

Summary
Background
A number of adhesion-mediated signaling pathways and cell cycle events have been identified that regulate cell proliferation, yet studies to date have been unable to determine which of these pathways control mitogenesis in response to physiologically relevant changes in tissue elasticity. In this report, we have used hydrogel-based substrata matched to biological tissue stiffness to investigate the effects of matrix elasticity on the cell cycle.
Results
We find that physiological tissue stiffness acts as a cell cycle inhibitor in mammary epithelial cells and vascular smooth muscle cells; subcellular analysis in these cells, mouse embryo fibroblasts, and osteoblasts shows that cell cycle control by matrix stiffness is widely conserved. Remarkably, most mitogenic events previously documented as ECM/integrin-dependent proceed normally when matrix stiffness is altered in the range that controls mitogenesis. These include ERK activity, immediately-early gene expression, and cdk inhibitor expression. In contrast, FAK-dependent Rac activation, Rac-dependent cyclin D1 gene induction, and cyclin D1-dependent Rb phosphorylation are strongly inhibited at physiological tissue stiffness and rescued when the matrix is stiffened in vitro. Importantly, the combined use of atomic force microscopy and fluorescence imaging in the mouse shows that comparable increases in tissue stiffness occur at sites of cell proliferation in vivo.
Conclusion
Matrix remodeling associated with pathogenesis is, in itself, a positive regulator of the cell cycle through a highly selective effect on integrin-dependent signaling to FAK, Rac, and cyclin D1.
Introduction
Soluble mitogens and anti-mitogens have long been viewed as important regulators of cell proliferation, but it is now accepted that non-soluble factors, especially components of the extracellular matrix (ECM), have equally essential roles in the proliferation of most non-transformed cell types. Soluble mitogens and ECM proteins jointly regulate activation of the G1 phase cyclin-dependent kinases (cdks), cdk4/6 and cdk2, required for S phase entry [1]. ECM proteins signal by binding and activating the integrin family of cell surface receptors, and many studies have identified integrin-dependent signaling events that support G1 phase progression, especially the induction of cyclin D1 mRNA and protein and the downregulation of the cip/kip family of cdk inhibitors [1]. These effects control activation of cdk4/6 and cdk2, respectively, which, in turn, catalyze the inactivating phosphorylations of the retinoblastoma protein (Rb) and related proteins p107 and p130. Rb phosphorylation results in the release of sequestered E2Fs and the induction of E2F-dependent genes required for S phase.
Integrin-dependent signaling events implicated upstream of cyclin D1 and the cip/kips include the activation of ERK MAP kinases, Rho family GTPases, and FAK [1]. Yet little is known about whether and how these pathways are activated in physiologically relevant mechanical microenvironments. This gap in knowledge arises, at least in part, because integrin-regulated events have typically been identified in cultured cells after complete blockade of ECM-integrin binding (e.g. by incubating cells in suspension or on poly-lysine coated dishes) or by preventing integrin clustering (e.g. by disrupting the actin cytoskeleton with depolymerizing drugs or inhibitors of Rho GTPase activation or signaling). These approaches result in much more severe changes in integrin occupancy, f-actin polymerization, and Rho GTPase activity than are likely to occur physiologically. Additionally, a pervasive shortcoming of traditional cell biological approaches to the study of ECM function is that cells are typically cultured on nondeformable substrata (culture dishes or glass coverslips) which have little relationship to the elastic (also called “compliant”) ECM that cells encounter in vivo. Since a hallmark of ECM-cell interactions is the ability to sense extracellular stiffness, tissue compliance may be an important determinant of downstream adhesion-dependent signaling events.
Some studies have used collagen gels to study the effect of a more compliant matrix on integrin signaling and the cell cycle. These studies showed that human foreskin fibroblasts in free-floating collagen gels have high levels of p27, do not phosphorylate ERK, and do not express cyclin D1 [2, 3]. Others have reported that increased p21cip1 or p15INK4B is responsible for G1 phase arrest that occurs when cells are plated on soft collagen gels [4, 5]. FAK autophosphorylation, ERK activity, and Rho activation are also impaired when cells are cultured on or in soft collagen matrices or Matrigels [2, 6]. Although these results suggest that changes in matrix stiffness recapitulate the effects seen upon complete adhesion blockade, collagen gels are much softer than many physiological tissues (elastic moduli of 10-50 Pa vs. 100-100,000 Pa) [7, 8]. Moreover, changing the stiffness of collagen gels by altering collagen concentration inherently affects the integrin ligand concentration, so observed effects can not be strictly attributed to changes in matrix elasticity. The same complication exists when cells are cultured in Matrigel, which is also less well-defined chemically.
Careful control of substratum elasticity is perhaps best achieved by seeding cells on ECM-coated biocompatible hydrogels because elasticity can be varied independently of matrix concentration [9, 10]. Hydrogels based on polyacrylamide have elastic moduli of ∼1500-150,000 Pa which encompass the stiffness of most physiological tissues [10]. Cells plated on soft hydrogels show a decrease in cell number [9, 11], but the molecular events that underlie cell cycle control by matrix stiffness remain undefined. In this report, we combine biophysical measurements of tissue elasticity with a molecular analysis of the cell cycle on compliance-appropriate hydrogels to elucidate the subcellular effects of matrix stiffness on cell proliferation.
Results
A small subset of adhesion-dependent signaling events accounts for cell cycle control by matrix stiffness
We adapted the use of deformable matrix protein-coated acrylamide hydrogels to a molecular analysis of the cell cycle. Quiescent mouse embryo fibroblasts (MEFs) were plated on fibronectin (FN)-coated hydrogels having elastic moduli within the physiological range [7, 12]. Serum-stimulated cell cycle entry was barely detected when MEFs were seeded on low stiffness FN substrata (E <2000 Pa), and the degree of S phase entry increased with matrix stiffness until optimal cycling was obtained at ∼24,000 Pa (Fig. 1A). Remarkably, the same range of matrix stiffness regulated S phase entry in MCF10A mammary epithelial cells (Fig. 1A), vascular smooth muscle cells (Fig. 1A; VSMCs) and osteoblastic cells (Suppl. Fig. 1). Thus, the effect of matrix stiffness on G1 and S phase progression is widely conserved and independent of the rigidity of individual tissue microenvironments. Mitogenesis (assessed as cyclin A mRNA induction; an E2F target required for S phase entry and progression) was dependent on matrix stiffness whether cells entered the cycle from G0 or G2/M (Fig. 1B), indicating that both cell cycle re-entry from quiescence and subsequent cycling are controlled by ECM stiffness. Subsequent experiments used hydrogels prepared at ∼2000 and ∼24,000 Pa; referred to as low and high stiffness, respectively.
Figure 1. Selective effect of ECM stiffness on cyclin D1 gene expression.
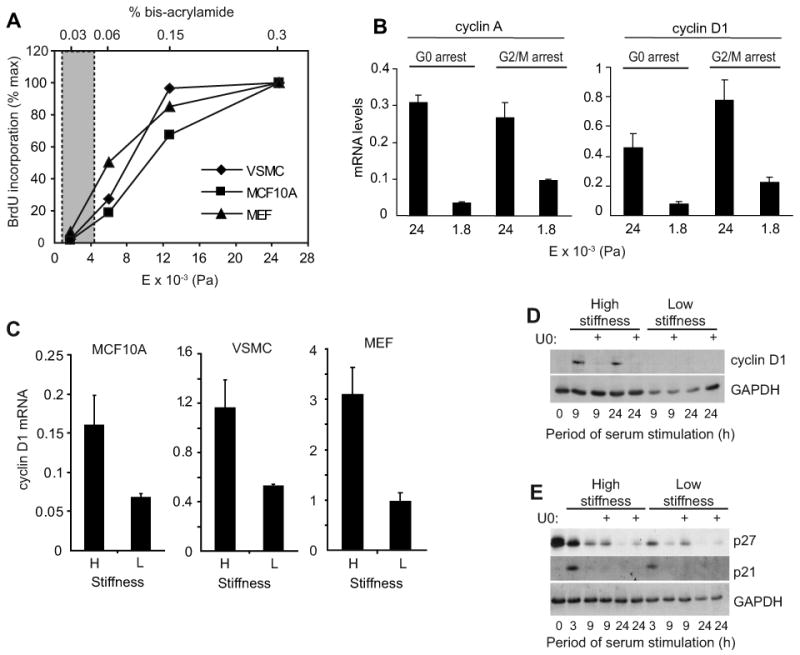
(A) Serum-starved cells were stimulated with mitogens, incubated with BrdU, and reseeded on hydrogels made with a constant 7.5% acrylamide. Bis-acrylamide varied from 0.03%-0.3%. After 24 h (MCF10A cells and MEFs) or 48 h (VSMCs), the cells were fixed and BrdU incorporation was determined. The graph compiles results from an individual experiment for each cell type and shows percent maximal BrdU incorporation compared to the stiffest hydrogel. The shaded area highlights the range of elastic moduli measured in mouse mammary glands and arteries as determined by milliprobe indentation and AFM; see Suppl. Table 2. (B) MEFs were synchronized at G0 (by 48 h serum-starvation) or G2/M (by treatment with 5 μg/ml nocodozole for 24 h). The cells were reseeded on hydrogels and stimulated with 10% FBS. RNA was isolated 24 h after reseeding and analyzed by QPCR for cyclin A or cyclin D1 mRNAs. (C) Serum-starved cells were reseeded on low (L) and high (H) stiffness hydrogels with mitogens. Cyclin D1 mRNA was measured by QPCR at times corresponding to optimal induction (12 h for MCF10A cells, 24 h for VSMCs, and 9 h for MEFs). (D and E) Serum-starved MEFs were pre-treated with DMSO (vehicle) or U0126 (U0) prior to reseeding on hydrogels and stimulation with 10% FBS. Reseeded cells were collected at the indicated times and analyzed by western blotting.
As in previous reports [13, 14], cell spreading and cytoskeletal organization were inhibited when either epithelial or mesenchymal cells were plated on the low stiffness acrylamide hydrogels (Suppl. Fig. 2). Since decreased cell spreading has been associated with decreased integrin signaling and S phase entry [15], we initially thought that the cell cycle inhibition reflected an overall decrease in integrin signaling. Overall inhibition of integrin signaling prevents the mitogen-dependent induction of cyclin D1 mRNA and the mitogen-dependent downregulation of the p21cip1 and p27kip1 cdk inhibitors [1, 16, 17]. In contrast, the soft substratum that prevents mitogenesis inhibited the induction of cyclin D1 mRNA and protein (Fig. 1C and D) but not the G1 phase downregulation of p21cip1 or p27kip1 (Fig. 1E). Thus, matrix compliance targets a discrete subset of adhesion-dependent cell cycle events. Cyclin D1 gene expression was similarly regulated by matrix stiffness in cells entering G1 phase from G0 or G2/M (Fig. 1B).
Previous studies on glass or plastic substrata indicate that the mid-G1 phase induction of cyclin D1 requires sustained ERK activity [18, 19]. In agreement with these results, we found that serum-stimulated MEFs plated on the high stiffness substratum used ERK activity to induce cyclin D1 protein as determined with the MEK inhibitor, U0126 (Fig. 1D). However, a comparison of ERK activation in cells cultured on low and high stiffness substrata showed that the magnitude and duration of ERK phosphorylation are not strongly affected by this range of ECM stiffness (Fig. 2A and Suppl. Fig. 3A) despite large differences in expression of cyclin D1 (Fig. 1C and D) and mitogenesis (Fig. 1A). Induction of the ERK-dependent immediate-early genes associated with cyclin D1 gene expression, Fra-1 and JunB [18, 19], was also similar on the high and low stiffness substrata (Fig. 2B-D and Suppl. Fig. 3B), indicating that ERK translocation to the nucleus and ERK-dependent transcription were proceeding normally. Thus, despite the fact that ERK activity is adhesion-dependent, as determined by the complete blockade of cell adhesion or actin depolymerization [1], changes in ERK activity are not responsible for the effect of matrix stiffness on cyclin D1 gene expression.
Figure 2. Regulation of mitogenesis and cyclin D1 gene expression by ECM stiffness is ERK-independent.
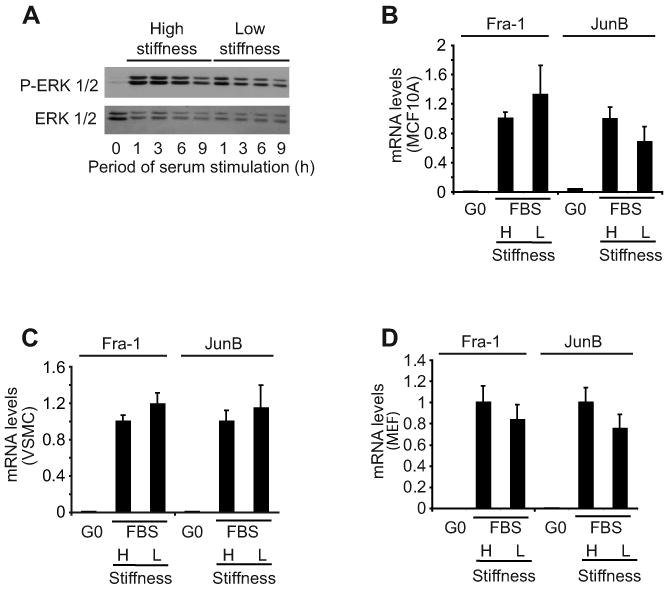
(A) Serum-starved MEFs were reseeded on hydrogels and stimulated with 10% FBS. Reseeded cells were collected at the indicated times and analyzed by western blotting. (B-D) Serum-starved cells were reseeded on high (H) and low (L) stiffness hydrogels with mitogens. RNA was collected from quiescent cells (G0) and cells stimulated with 10% FBS at times optimal for JunB and Fra1 mRNA induction (1 and 3 h, respectively) in the three cell types. Fra-1 and JunB mRNA levels were determined by QPCR, normalized to 18S rRNA, and plotted relative to the levels of the high stiffness samples.
Regulated FAK localization as a mechanosensor for cell cycle progression
The overall extent of immediate-early gene induction, cyclin D1 gene induction, and mitogenesis was similar in cells plated on glass vs. the high stiffness hydrogels (Suppl. Fig. 4), but focal adhesions were less robust than those seen on traditional glass coverslips as assessed by immunostaining for FAK, Y397-phosphorylated FAK (FAKpY397), vinculin, and paxillin (Suppl. Fig. 5). By these same criteria, focal adhesions were undetected in cells plated at low matrix stiffness (Suppl. Fig. 5). FAK phosphorylation has been linked to mechanotransduction and cyclin D1 gene expression [20-22], and, indeed, we found that FAK autophosphorylation at Y397 was reduced when cells were cultured on the low stiffness ECM (Fig. 3A). Conversely, ectopically expressed FRNK or non-phosphorylatable FAK (FAKY397F) suppressed cyclin D1 mRNA expression in MEFs incubated on the high stiffness ECM (Fig. 3B and C). This effect did not reflect a global disruption of integrin complexes because vinculin-containing focal adhesions persisted in the presence of FRNK or FAKY397F (Suppl. Fig. 6). Cyclin D1 induction was also impaired in FAK-null cells (Suppl. Fig. 7A). Thus, FAK inhibition or depletion on stiff substrata phenocopies the effect of low matrix stiffness on cyclin D1 gene expression.
Figure 3. FAK is linked to stiffness-dependent induction of cyclin D1 mRNA.
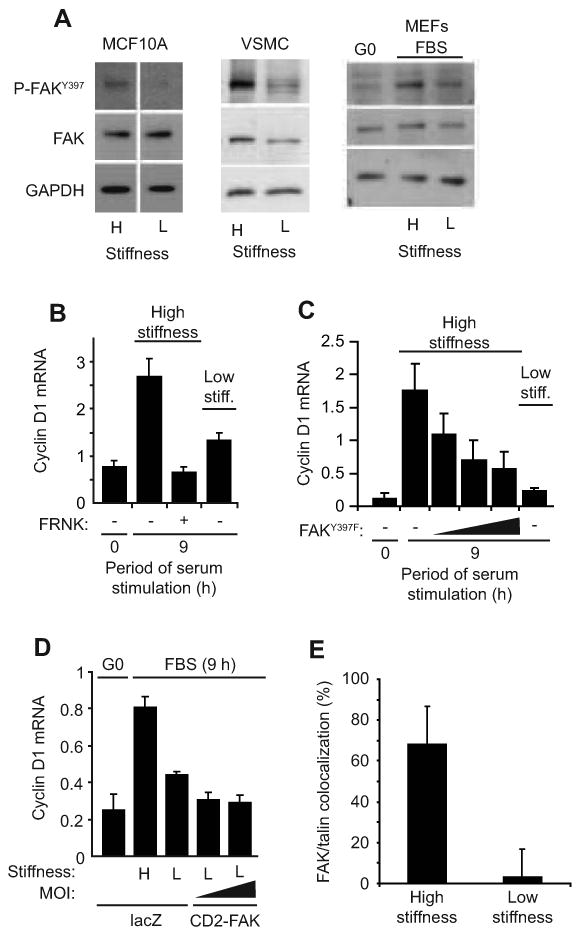
(A) Serum-starved cells were reseeded on high (H) and low (L) stiffness hydrogels with mitogens, collected 3 hr after reseeding, and analyzed by western blotting. The white vertical line in the MCF10A blot indicates where extraneous information was removed from the blot. (B) Starved MEFs infected with adenoviruses encoding LacZ (control; -) or GFP-FRNK were reseeded on hydrogels with 10% FBS. Cyclin D1 mRNA was measured by QPCR. (C) The experiment in panel B was repeated using an adenovirus encoding FAKY397F (at 250, 500, and 1500 MOI) rather than FRNK. (D) Starved MEFs infected with adenoviruses encoding LacZ or CD2-FAK (50 and 100 MOI) were incubated on hydrogels with 10% FBS, and cyclin D1 mRNA was measured by QPCR. CD2-FAK percent infection was 70-90% as determined from its GFP IRES. (E) Asynchronous MEFs were reseeded on hydrogels. Cells were fixed 24 h after reseeding and immunostained for FAK and talin. FAK and talin colocalization was determined as described in Experimental Procedures and the legend to Suppl. Fig. 9. Results are plotted as mean colocalization ± s.d, p<10-6.
However, we saw variable degrees of inhibition of Y397 phosphorylation (∼30-80%) when serum-stimulated cells were plated on low stiffness hydrogels despite consistent inhibitory effects on cyclin D1 mRNA. Additionally, an activated FAK allele, CD2-FAK [23], failed to rescue cyclin D1 gene expression (Fig. 3D) in MEFs plated on soft FN-coated hydrogels despite efficient ectopic expression (see Legend to Fig. 3D) and localization to focal adhesions on the high stiffness substratum (Suppl. Fig. 8). Thus, constitutive FAK activity is not sufficient to overcome the inhibitory effect of the low stiffness matrix on S phase entry. We therefore considered the possibility that matrix compliance was regulating the association of FAK with integrins. Indeed, confocal immunofluorescence microscopy showed that endogenous FAK and activated integrins (assessed by talin staining [24]) efficiently co-localized in MEFs cultured on the high stiffness ECM but not on the low stiffness ECM (Fig. 3E and Suppl. Fig. 9A and B). These results extend work of others showing that matrix stiffness controls formation of β1-integrin/FAK/talin complexes as determined by co-immunoprecipitation in ectopic expression paradigms [20]. They also distinguish the integrin complexes that form in response to low matrix stiffness from standard focal complexes, focal adhesions, and 3D adhesions (which are reported to have constitutive recruitment of FAK to integrins) and reveal a potential similarity to fibrillar adhesions [25]. A compliance-dependent regulation of FAK localization can explain why expression of CD2-FAK does not rescue cyclin D1 gene induction on a low stiffness substratum while mislocalization of endogenous FAK with FRNK or FAKY397F inhibits cyclin D1 gene expression on a stiff substratum. Collectively, these results indicate that cells respond to changes in extracellular stiffness by regulating the degree to which FAK is stably associated with activated integrins. In this model, compliance-dependent changes in FAK autophosphorylation are a secondary consequence of altered FAK-integrin association.
Mechanical signaling to the cell cycle transduced by the FAK-Rac-cyclin D1 pathway
Studies on glass or plastic substrata have indicated that FAK can regulate at least four effectors of cyclin D1 gene expression: ERK activity [26], Rho activity [27], expression of the transcription factor KLF8 [22], and Rac activity [28, 29]. We asked which, if any, of these effector pathways was responsible for the induction of cyclin D1 by matrix stiffness. We could discount ERK as the critical FAK effector because ERK activity was not strongly inhibited by functionally relevant changes in matrix stiffness (Fig. 2) or FRNK (Suppl. Fig. 7B). Rho activity is inhibited by the low stiffness substrata ([6] and Suppl. Fig. 10A), but we reasoned that Rho is not the major FAK effector, at least in terms of ECM compliance, because Rho has been linked to cyclin D1 gene expression through ERK and because expression of activated Rho did not rescue cell spreading (Suppl. Fig. 10B) or cyclin D1 gene expression in serum-stimulated MEFs cultured on a soft substratum in either the absence or presence of activated FAK (Suppl. Fig. 10C). KLF8 gene expression was barely detectable by quantitative real-time RT-PCR (QPCR) in MEFs (∼100-fold less than transcription factors Fra-1 or JunB; Suppl. Table 1) as expected from work by others demonstrating that FAK overexpression is required for KLF8 induction [22].
We recently reported that the mid-G1 phase induction of cyclin D1 mRNA requires Rac activity [30], and indeed we found that Rac inhibition blocked mitogen-stimulated cyclin D1 mRNA induction when cells were plated on the high stiffness substratum (Fig. 4A). Moreover, Rac GTP-loading in MEFs (Fig. 4B and C) and VSMCs (Suppl. Fig. 11) was strongly inhibited by the low stiffness substratum whereas expression of an activated Rac (RacV12) rescued cyclin D1 gene expression on the soft substratum (Fig. 4D). The effect of matrix stiffness on Rac-dependent cyclin D1 gene expression requires FAK because FRNK or FAKY397F expression blocked Rac activity on the high stiffness matrix (Fig. 4B and C). Collectively, these data causally link FAK-dependent Rac activation to the effect of ECM compliance on cyclin D1 expression. However, activated Rac did not rescue S phase entry in MEFs on the soft substratum (Fig. 4E), indicating that there must be an additional effect of matrix elasticity on mitogenesis.
Figure 4. Matrix stiffness regulates the FAK-Rac-cyclin D1 signaling pathway.

(A-D) Serum-starved MEFs were infected with adenoviruses encoding GFP (control), RacN17, RacV12, FRNK, or FAKY397F. Cells were reseeded on high (H) and low (L) stiffness hydrogels and stimulated with 10% FBS for 9 h to measure induction of cyclin D1 mRNA by QPCR (panels A and D) or 30 min to measure Rac GTP-loading by G-LISA (panels B and C). The data in panel D show the mean and SEM of 3 experiments. Statistical significance was determined by t-test (* p=0.029, ** p=0.015, # not significant p=0.19). (E) The experiment in panel D was repeated except that the cells were incubated with BrdU and fixed at 24 h for the determination of S phase entry.
Matrix stiffness regulates cyclin D1-dependent Rb phosphorylation
The mid/late-G1 phase phosphorylation of Rb results in the release of associated E2Fs and transcription of E2F-dependent genes required for S phase entry. Rb phosphorylation, as determined by gel-shift, and induction of cyclin A were inhibited by low matrix stiffness in MEFs (Fig. 5A). Ectopically expressed cyclin D1 rescued Rb phosphorylation, but efficient rescue required high levels of overexpression (Fig. 5B; >100 MOI). More moderate overexpression (30 MOI) poorly rescued Rb phosphorylation despite readily detected cdk4 (Fig. 5B). Similarly, cyclin D1 overexpression poorly rescued S phase entry (determined as Ki67-expressing nuclei) in serum-stimulated MEFs on the low stiffness ECM even though inactivation of Rb with HPV-E7 rescued mitogenesis efficiently (Fig. 5C). Similar results were seen with MCF10A cells (Suppl Fig. 12). Inefficient Rb phosphorylation could result from upregulated expression of inhibitory proteins for cdk4/6 (the INK4s) or cdk2 (the cip/kips) since these cdks jointly inactivate Rb. However, none of the INK4s (p15, p16, p18, and p19; Fig. 5D) nor the widely expressed cip/kips (p21 and p27; refer to Fig. 1E) were regulated by the range of matrix stiffness that controls mitogenesis. Collectively, these data indicate that ECM compliance regulates cyclin D1-dependent Rb phosphorylation most likely through an effect on cdk4/6 binding and/or activity. The integrin-signaling mechanism underlying the effect of matrix stiffness on cyclin D1-dependent Rb phosphorylation requires FAK because ectopically expressed FRNK inhibited S phase entry in serum-stimulated, cyclin D1-overexpressing MEFs cultured on high stiffness hydrogels (Fig. 5E). Conversely, CD2-FAK synergized with ectopically expressed cyclin D1 to promote S phase entry in cells on the low stiffness substratum (Fig. 5F), reaching about half of the level of BrdU incorporation seen on the high stiffness ECM.
Figure 5. Matrix stiffness regulates cyclin D1 function downstream of its expression.
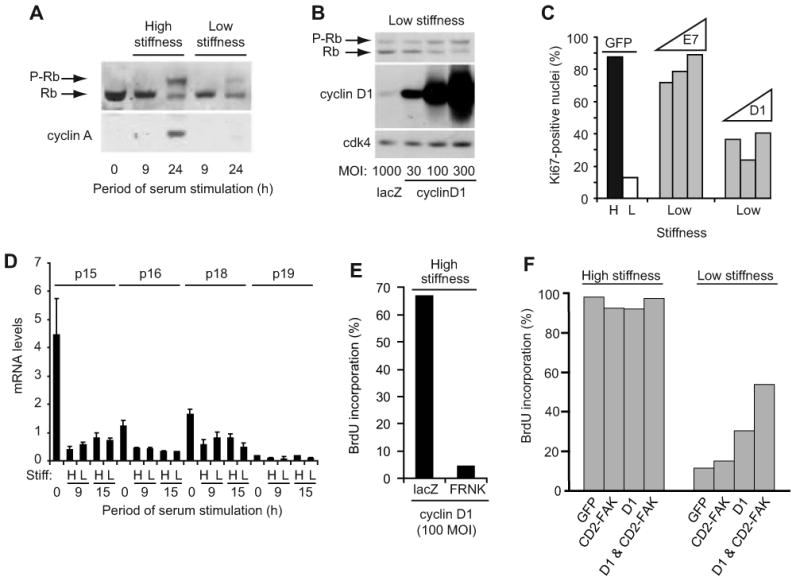
(A) Starved MEFs were reseeded on hydrogels with 10% FBS. Cell lysates were collected and analyzed by western blotting. (B) Starved MEFs infected with adenoviruses encoding LacZ or cyclin D1 (30-300 MOI) were reseeded on hydrogels with 10% FBS. Cells were collected at 24 h and analyzed by western blotting. (C) Starved MEFs infected with adenoviruses encoding GFP (1000 MOI), cyclin D1 (100, 300, and 1000 MOI), or HPV-E7 (100, 300, and 1000 MOI) were reseeded on high (H) and low (L) stiffness hydrogels and incubated with 10% FBS. S phase entry was measured 24 h after plating by immunostaining for Ki-67. (D) Starved MEFs were reseeded on hydrogels with 10% FBS. INK4 mRNA levels were measured by QPCR. (E) Starved MEFs infected with adenoviruses encoding cyclin D1 (100 MOI) and either LacZ or GFP-FRNK were reseeded in 10% FBS with BrdU. Cells were fixed at 24 h for analysis of BrdU incorporation. (F) MEFs infected with adenoviruses encoding GFP, CD2-FAK, cyclin D1 (100 MOI), or cyclin D1 & CD2-FAK were starved and plated on the high and low stiffness substrata and stimulated with 10% FBS for 24 h; S phase entry was determined by BrdU incorporation.
Tissue stiffening at sites of cell proliferation in vivo
To test the link between tissue stiffness and cell proliferation in vivo, we first isolated mammary glands, thoracic aortae, and femoral arteries from the mouse and compared their elasticities to the range of matrix stiffness that regulates mitogenesis as determined by the hydrogel analyses. Milliprobe indentation or atomic force microscopy indicated that these tissues have an elastic modulus range of 600-4300 Pa (Suppl. Table 2). This stiffness range strongly inhibits mitogenesis (refer to Fig. 1A), indicating that physiological compliance of the mammary gland and major arteries acts as a cell cycle inhibitor.
We then used fine-wire femoral artery injury in the mouse to examine potential changes in tissue stiffness occurring at sites of cell proliferation in vivo. The injury procedure disrupts the integrity of the intimal endothelium, allowing for platelet aggregation and the de-differentiation of medial VSMCs to a proliferative state. Although an intact endothelium is eventually restored, the transient denudation results in the formation of a “neointima” containing proliferating VSMCs. Immunohistochemistry confirmed VSMC proliferation, as assessed by BrdU incorporation, in the neointima of injured arteries (Fig. 6A). These regions of cell proliferation could be localized by monitoring smooth muscle-marker SMα-actin (SMA) expression, which is lost in both the neointima and underlying media as VSMCs de-differentiate to the proliferative state (Fig. 6A; box 3). Note, however, that the SMA staining pattern of the artery was mosaic, and positive staining regions were also detected. These SMA-positive regions likely reflect uninjured areas of the artery (refer to box 1) and sites of injury where VSMCs have dedifferentiated, proliferated, and then re-differentiated (boxes 2 and 4).
Figure 6. In vivo arterial stiffening at sites of cell proliferation.

Male, 6-mo C57BL/6 mice were subjected to fine-wire femoral artery injury and given BrdU. (A) The external and internal elastic laminae (EEL and IEL, respectively) were visualized by elastin staining, and overall arterial morphology was determined by staining with hematoxylin-eosin. These procedures allowed for the identification of the adventitia (AD), the medial layer of VSMCS (M) and the site of injury (neointima; NI). Adjacent sections were analyzed by immunohistochemistry with anti-BrdU (Roche, 1299964) to visualize proliferating cells (left) and anti-α-smooth muscle actin (clone 1A4, Sigma) to visualize regions of differentiated and de-differentiated VSMCs (right). Clockwise and counterclockwise arrows in the right panel mark the IEL and EEL, respectively. Boxed regions highlight distinct patterns of VSMC differentiation in the media and neointima. (B) Male, 6-mo SMA-GFP mice were subjected to fine-wire femoral artery injury. Uninjured and contralateral injured arteries were isolated, carefully opened, and imaged for GFP-fluorescence. Representative areas of the uninjured and injured arteries are shown for a single mouse. Scale bar = 200 μm. (C) Uninjured (control) and injured femoral arteries from SMA-GFP mice were collected 2 weeks after the fine-wire injury procedure. AFM was used to measure the elastic modulus of several GFP positive regions of uninjured arteries and GFP-negative regions of injured arteries. The figure compiles AFM measurements obtained from 4 mice. p=0.0029 (one-way t-test with Welch correction for unequal variance; p for unequal variance <0.0001).
We rendered the vascular injury model compatible with the analysis of stiffness in fresh tissue by performing the femoral artery injuries on transgenic mice in which the SMA promoter drives GFP. Vascular injury in this line resulted in discrete regions of GFP positive and negative fluorescence (Fig. 6B) whereas GFP fluorescence in the uninjured control artery was nearly uniform (Fig. 6B). We used AFM in force mode to probe the compliance of the GFP-negative regions (neointimas) and compared the results to those obtained for the uninjured contralateral artery of the same SMA-GFP mouse. We excluded GFP positive regions of injured arteries from the analysis to eliminate the confounding effects of VSMC re-differentiation (see above). The results revealed regions of clear arterial stiffening 2 weeks after injury (the time at which BrdU incorporation is detected in vivo), with an increase in both the mean stiffness and variance at sites of VSMC proliferation (Fig. 6C). Remarkably, the degree of stiffening seen at sites of injury and VSMC proliferation in the mouse reached the range of elastic moduli that support mitogenesis in the hydrogel studies (Fig. 1A).
Discussion
Matrix stiffness and the cell cycle
Our study describes cellular and molecular effects of matrix compliance on the cell cycle. The elastic moduli that regulate mitogenesis in MEFs, mammary epithelial cells, vascular SMCs, and osteoblastic cells are strikingly similar, indicating that the effect of tissue compliance on the cell cycle is widely conserved and independent of a cell's particular microenvironment. The mitogen-dependent induction of cyclin D1 mRNA is strongly affected by the range of matrix elasticity that controls mitogenesis whereas other mitogenic events previously identified as being integrin-dependent (the expression of cdk inhibitory proteins and immediate-early genes, as well as the activation of ERK) are relatively resistant. Thus, adhesion-mediated cell cycle events in G1 phase are not binary but have distinct compliance thresholds, with many of the previously identified events proceeding normally in the range of matrix stiffness that strongly inhibits mitogenesis.
In addition to regulating the expression of cyclin D1, matrix stiffness controls the ability of ectopically expressed cyclin D1 to stimulate Rb phosphorylation and S phase entry. This effect is of particular interest in mammary cells given the frequent overexpression of cyclin D1 in breast cancer [31, 32]. A common idea is that overexpressed cyclin D1 acts cooperatively with other oncogenic events, and our results indicate that tissue stiffening, a hallmark of mammary tumors, can be considered as one of these complementing oncogenic events since it is required for the efficient function of overexpressed cyclin D1. Additionally, our data indicate that oncogenic events inactivating Rb, such as expression of HPV-E7, can override the inhibitory effect of physiological ECM compliance on the cell cycle without increasing ECM stiffness because they deregulate the molecular events targeted by tissue compliance.
FAK localization as a mechanosensor for the cell cycle
Paszek et al. [6] reported that FAK phosphorylation at Y397, but not its localization to integrins, is regulated by matrix stiffness in mammary epithelial cells and fibroblasts. While we also see changes in FAK autophosphorylation in response to changing extracellular stiffness and intracellular tension, our data and those of Wei et al [20] support a model in which FAK localization, rather than its autophosphorylation at Y397, represents the primary compliance effect. Wozniak et al. [33] also described a compliance-dependent localization of FAK in mammary cells undergoing tubulogenesis.
Although CD2-FAK is unable to rescue cyclin D1 expression in cells plated on a low stiffness substrate, CD2-FAK is able to synergize with ectopically expressed cyclin D1 to rescue a large part of S phase entry. These results suggest that the inhibitory effects of low matrix stiffness on cyclin D1 mRNA and cyclin D1-dependent mitogenesis are mechanistically distinct downstream of FAK. We envision that differential localization of the relevant FAK substrates will ultimately account for these differential effects of CD2-FAK on compliance-dependent cell cycling. The focal adhesion proteins paxillin and p130Cas have the potential to regulate the FAK-dependent activation of Rac through CrkII-DOCK180/Elmo or Pkl-Pix-Pak, and both proteins have been implicated in mechanosensing [34, 35]. It will be interesting to determine whether FAK and these focal adhesion components have common or distinct compliance thresholds for localization to activated integrins. Since activated Rac does not rescue S phase entry in cells plated on the low stiffness substratum, the FAK effectors pathway(s) regulating cyclin D1-dependent Rb phosphorylation extends beyond Rac.
Tissue stiffness and cell cycling in vivo
Direct measurement of arterial elasticity at sites of proliferation in the mouse demonstrates that tissue stiffness increases at sites of VSMC proliferation in vivo and that regions of the remodeled matrix attain elastic moduli compatible with cell cycling. Although the relationship between arterial compliance and VSMC proliferation in vivo is complicated by pulsatile blood flow, the plasticity of VSMC differentiation, and the effects of anti-proliferative signals derived from adjacent intimal endothelial cells [36], our studies indicate that injury-associated change in the elasticity of the VSMC microenvironment is a critical regulator of proliferation in cardiovascular disease. While we studied tissue stiffness at sites of vascular injury, a comparable elastic modulus has been reported for atherosclerotic lesions [37]. Similarly, the reported elastic moduli of mammary tumors [38] should be sufficient to support cyclin D1 gene induction and mitogenesis according to our hydrogel analysis. Thus, control of the cell cycle by tissue compliance may contribute widely to the absence of cell proliferation in normal tissues as well as to the increased proliferation of cells seen during pathological ECM remodeling and stiffening of the microenvironment.
Experimental Procedures
Serum-starved cells were trypsinized and replated on matrix protein-coated acrylamide hydrogels [39] and stimulated with mitogens. Cells were lysed and analyzed for the expression of mRNA and protein by QPCR and western blotting, respectively. Rac and Rho GTPase activity were determined by G-LISA (Cytoskeleton). Epifluorescence microscopy was used to measure BrdU incorporation, Ki67, and to visualize f-actin [39]. Focal adhesion formation was determined by confocal immunofluorescence microscopy, and FAK-talin colocalization was quantified by morphometric analysis of confocal images. The elastic moduli of mouse mammary glands, aortae, and femoral arteries were determined using a custom-made microindentor or by AFM in force mode. Some experiments used cells infected with adenoviruses encoding LacZ, GFP, cyclin D1, HPV-E7, dominant negative and activated alleles of FAK and Rac. See supporting materials for detailed Experimental Procedures.
Supplementary Material
Acknowledgments
We thank Sanai Sato for the SMA-GFP mouse, James Hayden at the Wistar Institute for his help with confocal microscopy, and Keith Burridge, Tom Parsons, Craig Henke, Jeffrey Albrecht, Meenhard Herlyn, Kurt Hankenson, and Chris Chen for reagents. We thank Janice Walker for data on FAK KO MEFs. Supported by NIH grants CA72639 and HL083367 to R.K.A. and NSF MRSEC Grant #05-20020 to P.A.J.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Assoian RK, Schwartz MA. Coordinate signaling by integrins and receptor tyrosine kinases in the regulation of G1 phase cell-cycle progression. Curr Opin Genet Dev. 2001;11:48–53. doi: 10.1016/s0959-437x(00)00155-6. [DOI] [PubMed] [Google Scholar]
- 2.Fringer J, Grinnell F. Fibroblast quiescence in floating or released collagen matrices: contribution of the ERK signaling pathway and actin cytoskeletal organization. J Biol Chem. 2001;276:31047–31052. doi: 10.1074/jbc.M101898200. [DOI] [PubMed] [Google Scholar]
- 3.Rosenfeldt H, Grinnell F. Fibroblast quiescence and the disruption of ERK signaling in mechanically unloaded collagen matrices. J Biol Chem. 2000;275:3088–3092. doi: 10.1074/jbc.275.5.3088. [DOI] [PubMed] [Google Scholar]
- 4.Koyama H, Raines EW, Bornfeldt KE, Roberts JM, Ross R. Fibrillar collagen inhibits arterial smooth muscle proliferation through regulation of Cdk2 inhibitors. Cell. 1996;87:1069–1078. doi: 10.1016/s0092-8674(00)81801-2. [DOI] [PubMed] [Google Scholar]
- 5.Wall SJ, Zhong ZD, DeClerck YA. The cyclin-dependent kinase inhibitors p15INK4B and p21CIP1 are critical regulators of fibrillar collagen-induced tumor cell cycle arrest. J Biol Chem. 2007;282:24471–24476. doi: 10.1074/jbc.M702697200. [DOI] [PubMed] [Google Scholar]
- 6.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 7.Bao G, Suresh S. Cell and molecular mechanics of biological materials. Nat Mater. 2003;2:715–725. doi: 10.1038/nmat1001. [DOI] [PubMed] [Google Scholar]
- 8.Willits RK, Skornia SL. Effect of collagen gel stiffness on neurite extension. Journal of Biomaterials Science. (Polymer) 2004;15:1521–1531. doi: 10.1163/1568562042459698. [DOI] [PubMed] [Google Scholar]
- 9.Peyton SR, Raub CB, Keschrumrus VP, Putnam AJ. The use of poly(ethylene glycol) hydrogels to investigate the impact of ECM chemistry and mechanics on smooth muscle cells. Biomaterials. 2006;27:4881–4893. doi: 10.1016/j.biomaterials.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 10.Yeung T, Georges PC, Flanagan LA, Marg B, Ortiz M, Funaki M, Zahir N, Ming W, Weaver V, Janmey PA. Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil Cytoskeleton. 2005;60:24–34. doi: 10.1002/cm.20041. [DOI] [PubMed] [Google Scholar]
- 11.Khatiwala CB, Peyton SR, Putnam AJ. Intrinsic mechanical properties of the extracellular matrix affect the behavior of pre-osteoblastic MC3T3-E1 cells. Am J Physiol Cell Physiol. 2006;290:C1640–1650. doi: 10.1152/ajpcell.00455.2005. [DOI] [PubMed] [Google Scholar]
- 12.Zajac AL, Discher DE. Cell differentiation through tissue elasticity-coupled, myosin-driven remodeling. Curr Opin Cell Biol. 2008;20:609–615. doi: 10.1016/j.ceb.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pelham RJ, Jr, Wang Y. Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc Natl Acad Sci U S A. 1997;94:13661–13665. doi: 10.1073/pnas.94.25.13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Solon J, Levental I, Sengupta K, Georges PC, Janmey PA. Fibroblast adaptation and stiffness matching to soft elastic substrates. Biophys J. 2007;93:4453–4461. doi: 10.1529/biophysj.106.101386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen CS, Mrksich M, Huang S, Whitesides GM, Ingber DE. Geometric control of cell life and death. Science. 1997;276:1425–1428. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- 16.Huang S, Chen CS, Ingber DE. Control of cyclin D1, p27(Kip1), and cell cycle progression in human capillary endothelial cells by cell shape and cytoskeletal tension. Mol Biol Cell. 1998;9:3179–3193. doi: 10.1091/mbc.9.11.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olson MF, Paterson HF, Marshall CJ. Signals from Ras and Rho GTPases interact to regulate expression of p21Waf1/Cip1. Nature. 1998;394:295–299. doi: 10.1038/28425. [DOI] [PubMed] [Google Scholar]
- 18.Balmanno K, Cook SJ. Sustained MAP kinase activation is required for the expression of cyclin D1, p21Cip1 and a subset of AP-1 proteins in CCL39 cells. Oncogene. 1999;18:3085–3097. doi: 10.1038/sj.onc.1202647. [DOI] [PubMed] [Google Scholar]
- 19.Cook SJ, Aziz N, McMahon M. The repertoire of fos and jun proteins expressed during the G1 phase of the cell cycle is determined by the duration of mitogen-activated protein kinase activation. Mol Cell Biol. 1999;19:330–341. doi: 10.1128/mcb.19.1.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wei WC, Lin HH, Shen MR, Tang MJ. Mechanosensing machinery for cells under low substratum rigidity. Am J Physiol Cell Physiol. 2008;295:C1579–1589. doi: 10.1152/ajpcell.00223.2008. [DOI] [PubMed] [Google Scholar]
- 21.Wozniak MA, Modzelewska K, Kwong L, Keely PJ. Focal adhesion regulation of cell behavior. Biochim Biophys Acta. 2004;1692:103–119. doi: 10.1016/j.bbamcr.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 22.Zhao J, Bian ZC, Yee K, Chen BP, Chien S, Guan JL. Identification of transcription factor KLF8 as a downstream target of focal adhesion kinase in its regulation of cyclin D1 and cell cycle progression. Mol Cell. 2003;11:1503–1515. doi: 10.1016/s1097-2765(03)00179-5. [DOI] [PubMed] [Google Scholar]
- 23.Frisch SM, Vuori K, Ruoslahti E, Chan-Hui PY. Control of adhesion-dependent cell survival by focal adhesion kinase. J Cell Biol. 1996;134:793–799. doi: 10.1083/jcb.134.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tadokoro S, Shattil SJ, Eto K, Tai V, Liddington RC, de Pereda JM, Ginsberg MH, Calderwood DA. Talin binding to integrin beta tails: a final common step in integrin activation. Science. 2003;302:103–106. doi: 10.1126/science.1086652. [DOI] [PubMed] [Google Scholar]
- 25.Cukierman E, Pankov R, Yamada KM. Cell interactions with three-dimensional matrices. Curr Opin Cell Biol. 2002;14:633–639. doi: 10.1016/s0955-0674(02)00364-2. [DOI] [PubMed] [Google Scholar]
- 26.Schwartz MA, Assoian RK. Integrins and cell proliferation: regulation of cyclin-dependent kinases via cytoplasmic signaling pathways. J Cell Sci. 2001;114:2553–2560. doi: 10.1242/jcs.114.14.2553. [DOI] [PubMed] [Google Scholar]
- 27.Zhai J, Lin H, Nie Z, Wu J, Canete-Soler R, Schlaepfer WW, Schlaepfer DD. Direct interaction of focal adhesion kinase with p190RhoGEF. J Biol Chem. 2003;278:24865–24873. doi: 10.1074/jbc.M302381200. [DOI] [PubMed] [Google Scholar]
- 28.Chang F, Lemmon CA, Park D, Romer LH. FAK potentiates Rac1 activation and localization to matrix adhesion sites: a role for betaPIX. Mol Biol Cell. 2007;18:253–264. doi: 10.1091/mbc.E06-03-0207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sundberg LJ, Galante LM, Bill HM, Mack CP, Taylor JM. An endogenous inhibitor of focal adhesion kinase blocks Rac1/JNK but not Ras/ERK-dependent signaling in vascular smooth muscle cells. J Biol Chem. 2003;278:29783–29791. doi: 10.1074/jbc.M303771200. [DOI] [PubMed] [Google Scholar]
- 30.Klein EA, Campbell LE, Kothapalli D, Fournier AK, Assoian RK. Joint requirement for Rac and ERK activities underlies the mid-G1 phase induction of cyclin D1 and S phase entry in both epithelial and mesenchymal cells. J Biol Chem. 2008;283:30911–30918. doi: 10.1074/jbc.M804537200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bartkova J, Lukas J, Muller H, Lutzhoft D, Strauss M, Bartek J. Cyclin D1 protein expression and function in human breast cancer. Int J Cancer. 1994;57:353–361. doi: 10.1002/ijc.2910570311. [DOI] [PubMed] [Google Scholar]
- 32.Buckley MF, Sweeney KJ, Hamilton JA, Sini RL, Manning DL, Nicholson RI, deFazio A, Watts CK, Musgrove EA, Sutherland RL. Expression and amplification of cyclin genes in human breast cancer. Oncogene. 1993;8:2127–2133. [PubMed] [Google Scholar]
- 33.Wozniak MA, Desai R, Solski PA, Der CJ, Keely PJ. ROCK-generated contractility regulates breast epithelial cell differentiation in response to the physical properties of a three-dimensional collagen matrix. J Cell Biol. 2003;163:583–595. doi: 10.1083/jcb.200305010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sawada Y, Tamada M, Dubin-Thaler BJ, Cherniavskaya O, Sakai R, Tanaka S, Sheetz MP. Force Sensing by Mechanical Extension of the Src Family Kinase Substrate p130Cas. Cell. 2006;127:1015–1026. doi: 10.1016/j.cell.2006.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zaidel-Bar R, Kam Z, Geiger B. Polarized downregulation of the paxillin-p130CAS-Rac1 pathway induced by shear flow. J Cell Sci. 2005;118:3997–4007. doi: 10.1242/jcs.02523. [DOI] [PubMed] [Google Scholar]
- 36.Rzucidlo EM, Martin KA, Powell RJ. Regulation of vascular smooth muscle cell differentiation. J Vasc Surg. 2007;45 A:A25–32. doi: 10.1016/j.jvs.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 37.Lee RT, Richardson SG, Loree HM, Grodzinsky AJ, Gharib SA, Schoen FJ, Pandian N. Prediction of mechanical properties of human atherosclerotic tissue by high-frequency intravascular ultrasound imaging. An in vitro study. Arterioscler Thromb. 1992;12:1–5. doi: 10.1161/01.atv.12.1.1. [DOI] [PubMed] [Google Scholar]
- 38.Samani A, Zubovits J, Plewes D. Elastic moduli of normal and pathological human breast tissues: an inversion-technique-based investigation of 169 samples. Phys Med Biol. 2007;52:1565–1576. doi: 10.1088/0031-9155/52/6/002. [DOI] [PubMed] [Google Scholar]
- 39.Klein EA, Yung Y, Castagnino P, Kothapalli D, Assoian RK. Cell adhesion, cellular tension, and cell cycle control. Methods Enzymol. 2007;426:155–175. doi: 10.1016/S0076-6879(07)26008-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.