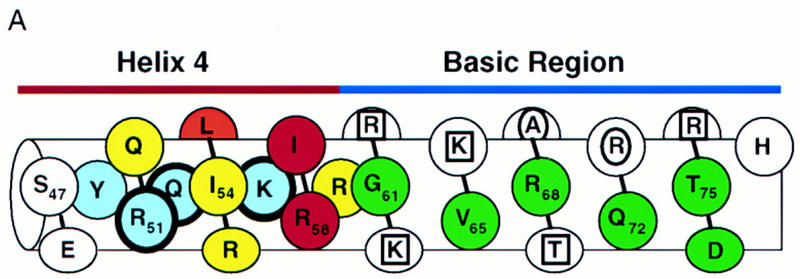





Stabilization of the BR–DNA complex is mediated through helix 4. (A) A continuous helix consisting of the BR and helix 4, and indicating the results of alanine-scanning mutagenesis (E,F; Fig. 5C). This helix is viewed looking approximately toward the BR back side. Residues corresponding to GCN4 residues that contact DNA are indicated as in Fig. 5B. Green indicates BR residues that could be substituted simultaneously without impairing DNA-binding affinity. Blue indicates helix 4 residues at which Ala substitution was similarly allowed, and those that were substituted simultaneously are circled by thick lines. Substitutions at yellow residues impaired binding at room temperature, but not at 0°C. The substitution indicated by orange bound weakly at room temperature, but those indicated by red did not bind at detectable levels either at room temperature or 0°C. Residues on the other side of this helix are shown only within helix 4 and are depicted underneath those facing the viewer. (B) Helix 4 mutants that were constructed in SknT (Fig. 1). Only the helix 4 sequences (Skn domain residues 47–61; Figs. 1 and 6A) are shown. Dashes indicate residues that are identical to SknT. (C) An EMSA comparing binding of the Skn domain, Δ1–9, SknT, and the indicated helix 4 mutants (described in A) to the SK1 site (Fig. 5C). (D) An EMSA identical to that shown in C but performed at 0°C. (E) EMSA of binding of the indicated helix 4 mutants to the SK1 site. (F) An EMSA identical to that shown in E but performed at 0°C.
Model 2 (Fig. 4) also predicts that, unlike the BR, helix 4 would be stabilized and/or oriented by packing interactions with other support segment residues. Alanine scanning of helix 4 revealed that DNA binding was not impaired by substitution of multiple individual residues (Y49, R51, Q52, and K56, Fig. 7B; Fig. 7, E and F, respectively, lanes 4,6,7,11) that are located either distal to the BR, or along the same side of helix 4 as the DNA (Fig. 7A). Other single Ala substitution mutants (at Q50, L53, I54, R55, and R59) bound with lower affinities (Fig. 7E, lanes 5,8–10,14), but at 0°C their binding affinities were more comparable with that of Skn T (Fig. 7F, lanes 5,8–10,14). Ala substitution of I57 or R58 eliminated detectable binding, even at 0°C, however, indicating affinities lower than that of Δ1–9 (Fig. 7, E and F, respectively, lanes 3, 12, and 13). The importance of multiple individual helix 4 side chains for DNA binding contrasts markedly with the variability allowed on the BR back side (Figs. 5C and 7A). These critical helix 4 side chains are located primarily in a patch (Fig. 7A) that includes a small hydrophobic cluster (residues L53, I54, and I57) and is oriented away from the DNA, suggesting that they are involved in intramolecular packing interactions. Presumably, these residues promote BR–DNA binding by stabilizing and/or orienting helix 4 when the Skn domain is folded on DNA (Fig. 4, model 2). Simultaneous Ala substitution of nonessential residues R51, Q52, and K56 (51, 52, 56A; Fig. 7B) increases DNA-binding affinity slightly (Fig. 7, C and D, respectively, lane 4). In contrast, the corresponding glycine (Gly) substitution mutant (51, 52, 56G; Fig. 7B) binds DNA detectably only at 0°C (Fig. 7, C and D, respectively, lane 5), indicating that the helical character of helix 4 is also important for BR–DNA binding.
Discussion
In sharp contrast to binding of isolated BR peptides to DNA, which occurs only at micromolar peptide concentrations (Park et al. 1996) or when the BR is tethered directly to the DNA (Stanojevic and Verdine 1995), the Skn domain monomer binds DNA at high affinity (see above). The amino-terminal arm contributes binding energy, but the support segment (Fig. 1) can independently promote BR–DNA binding. Although the support segment is composed of α-helices, the Skn domain differs from other monomeric helical DNA-binding domains (Harrison 1991; Gehring et al. 1994) in that its BR recognition helix is not part of a globular helical bundle (Fig. 4, model 1). Instead, remarkably, the BR helix is exposed in the major groove and is stabilized entirely through its extension from support segment helix 4 (Fig. 4, model 2; Fig. 7A).
It was expected that the Skn domain amino-terminal arm and BR segments would be unstructured off DNA (Ellenberger 1994; Gehring et al. 1994), but it is surprising that the support segment helices do not fold cooperatively. A secondary structure in the absence of a tertiary fold is characteristic of a molten globule (Kuwajima 1989; Ptitsyn 1996). Molten globules appear to mimic folding intermediates that are subject to some native-like tertiary interactions (Kuwajima 1989; Jennings and Wright 1993; Peng et al. 1995; Kay and Baldwin 1996; Ptitsyn 1996; Wu et al. 1996). The molten globule state is most commonly observed in partially denatured protein fragments, but is also seen in native sequences (Seeley et al. 1996). The Skn domain is a native molten globule that folds to perform a specific function (BR stabilization). This is consistent with proposals that the molten globule state is involved in some molecular recognition and membrane insertion events (van der Goot et al. 1991; González-Mañas et al. 1992; Mach and Middaugh 1995; Tortorella et al. 1995; Boniface et al. 1996; De Filippis et al. 1996; Evans et al. 1996; Runnels et al. 1996). The Skn domain binds DNA with an affinity comparable to that of full-length SKN-1 (Blackwell et al. 1994), indicating that the remainder of SKN-1 is dispensable for binding, but other SKN-1 residues (or another protein) could potentially stabilize a Skn-domain fold off DNA. SKN-1 diverges from its close C. elegans relative SRG-1 17 residues amino-terminal to the Skn domain (B. Bowerman, pers. comm.), however, and NMR evidence shows that this conserved motif of 102 SKN-1 residues also lacks a defined tertiary structure (not shown).
Specific DNA binding may generally involve an induced fit (Spolar and Record, Jr. 1994), in which the DNA, protein, or both, undergo a structural accommodation when they dock together. The Skn domain is an extreme example of this phenomenon, because DNA binding drives folding of the entire motif. This is unusual, because the DNA-binding domains studied so far all adopt some tertiary structure off DNA (Harrison 1991; Ellenberger 1994; Gehring et al. 1994; Berg and Shi 1996). The flexibility of the free Skn domain could be advantageous, if the support segment helices adopt an extended arrangement to place both the amino-terminal arm and the BR on DNA (Fig. 4, Model 2). Complete folding apparently is not required for the Skn domain to bind DNA nonspecifically (not shown), and thus to sample potential binding sites. These observations are consistent with recent proposals that some bZIP proteins bind DNA initially as monomers, then dimerize and adopt both secondary and tertiary structure on DNA (Park et al. 1996; Metallo and Schepartz 1997).
The Skn domain is notably versatile, in that the support segment can accommodate BRs from bZIP proteins with distinct binding specificities (Fig. 5C, lanes 5,9,15,16). Base contact residues are conserved among bZIP proteins (Fig. 5A) (Ellenberger et al. 1992), implying that variations in positioning of these residues mediate differences in binding specificity. BR monomers bind with native half-site specificity when substituted into the Skn domain (Fig. 5C, lanes 5,9,15,16), indicating that base contact residue positioning is intrinsic to the BR segment. Members of a bZIP protein subfamily (the CNC proteins, Fig. 1) are defined by residues that are related to the Skn domain support segment, but these proteins lack an adjacent amino-terminal arm. The corresponding residues of the NF–E2 p45 protein (Fig. 1) contribute to binding of the NF–E2 bZIP dimer to DNA (K. Kotkow and S. Orkin, pers. comm.) and, when linked to the Skn domain amino-terminal arm, can promote monomeric BR–DNA binding at low temperature (not shown). These residues of CNC-type proteins (Fig. 1) thus appear to constitute a support segment that is functionally related to that of the Skn domain.
By forming an extension of support segment helix 4, the Skn domain BR helix is stabilized on DNA through two mechanisms. First, other support segment helices (Fig. 6A) are likely to pack against the DNA backbone, as well as against helix 4 (Fig. 4, model 2; Fig. 7A), and through helix 4 could anchor the BR helix in the major groove. The intense hydroxyl radical footprinting between top strand residues −1 and +1 (Fig. 3A,B,E) is consistent with this model. BR positioning is probably of general importance for DNA binding, because the BR cannot completely fill the wide major groove, and does not bind parallel to it (Ellenberger 1994). In contrast, RNA hairpins are more flexible than DNA, and thus can provide a snug fit for an α-helix, and can be bound more stably by short helical peptides (Tan et al. 1993; Harada et al. 1996). In a second mechanism, the BR is stabilized directly by being coupled to the more stable helix 4 (Fig. 6A), although presence of a kink in this uninterrupted helix cannot be ruled out. α-helices are stabilized by increased length, through cooperative hydrogen bonding of main chain atoms (Zimm and Bragg 1959), and also by having appropriate terminal residues (Presta and Rose 1988; Richardson and Richardson 1988; Serrano and Fersht 1989), particularly at the amino terminus (Scholtz and Baldwin 1995). The BR has intrinsic helical propensity (Weiss 1990; Saudek et al. 1991; Krebs et al. 1995), but in the Skn domain it is stabilized further by the additional helix length contributed by helix 4, as well as by the helix 4 amino-terminal cap. The impairment of DNA binding that resulted from G substitutions at three nonessential helix 4 positions (Fig. 7, C and D, respectively, lane 5), is consistent with both BR stabilization mechanisms, particularly the second.
The Skn domain provides stability to the BR monomer that is lacking in tethered BR peptide dimers, which are missing the ZIP segment, and generally bind DNA only at lower temperatures, and/or when stabilized by terminal modifications (Talanian et al. 1990; Park et al. 1992; Talanian et al. 1992; Cuenoud and Schepartz 1993a,b; Stanojevic and Verdine 1995; Pellegrini and Ebright 1996). In bZIP and bHLH proteins, the BR helix extends directly from the amino terminus of the respective dimerization segment helix (Ellenberger 1994). This arrangement is analogous, in reverse, to how the Skn domain BR helix extends from the carboxyl terminus of helix 4. Presumably, then, the ZIP and HLH dimerization segments also stabilize the BR through both of the mechanisms described above. In bZIP and bHLH dimers, each BR is positioned on the DNA by its extension from the dimerization segment complex, which in turn is anchored by the other BR. In addition, these dimerization segments increase BR helix length, and provide the carboxyl terminus of the continuous helix. Our findings show that helix extension is a conserved means of supporting DNA binding by short, exposed BRs.
Materials and methods
Protein expression and DNA binding assays
The Skn domain and Δ1–9 proteins consist of the residues indicated in Figure 1, preceded by a methionine. They were expressed in Escherichia coli BL21 cells from T7 expression (Studier 1991) plasmid vectors (T7Skn and T7Δ1–9). Coding region inserts were produced by PCR, and their fidelity was confirmed by DNA sequencing. Proteins were expressed by IPTG induction (Studier 1991) and purified to >95% homogeneity by ion exchange chromatography. Their concentrations were determined by tyrosine fluorescence (Edelhoch 1967). The Skn domain concentration was confirmed by quantitative amino acid analysis (Harvard University Microchemistry Facility).
Kd values for binding to specific and nonspecific DNA were estimated by EMSA titration, as the protein concentration at which 50% of the DNA is bound under conditions of vast protein excess (Carey 1991). Specific DNA binding was measured with the 22-bp double-stranded oligonucleotide SK1 (Blackwell et al. 1994), which contains a consensus SKN-1-binding site, and nonspecific binding was assayed with the MSK1 oligonucleotide, in which the bZIP half-site in SK1 was changed to CGTGT. For each Kd measurement, annealed DNA was freshly diluted in 100 mm NaCl to 1 × 10−11 m. Protein dilutions were made in 200 mm NaOAc, 5 mm DTT, and added at a 1:10 ratio to the binding cocktails. DNA labeling with 32P and EMSA analyses were performed as described (Blackwell et al. 1994), except that the binding cocktail salt consisted of 110 mm KCl. Error ranges given indicate the approximate upper and lower limits predicted by a plot of four EMSA titrations that were quantitated by a PhosphorImager. These Kd values were corrected to account for the fraction of each protein that participated in binding, as estimated by titrations performed at DNA and protein concentrations both vastly higher than the Kd (Carey 1991). The on-rate for the Skn domain-DNA complex is so rapid that a binding cocktail that is mixed and loaded immediately onto a running EMSA gel is at equilibrium before the bound and free fractions can be separated. In off-rate measurements, addition of excess unlabeled specific competitor rapidly disrupts Skn domain–DNA complexes so rapidly that they are undetectable even if the gel is loaded immediately.
BR and helix 4 mutants were constructed by PCR as derivatives of SknT, in the T7Skn expression plasmid (Figs. 1 and 5A). EMSAs of in vitro-translated proteins were performed by use of 32P-labeled probes and approximately equal (within twofold accuracy) protein concentrations in each sample (0.3 × 10−10 to 1.0 × 10−10 m), as described (Blackwell et al. 1994). The C/EBP half swap site was identical to SK1, except that the bZIP half site was GCAAT (Johnson 1993; Suckow et al. 1993). In EMSAs performed at 0°C, samples were mixed and incubated for 20 min on ice, and run on a prechilled gel in the cold room.
CD spectroscopy
CD spectra were obtained with an AVIV 62DS spectrometer by use of a 1-mm cell. Samples contained the Skn domain at 1.6 × 10−5 m and, when appropriate, DNA at 2.0 × 10−5 m. The scans shown were performed at 0.4 m NaCl, but the Skn domain wavelength spectra did not vary substantially between NaCl concentrations of 0.1 and 1 m. These samples also contained 0.1 mm DTT and either 20 mm NH4OAc (pH 7.0) or 20 mm phosphate buffer (pH 6.5). Both buffers gave comparable results. Each spectra represents the average of 10 scans, and has been baseline-corrected with spectra of buffer alone (for the Skn domain alone) or of the DNA fragment in buffer (for Skn domain–DNA complex scans). The ellipticity of the Skn domain alone was linearly proportional to its concentration (not shown). The midpoint of the Skn domain:DNA complex folding transition was obtained by plotting the first derivative of the plot shown in Figure 2B. The double-stranded DNA fragment used (SK2101) was ATGACCATTGTCATCCCACTG. The percent helix is estimated from the mean residue ellipticity at 222 nm, assuming a value of −33,000°/cm2 per dmole for a 100% helical peptide at 0°C, and a correction of 0.3% per °C (to 30,500 at 25°C) (Weiss et al. 1990).
Hydroxyl radical footprinting and interference
Hydroxyl radical footprinting and interference assays were performed essentially as described (Blackwell et al. 1994), except that the footprints were obtained without separation of bound and free fractions. Previously, after hydroxyl radical cleavage, bound and free DNA fractions were separated by EMSA to maximize contrast (Blackwell et al. 1994). The extremely rapid on and off rates of the Skn domain–DNA complex, however, suggested that such footprints might be influenced by binding interference patterns (Dixon et al. 1991), because these complexes could repeatedly dissociate and reform during the incubation. The footprints shown in Figure 2, therefore, were performed by incubating labeled DNA with a protein concentration that yielded maximal specific (but minimal nonspecific) binding, then cleaving with hydroxyl radicals. Because the binding affinities were lower under these conditions than in the EMSA assay of SK1 oligonucleotide binding, the final concentrations of the Skn domain and Δ1–9 used averaged ∼20 and 200 nm, respectively. The resulting Skn domain footprint was reproducibly broader along the bottom strand than the previous footprint of full-length SKN-1 (Fig. 3A) (Blackwell et al. 1994), but was not distinguishable from a bottom strand full-length SKN-1 footprint obtained by this method (not shown). Quantitative analysis of PhosphorImager (Bio-Rad) data was performed with Molecular Analyst and Microsoft Excel. PhosphorImaging of the top strand samples was performed on a duplicate gel that lacked the small spot at position −4 of the Δ1–9 footprint (Fig. 3A).
NMR spectroscopy
NOE data were obtained from a two-dimensional NOESY spectrum acquired at 750 MHz and a three-dimensional NOESY–HSQC acquired at 600 MHz. Residues 9–66 were assigned based on two-dimensional NOESY and three-dimensional 15N NOESY–HSQC and 15N total correlation spectroscopy (TOCSY)–HSQC spectra. Samples for these NOESY spectra contained 2 mm Skn domain in 20 mm phosphate (pH 5), 0.1 m NaCl, 10 mm DTT. They were degassed in the NMR tube and blanketed with argon or nitrogen to prevent oxidation of the free cysteine. 15N-Labeled Skn domain was purified from E. coli on M9 medium with 15NH4Cl as the sole nitrogen source. The three-dimensional NOESY–HSQC was recorded on a Bruker AMX600 spectrometer and the two-dimensional NOESY acquired on a Varian Unity plus 750 MHz spectrometer. Both NOESY spectra had mixing times of 100 msec. HSQC samples contained 0.2 mm Skn domain in 20mm phosphate (pH 6.5), 0.1 m NaCl, and 10 mm DTT. The sample of the specific Skn domain/DNA complex also contained 0.2 mm duplex DNA (TACATTGTCATCCCTCA). For the corresponding spectrum with 0.2 mm nonspecific DNA, the oligonucleotide CGTCGGAGGACTGTCCTCCGACG was annealed to create a duplex with a single T:T mismatch. One thousand twenty-four complex points were acquired for 256 complex points in the indirect dimension. The final size of each data set was 512 × 512 points. All NOESY and HSQC spectra were acquired with Watergate for water suppression (Sklenar et al. 1993) and all data processed with Felix 2.3 (Biosym Technologies).
Acknowledgments
We thank Lew Cantley for use of his Bio-Rad PhosphorImager, Hans Wendt for helpful discussions, and Cary Gunther and Thip Kophengnavong for contributing to the project. For reading the manuscript, we thank members of the Blackwell laboratory, Phil Auron, and Steve Harrison, whom we also thank for use of his CD spectrometer. T.K.B. is grateful to the late Harold Weintraub for invaluable discussions, insights, and for his boundless enthusiasm, all of which are sorely missed. This work was supported by grants from the National Institutes of Health to T.K.B. (RO1GM50900) and G.W. (PO1GM47467). T.E.E. is supported by the Lucille P. Markey Charitable Trust, and T.K.B. is a Searle Scholar.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Note added in proof
After submission of this manuscript, Pal et al. (Proc. Natl. Acad. Sci. 94: 5556–5561) reported that the SKN-1 BR folds upon binding DNA and showed NMR evidence that residues 49–59 (the core of helix 4) are helical in solution. They also reported that a Skn domain version that contains a Cys → Ser substitution at position 70 and lacks the four most amino-terminal residues forms a complex with DNA that melts at 71°C.
Footnotes
E-MAIL blackwell@cbr.med.harvard.edu; FAX (617) 278-3131.
References
- Anthony-Cahill SJ, Benfield PA, Fairman R, Wasserman ZR, Brenner SL, Stafford WF, Altenbach C, Hubbell WL, DeGrado WF. Molecular characterization of helix-loop-helix peptides. Science. 1992;255:979–983. doi: 10.1126/science.1312255. [DOI] [PubMed] [Google Scholar]
- Aurora R, Srinivasan R, Rose GD. Rules for α-helix termination by glycine. Science. 1994;264:1126–1130. doi: 10.1126/science.8178170. [DOI] [PubMed] [Google Scholar]
- Berg JM, Shi Y. The galvanization of biology: A growing appreciation for the roles of zinc. Science. 1996;271:1081–1085. doi: 10.1126/science.271.5252.1081. [DOI] [PubMed] [Google Scholar]
- Blackwell TK, Bowerman B, Priess J, Weintraub H. Formation of a monomeric DNA binding domain by Skn-1 bZIP and homeodomain elements. Science. 1994;266:621–628. doi: 10.1126/science.7939715. [DOI] [PubMed] [Google Scholar]
- Boniface JJ, Lyons DS, Wettstein DA, Allbritton NL, Davis MM. Evidence for a conformational change in a class II major histocompatibility complex molecule occurring in the same pH range where antigen binding is enhanced. J Exp Med. 1996;183:119–126. doi: 10.1084/jem.183.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowerman B, Eaton BA, Priess JR. skn-1, a maternally expressed gene required to specify the fate of ventral blastomeres in the early C. elegans embryo. Cell. 1992;68:1061–1075. doi: 10.1016/0092-8674(92)90078-q. [DOI] [PubMed] [Google Scholar]
- Bowerman B, Draper BW, Mello C, Priess J. The maternal gene skn-1 encodes a protein that is distributed unequally in early C. elegans embryos. Cell. 1993;74:443–452. doi: 10.1016/0092-8674(93)80046-h. [DOI] [PubMed] [Google Scholar]
- Bradley EK, Thomason JF, Cohen FE, Kosen PA, Kuntz ID. Studies of synthetic helical peptides using circular dichroism and nuclear magnetic resonance. J Mol Biol. 1990;215:607–622. doi: 10.1016/S0022-2836(05)80172-X. [DOI] [PubMed] [Google Scholar]
- Carey J. Gel retardation. Methods Enzymol. 1991;208:103–117. doi: 10.1016/0076-6879(91)08010-f. [DOI] [PubMed] [Google Scholar]
- Cuenoud B, Schepartz A. Altered specificity of DNA-binding proteins with transition metal dimerization domains. Science. 1993a;259:510–513. doi: 10.1126/science.8424173. [DOI] [PubMed] [Google Scholar]
- ————— Design of a metallo-bZIP protein that discriminates between CRE and AP1 target sites: Selection against AP1. Proc Natl Acad Sci. 1993b;90:1154–1159. doi: 10.1073/pnas.90.4.1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Filippis V, Polverino de Laureto P, Toniutti N, Fontana A. Acid-induced molten globule state of a fully active mutant of human interleukin-6. Biochemistry. 1996;35:11503–11511. doi: 10.1021/bi9604587. [DOI] [PubMed] [Google Scholar]
- Dixon W, Hayes JJ, Levin JR, Weidner MF, Dombrowski BA, Tullius TD. Hydroxyl radical footprinting. Methods Enzymol. 1991;208:380–413. doi: 10.1016/0076-6879(91)08021-9. [DOI] [PubMed] [Google Scholar]
- Edelhoch H. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry. 1967;6:1948–1954. doi: 10.1021/bi00859a010. [DOI] [PubMed] [Google Scholar]
- Ellenberger TE. Getting a grip on DNA recognition: Structures of the basic region leucine zipper, and the basic region helix-loop-helix DNA-binding domains. Curr Opin Struct Biol. 1994;4:12–21. [Google Scholar]
- Ellenberger TE, Brandl CJ, Struhl K, Harrison SC. The GCN4 BR-leucine zipper binds DNA as a dimer of uninterrupted α-helices: Crystal structure of the protein-DNA complex. Cell. 1992;71:1223–1237. doi: 10.1016/s0092-8674(05)80070-4. [DOI] [PubMed] [Google Scholar]
- Evans LJA, Goble ML, Hales KA, Lakey JH. Different sensitivities to acid denaturation within a family of proteins: Implications for acid unfolding and membrane translocation. Biochemistry. 1996;35:13180–13185. doi: 10.1021/bi960990u. [DOI] [PubMed] [Google Scholar]
- Gehring WJ, Qian YQ, Billeter M, Furukubo-Tokunaka K, Schier AF, Resendez-Perez D, Affolter M, Otting G, Wuthrich K. Homeodomain-DNA recognition. Cell. 1994;78:211–223. doi: 10.1016/0092-8674(94)90292-5. [DOI] [PubMed] [Google Scholar]
- Glover JN, Harrison SC. Crystal structure of the heterodimeric bZIP transcription factor c-Fos–c-Jun bound to DNA. Nature. 1995;373:257–261. doi: 10.1038/373257a0. [DOI] [PubMed] [Google Scholar]
- González-Mañas JM, Lakey JH, Pattus F. Brominated phospholipids as a tool for monitoring the membrane insertion of colicin A. Biochemistry. 1992;31:7294–7300. doi: 10.1021/bi00147a013. [DOI] [PubMed] [Google Scholar]
- Harada K, Martin SS, Frankel AD. Selection of RNA-binding peptides in vivo. Nature. 1996;380:175–179. doi: 10.1038/380175a0. [DOI] [PubMed] [Google Scholar]
- Harper ET, Rose GD. Helix stop signals in proteins and peptides: The capping box. Biochemistry. 1993;32:7605–7609. doi: 10.1021/bi00081a001. [DOI] [PubMed] [Google Scholar]
- Harrison SC. A structural taxonomy of DNA-binding proteins. Nature. 1991;353:715–719. doi: 10.1038/353715a0. [DOI] [PubMed] [Google Scholar]
- Jennings PA, Wright PE. Formation of a molten globule intermediate early in the kinetic folding pathway of apomyoglobin. Science. 1993;262:892–896. doi: 10.1126/science.8235610. [DOI] [PubMed] [Google Scholar]
- Johnson PF. Identification of C/EBP basic region residues involved in DNA sequence recognition and half-site spacing preference. Mol Cell Biol. 1993;13:6919–6930. doi: 10.1128/mcb.13.11.6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WC. Secondary structure of proteins through circular dichroism spectroscopy. Annu Rev Biophys Biophys Chem. 1988;17:145–166. doi: 10.1146/annurev.bb.17.060188.001045. [DOI] [PubMed] [Google Scholar]
- Kay MS, Baldwin RL. Packing interactions in the apomyoglobin folding intermediate. Nature Struct Biol. 1996;3:439–445. doi: 10.1038/nsb0596-439. [DOI] [PubMed] [Google Scholar]
- Keller W, König P, Richmond TJ. Crystal structure of a bZIP/DNA complex at 2.2å: Determinants of DNA specific recognition. J Mol Biol. 1995;254:657–667. doi: 10.1006/jmbi.1995.0645. [DOI] [PubMed] [Google Scholar]
- Kissinger CR, Liu B, Martin-Blanco E, Kornberg TB, Pabo C. Crystal structure of an engrailed homeodomain-DNA complex at 2.8 Å resolution: A framework for understanding homeodomain-DNA interactions. Cell. 1990;63:579–580. doi: 10.1016/0092-8674(90)90453-l. [DOI] [PubMed] [Google Scholar]
- König P, Richmond TJ. The x-ray structure of the GCN4-bZIP bound to ATF/CREB site DNA shows the complex depends on DNA flexibility. J Mol Biol. 1993;233:139–154. doi: 10.1006/jmbi.1993.1490. [DOI] [PubMed] [Google Scholar]
- Krebs D, Dahmani B, El Antri S, Monnot M, Convert O, Mauffret O, Troalen F, Fermandjian S. The basic subdomain of the c-Jun oncoprotein: A joint CD, Fourier-transform infrared and NMR study. Eur J Biochem. 1995;231:370–380. doi: 10.1111/j.1432-1033.1995.tb20709.x. [DOI] [PubMed] [Google Scholar]
- Kuwajima K. The molten globule as a clue for understanding the folding and cooperativity of globular-protein structure. Proteins: Struct Function Genet. 1989;6:87–103. doi: 10.1002/prot.340060202. [DOI] [PubMed] [Google Scholar]
- Landschulz WH, Johnson PF, Adashi EY, Graves BJ, McKnight SL. Isolation of a recombinant copy of the gene encoding C/EBP. Genes & Dev. 1988;2:786–800. doi: 10.1101/gad.2.7.786. [DOI] [PubMed] [Google Scholar]
- Mach H, Middaugh CR. Interaction of partially structured states of acidic fibroblast growth factor with phospholipid membranes. Biochemistry. 1995;34:9913–9920. doi: 10.1021/bi00031a013. [DOI] [PubMed] [Google Scholar]
- Metallo SJ, Schepartz A. Certain bZIP peptides bind DNA sequentially as monomers and dimerize on the DNA. Nature Struct Biol. 1997;4:115–117. doi: 10.1038/nsb0297-115. [DOI] [PubMed] [Google Scholar]
- O’Neil KT, Hoess RH, DeGrado WF. Design of DNA-binding peptides based on the leucine zipper motif. Science. 1990;249:774–778. doi: 10.1126/science.2389143. [DOI] [PubMed] [Google Scholar]
- O’Shea EK, Rutkowski R, Kim PS. Evidence that the leucine zipper is a coiled coil. Science. 1989;243:538–542. doi: 10.1126/science.2911757. [DOI] [PubMed] [Google Scholar]
- Park C, Campbell JL, Goddard WAI. Protein stitchery: Design of a protein for selective binding to a specific DNA sequence. Proc Natl Acad Sci. 1992;89:9094–9096. doi: 10.1073/pnas.89.19.9094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Can the monomer of the leucine zipper proteins recognize the dimer binding site without dimerization? J Am Chem Soc. 1996;118:4235–4239. [Google Scholar]
- Patel L, Abate C, Curran T. Altered protein conformation on DNA binding by Fos and Jun. Nature. 1990;347:572–575. doi: 10.1038/347572a0. [DOI] [PubMed] [Google Scholar]
- Pellegrini M, Ebright RH. Artificial sequence-specific DNA binding peptides: Branched-chain basic regions. J Am Chem Soc. 1996;118:5831–5835. [Google Scholar]
- Peng Z-Y, Wu LC, Schulman BA, Kim PS. Does the molten globule have a native-like tertiary fold? Phil Trans R Soc Lond B. 1995;348:43–47. doi: 10.1098/rstb.1995.0044. [DOI] [PubMed] [Google Scholar]
- Percival-Smith A, Müller M, Affolter M, Gehring WJ. The interaction with DNA of wild-type and mutant fushi tarazu homeodomains. EMBO J. 1990;9:3967–3974. doi: 10.1002/j.1460-2075.1990.tb07617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presta LG, Rose GD. Helix signals in proteins. Science. 1988;240:1632–1641. doi: 10.1126/science.2837824. [DOI] [PubMed] [Google Scholar]
- Ptitsyn O. How molten is the molten globule? Nature Struct Biol. 1996;3:488–490. doi: 10.1038/nsb0696-488. [DOI] [PubMed] [Google Scholar]
- Richardson JS, Richardson DC. Amino acid preferences for specific locations at the ends of α helices. Science. 1988;240:1648–1652. doi: 10.1126/science.3381086. [DOI] [PubMed] [Google Scholar]
- Runnels HA, Moore JC, Jensen PE. A structural transition in class II major histocompatibility complex proteins at mildly acidic pH. J Exp Med. 1996;183:127–136. doi: 10.1084/jem.183.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saudek V, Pasley HS, Gibson T, Gausepohl H, Frank R, Pastore A. Solution structure of the basic region from the transcriptional activator GCN4. Biochemistry. 1991;30:1310–1317. doi: 10.1021/bi00219a022. [DOI] [PubMed] [Google Scholar]
- Scholtz JM, Baldwin RL. α-Helix formation by peptides in water. In: Gotte B, editor. Peptides: Synthesis, structures, and applications. San Diego, CA: Academic Press; 1995. pp. 171–192. [Google Scholar]
- Seeley SK, Weis RM, Thompson LK. The cytoplasmic fragment of the aspartate receptor displays globally dynamic behavior. Biochemistry. 1996;35:5199–5206. doi: 10.1021/bi9524979. [DOI] [PubMed] [Google Scholar]
- Serrano L, Fersht AR. Capping and α-helix stability. Nature. 1989;342:296–299. doi: 10.1038/342296a0. [DOI] [PubMed] [Google Scholar]
- Shuman JD, Vinson CR, McKnight SL. Evidence of changes in protease sensitivity and subunit exchange rate on DNA binding by C/EBP. Science. 1990;249:771–774. doi: 10.1126/science.2202050. [DOI] [PubMed] [Google Scholar]
- Sklenar V, Piotto M, Leppik R, Saudek V. Gradient-tailored water suppression of 1H-15N HSQC experiments optimized to retain full sensitivity. J Magnet Res. 1993;102:241–245. [Google Scholar]
- Spolar RS, Record MT., Jr Coupling of local folding to site-specific binding of proteins to DNA. Science. 1994;263:777–784. doi: 10.1126/science.8303294. [DOI] [PubMed] [Google Scholar]
- Stanojevic D, Verdine GL. Deconstruction of GCN4/GCRE into a monomeric peptide-DNA complex. Nature Struct Biol. 1995;2:450–457. doi: 10.1038/nsb0695-450. [DOI] [PubMed] [Google Scholar]
- Studier FW. Use of bacteriophage T7 lysozyme to improve an inducible T7 expression system. J Mol Biol. 1991;219:37–44. doi: 10.1016/0022-2836(91)90855-z. [DOI] [PubMed] [Google Scholar]
- Suckow M, von Wilcken-Bergmann B, Muller-Hill B. Identification of three residues in the BRs of the bZIP proteins GCN4, C/EBP, and TAF-1 that are involved in specific DNA-binding. EMBO J. 1993;12:1193–1200. doi: 10.1002/j.1460-2075.1993.tb05760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talanian R, McKnight CJ, Kim P. Sequence-specific DNA binding by a short peptide dimer. Science. 1990;249:769–771. doi: 10.1126/science.2389142. [DOI] [PubMed] [Google Scholar]
- Talanian RV, McKnight CJ, Rutkowski R, Kim PS. Minimum length of a sequence-specific DNA binding peptide. Biochemistry. 1992;31:6871–6875. doi: 10.1021/bi00145a002. [DOI] [PubMed] [Google Scholar]
- Tan R, Chen L, Buettner JA, Hudson D, Frankel AD. RNA recognition by an isolated α-helix. Cell. 1993;73:1031–1040. doi: 10.1016/0092-8674(93)90280-4. [DOI] [PubMed] [Google Scholar]
- Tortorella D, Sesardic D, Dawes CS, London E. Immunochemical analysis of the structure of diptheria toxin shows all three domains undergo structural changes at low pH. J Biol Chem. 1995;270:27439–27445. doi: 10.1074/jbc.270.46.27439. [DOI] [PubMed] [Google Scholar]
- van der Goot FG, González-Mañas JM, Lakey JH, Pattus F. A “molten-globule” membrane-insertion intermediate of the pore-forming domain of colicin A. Nature. 1991;354:408–410. doi: 10.1038/354408a0. [DOI] [PubMed] [Google Scholar]
- Weiss MA. Thermal unfolding studies of a leucine zipper domain and its specific DNA complex: Implications for scissors grip recognition. Biochemistry. 1990;29:8020–8024. doi: 10.1021/bi00487a004. [DOI] [PubMed] [Google Scholar]
- Weiss MA, Ellenberger T, Wobbe CR, Lee JP, Harrison SC, Struhl K. Folding transition in the DNA-binding domain of GCN4 on specific binding to DNA. Nature. 1990;347:575–578. doi: 10.1038/347575a0. [DOI] [PubMed] [Google Scholar]
- Wishart DS, Sykes BD, Richards FM. The chemical shift index: A fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry. 1992;31:1647–1651. doi: 10.1021/bi00121a010. [DOI] [PubMed] [Google Scholar]
- Wolberger C, Vershon AK, Liu B, Johnson AD, Pabo CO. Crystal structure of a MATα2 homeodomain-operator complex suggests a general model for homeodomain-DNA interactions. Cell. 1991;67:517–528. doi: 10.1016/0092-8674(91)90526-5. [DOI] [PubMed] [Google Scholar]
- Wu LC, Schulman BA, Peng Z-Y, Kim PS. Disulfide determinants of calcium-induced packing in α-lactalbumin. Biochemistry. 1996;35:859–863. doi: 10.1021/bi951408p. [DOI] [PubMed] [Google Scholar]
- Zimm BH, Bragg JK. Theory of the phase transition between helix and random coil in polypeptide chains. J Chem Phys. 1959;31:526–535. [Google Scholar]