

APS kinase from Thiobacillus denitrificans contains an inactive N-terminal ATP sulfurylase domain. The structure presented unveils the first hexameric assembly for an APS kinase, and reveals that structural changes in the N-terminal domain disrupt the ATP sulfurylase active site thus prohibiting activity.
Keywords: APS kinases, adenylylsulfate kinases, ATP sulfurylases, sulfate metabolism, nucleotide kinase, Thiobacillus denitrificans
Abstract
The Tbd_0210 gene of the chemolithotrophic bacterium Thiobacillus denitrificans is annotated to encode a 60.5 kDa bifunctional enzyme with ATP sulfurylase and APS kinase activity. This putative bifunctional enzyme was cloned, expressed and structurally characterized. The 2.95 Å resolution X-ray crystal structure reported here revealed a hexameric assembly with D 3 symmetry. Each subunit contains a large N-terminal sulfurylase-like domain and a C-terminal APS kinase domain reminiscent of the two-domain fungal ATP sulfurylases of Penicillium chrysogenum and Saccharomyces cerevisiae, which also exhibit a hexameric assembly. However, the T. denitrificans enzyme exhibits numerous structural and sequence differences in the N-terminal domain that render it inactive with respect to ATP sulfurylase activity. Surprisingly, the C-terminal domain does indeed display APS kinase activity, indicating that this gene product is a true APS kinase. Therefore, these results provide the first structural insights into a unique hexameric APS kinase that contains a nonfunctional ATP sulfurylase-like domain of unknown function.
1. Introduction
Inorganic sulfate is converted to its biologically active form, 3′-adenosine 5′-phosphosulfate (PAPS), in a two-step enzymatic process. In the first step, ATP sulfurylase (MgATP:sulfate adenylyltransferase; EC 2.7.7.4) adenylates sulfate, forming adenosine 5′-phosphosulfate (APS), which is then phosphorylated on the 3′-hydroxyl by APS kinase (MgATP:APS 3′-phosphotransferase; EC 2.7.1.25) to produce PAPS,
PAPS serves as the sulfuryl donor for the formation of sulfate esters by sulfotransferases (Kusche et al., 1991 ▶; Suiko et al., 1992 ▶; Varin et al., 1992 ▶). Sulfation of biological molecules by sulfotransferases controls numerous cellular processes including activation/deactivation of xenobiotics, inactivation of hormones and functional regulation of many macromolecules (Klaassen & Boles, 1997 ▶). In fungi, yeast and most bacteria, PAPS is the substrate for an NADPH-dependent reduction that yields sulfite through the utilization of thioredoxin. Sulfite is then further reduced to form sulfide and condensed with O-acetylserine to produce the amino acid cysteine. In other organisms, such as higher plants, algae and some bacteria, APS is the starting substrate for the assimilatory sulfate-reduction pathway. Organisms that utilize APS in this process still contain an APS kinase gene, probably to produce a reserve of PAPS from which more APS can be formed; alternatively, PAPS may serve as a sulfate donor for sulfotransferases in sulfate-ester formation. Both APS and PAPS are considered to be ‘activated’ forms of sulfate and act as the precursors of all sulfur-containing biological molecules under aerobic conditions.
Chemolithotrophic bacteria are able to use inorganic reduced sulfur compounds as energy/electron sources. Following an AMP-dependent oxidation, ATP sulfurylase catalyzes the terminal step in the overall sulfur-oxidation process, producing ATP and sulfate. That is, chemolithotrophic ATP sulfurylases work in the reverse direction compared with sulfate assimilators. Analysis of the genomes of these organisms reveals the presence of APS kinase genes. While they do not require the enzyme for sulfur assimilation, it is hypothesized that the product, PAPS, is still required for the formation of sulfate esters.
In Penicillium chrysogenum (Lansdon et al., 2002 ▶; MacRae et al., 2000 ▶; Renosto et al., 1985 ▶), Saccharomyces cerevisiae (Schriek & Schwenn, 1986 ▶; Ullrich et al., 2001 ▶; Ullrich & Huber, 2001 ▶), Arabidopsis thaliana (Lillig et al., 2001 ▶) and Escherichia coli (Satishchandran et al., 1992 ▶; Satishchandran & Markham, 1989 ▶; Schriek & Schwenn, 1986 ▶), APS kinase is found as a single-domain peptide that displays solely APS kinase activity. However, APS kinase can also be found as part of a bifunctional enzyme that also contains ATP sulfurylase activity as in Aquifex aeolicus (Hanna et al., 2002 ▶; Yu et al., 2007 ▶), rats (Hommes et al., 1987 ▶; Lyle et al., 1994 ▶; Yu et al., 1989 ▶) and humans (Harjes et al., 2005 ▶; Lansdon et al., 2004 ▶). Regardless of its source, all structural data gathered to date reveal the active form of APS kinase to be a homodimer.
The ATP sulfurylases of some organisms possess a C-terminal domain that displays sequence and structural homology to APS kinase (MacRae et al., 2001 ▶, 2002 ▶; Ullrich et al., 2001 ▶; Ullrich & Huber, 2001 ▶). In the hexameric ATP sulfurylase from P. chrysogenum, this domain acts as an allosteric regulatory domain that binds PAPS (MacRae & Segel, 1997 ▶; Renosto et al., 1989 ▶). In the hexameric ATP sulfurylase from S. cerevisiae, this domain still retains structural homology to the APS kinase core, but does not contain many of the loops required to bind the allosteric effector PAPS (MacRae & Segel, 1997 ▶).
A genomic analysis of Thiobacillus denitrificans (Beller et al., 2006 ▶) revealed a gene (Tbd_0210) containing a single coding sequence with an N-terminal ATP sulfurylase domain and a C-terminal APS kinase domain and led to the gene being annotated as an ATP sulfurylase/APS kinase bifunctional enzyme. However, enzymatic kinetic studies have shown this enzyme to serve solely as an APS kinase and to have no detectable ATP sulfurylase activity (Gay et al., manuscript in preparation). Its large inactive N-terminal domain, which displays high sequence similarity to other ATP sulfurylases, raised questions about its function. [The organism does possess a second single-domain ATP sulfurylase gene (Tbd_0874), which indeed displays ATP sulfurylase activity (Gay et al., manuscript in preparation).] In order to investigate the structural properties of this unique APS kinase from T. denitrificans and to gain insight into the function of the APS kinase domain as well as the potential function of the inactive ATP sulfurylase domain, the enzyme was crystallized and the structure was solved. The present study summarizes the results of the 2.95 Å resolution crystal structure determination of a hexameric APS kinase from T. denitrificans.
2. Experimental procedures
2.1. Cloning, expression and purification
T. denitrificans ATCC 25259 genomic DNA was a kind gift from Drs Sherry Huston and Doug Nelson (University of California, Davis). The open reading frame encoding the putative bifunctional ATP sulfurylase/APS kinase (Tbd_0210) was amplified by PCR using primers 5′-ACGTATTCATATGGTGAATCAACTGATCGAGCC-3′ and 5′-ATCATCTCGAGTCAGCGCAGATAGCCTTCGTGTTCG-3′ (Invitrogen). The 30 µl PCR mixture included two units of PfuTurbo Cx (Stratagene), 3 µl 10× Vent DNA polymerase buffer (New England Biolabs), 84 µM each of dATP, dCTP, dTTP and dGTP (Promega), approximately 1 µM of each primer and approximately 3.33 ng ml−1 template DNA and 5% DMSO. This mixture was exposed to 30 cycles of 1 min melting at 368 K, 1 min annealing at 328 K and 3 min extension at 345 K. This was followed by an additional 10 min of extension at 345 K to ensure that any partially extended products were completed. This process introduced NdeI (5′) and XhoI (3′) restriction sites. To hybridize with the stretch of thymine bases in the TOPO TA cloning vector (Invitrogen), a polyadenine tail was added to the DNA by adding Taq polymerase (Qiagen) and incubating at 345 K for 10 min. The gene was then ligated into pCR2.1 (Invitrogen) using a standard protocol. Plasmid DNA was isolated from colonies using a QIAquick Spin Miniprep Kit (Qiagen). DNA sequencing using T7 and M13 reverse primers confirmed the gene sequence (Davis Sequencing, Davis, California, USA). The gene was then subcloned as an NdeI/XhoI fragment into pET-22b(+) (Novagen). This plasmid was chemically transformed into BL21 (DE3) (Stratagene) for protein expression.
1 ml overnight culture grown at 310 K in LB medium containing 100 µg ml−1 ampicillin was added to each of six 2 l baffled flasks containing 1 l LB and 100 µg ml−1 ampicillin. Inoculated media were incubated at 310 K with shaking until OD600nm reached approximately 1. Cultures were cooled to 288 K and protein expression was induced with 2 mM α-lactose. Protein expression was allowed to continue for 12–15 h at 288 K. Cells were harvested by centrifugation at 5000g for 15 min using a Beckman JLA 8.1000 rotor in a Beckman Avanti J-20 centrifuge. Cell pellets were resuspended in 100 ml buffer A (50 mM sodium phosphate, 300 mM sodium chloride, 10 mM imidazole pH 8) at 277 K and passed through a Watts Fluidair Microfluidizer three or four times to lyse the cells. Insoluble material was removed by centrifugation at 39 000g for 20 min using a Beckman JLA 16.250 rotor in the same centrifuge as above. All subsequent steps were performed at 277 K. The soluble portion was loaded onto a Sigma His-Select column (equilibrated with buffer A). The column was washed with buffer A for approximately 15 column volumes, followed by 30 column volumes of wash buffer B (50 mM sodium phosphate, 300 mM sodium chloride, 20 mM imidazole pH 8) until A 280nm was below 0.02. The enzyme was eluted with buffer C (50 mM sodium phosphate, 300 mM sodium chloride, 250 mM imidazole pH 8) into 2 ml fractions over a total of 15 column volumes. Fractions corresponding to the major A 280nm peak were analyzed by SDS–PAGE and then pooled. The pooled fractions were dialyzed against 2 l 40 mM Tris–HCl pH 8.0 at 277 K. Purified protein was concentrated in Millipore spin concentrators to approximately 15 mg ml−1. The protein concentration was determined by absorbance at 280 nm. A molar extinction coefficient of 56 150 M −1 cm−1 was used as determined by Vector NTI Advance 10.3 (Invitrogen).
2.2. Crystallization
Concentrated T. denitrificans APS kinase was crystallized by hanging-drop vapor diffusion at room temperature by mixing a solution containing 10 mg ml−1 protein, 1 mM TCEP, 5 mM APS and 5 mM MgAMP-PNP with a crystallization solution containing 0.1 M bis-tris propane pH 7, 0.1 M sodium chloride and 1.0–1.5 M sodium/potassium tartrate in a 1:1 ratio. This 1:1 mixture drop was then equilibrated via vapor diffusion over the tartrate crystallization solution. C-centered orthorhombic crystals belonging to space group C2221 grew overnight and continued to enlarge over the course of 7–10 d. Crystals were dragged through Paratone-N oil to remove exterior aqueous buffer before freezing in liquid nitrogen. These crystals had unit-cell parameters a = 161.1, b = 227.2, c = 106.8 Å, α = β = γ = 90.0°. The Matthews coefficient (V M) was calculated to be 2.65 Å3 Da−1, corresponding to a solvent content of 53.5%, assuming the presence of three monomers per asymmetric unit. Numerous crystals with poor or no diffraction were screened before one was found that diffracted to 2.95 Å resolution.
2.3. Data collection and phase determination
A 2.95 Å resolution native data set was collected on beamline 9-2 at SSRL and processed with DENZO and SCALEPACK (Otwinowski & Minor, 1997 ▶). Table 1 ▶ lists the data-collection statistics. After several attempts using numerous search models and algorithms, a molecular-replacement solution was obtained with EPMR (Kissinger et al., 1999 ▶) using a polyalanine model of the R-state P. chrysogenum ATP sulfurylase trimer structure (half of the overall hexamer; PDB code 1i2d). The best solution resulted in a correlation coefficient of 37.2% and an R factor of 62.9%. The next highest peak had a correlation coefficient of 37.1% and an R factor of 62.8% and differed by only 0.12°. Most other peaks were closer to 33–34% and 64–66%, respectively, and differed by much larger angles.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| X-ray source | SSRL BL 9-2 |
| Wavelength () | 0.979 |
| Space group | C2221 |
| Unit-cell parameters | |
| a () | 161.1 |
| b () | 227.2 |
| c () | 106.8 |
| = = () | 90 |
| Resolution () | 35.02 (2.95) |
| No. of reflections | 116293 |
| No. of unique reflections | 38467 |
| Completeness (%) | 92.1 (95.8) |
| Redundancy | 3.0 (3.3) |
| R merge † (%) | 10.7 (35.7) |
| I/(I) | 7.32 (1.95) |
| Refinement statistics | |
| Resolution () | 35.032.95 |
| No. of reflections (F 0) | 38315 |
| R factor‡ (%) | 24.29 |
| R free ‡ (%) | 28.22 |
| R.m.s.d. bond lengths () | 0.014 |
| R.m.s.d. bond angles () | 1.867 |
| Ramachandran plot statistics | |
| Residues§ | 1259 |
| Most favorable regions | 1075 |
| Allowed regions | 184 |
| Generously allowed regions | 0 |
| Disallowed regions | 0 |
| Asymmetric unit content | |
| Non-H protein atoms | 11285 |
| Waters | 86 |
R
merge = 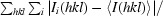
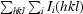 , where I(hkl) is the mean of i observations of reflection hkl.
, where I(hkl) is the mean of i observations of reflection hkl.
R factor and R
free = 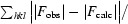
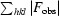 for 95% of the recorded data (R factor) or 5% of the data (R
free).
for 95% of the recorded data (R factor) or 5% of the data (R
free).
Number of nonproline and nonglycine residues used for calculation.
2.4. Model building and refinement
The solution model was first modified to reflect gaps in the T. denitrificans APS kinase sequence compared with that of P. chrysogenum ATP sulfurylase. Refinement began with a rigid-body refinement in REFMAC (Collaborative Computational Project, Number 4, 1994 ▶) using residues 5–363 (ATP sulfurylase domain) and 364–544 (APS kinase domain) as separate entities. To improve the side-chain electron density, the electron density was averaged with respect to threefold noncrystallographic symmetry using DM (Cowtan, 1994 ▶). Model building was performed using Coot (Emsley & Cowtan, 2004 ▶) followed by restrained refinement using noncrystallographic symmetry restraints in REFMAC after each round of modeling until a minimum R factor of 34.1% and an R free of 41.7% were reached. After this point a combination of model building and refinement with REFMAC (Collaborative Computational Project, Number 4, 1994 ▶) and simulated annealing in both PHENIX (Adams et al., 2002 ▶) and CNS (Brünger et al., 1998 ▶) was used, gradually releasing the noncrystallographic symmetry restraints until a final R factor of 24.3% and an R free of 28.2% were reached. The quality of the final model was checked with PROCHECK (Laskowski et al., 1993 ▶) and the results are summarized in Table 1 ▶. Protein coordinates have been deposited in the Protein Data Bank (PDB code 3cr8). All structure figures were generated using PyMOL (DeLano, 2002 ▶).
3. Results
3.1. Overall protein structure
The crystals contained one trimer per asymmetric unit, labeled chains A, B and C (Fig. 1 ▶ a). The overall structure is a hexamer that is generated through the twofold crystallographic symmetry (Fig. 2 ▶ a). The model includes residues 3–263, 286–477, 507–546 in chain A, 4–264, 284–477, 507–544 in chain B and 5–265, 286–480, 509–545 in chain C. Chain C had the largest number of residues that could be modeled of the three. There are two regions of poor to no density for all three chains. These correspond to a loop in the ATP sulfurylase-like domain and the mobile lid of the APS kinase active site. (Fig. 3 ▶ b).
Figure 1.

Ribbon diagram of the T. denitrificans APS kinase. (a) The asymmetric unit is shown looking down the noncrystallographic threefold axis. Chains A (yellow), B (teal) and C (aqua) comprise one half of the hexamer. The biologically relevant hexamer is created through crystallographic twofold symmetry. (b) The monomer is shown with its N-terminal sulfurylase-like domain colored green. The APS kinase functional domain is colored blue. The black dashed lines signify the two stretches of undefined electron density.
Figure 2.

Ribbon diagrams of the T. denitrificans APS kinase (a) and P. chrysogenum ATP sulfurylase (b) (PDB code 1i2d) hexamers. (a) Each chain of T. denitrificans APS kinase contains an inactive N-terminal domain that assembles into a hexamer that resembles fungal ATP sulfurylase and has an active APS kinase domain at the C-terminus. The top view is looking down the noncrystallographic threefold axis, with the crystallographic twofold shown as a dashed line. The bottom view is rotated 90°, looking down the crystallographic twofold axis. The overlaid black, cyan and red boxes represent the intersubunit interactions described in the text and shown in detail in Fig. 6 ▶. (b) For comparison, ATP sulfurylase from P. chrysogenum (PDB code 1i2d) is shown in similar orientations as in (a). Here, each chain contains an active ATP sulfurylase domain at its N-terminus and an inactive C-terminal APS kinase-like domain, which allosterically down-regulates the ATP sulfurylase domain upon binding PAPS.
Figure 3.


Domain and sequence alignment of sulfur-activating enzymes from different organisms. (a) General domain arrangement of the ATP sulfurylase and APS kinase enzymes/domains from various species presented/discussed here. Green and blue represent ATP sulfurylase and APS kinase domains, respectively. Domains with vertical stripes indicate no enzymatic activity. (b) Sequence alignment. The bifunctional human PAPSS1 gene has been separated into two sequences owing to differences in enzyme architecture (its APS kinase domain precedes its ATP sulfurylase domain). Also shown are APS kinase and ATP sulfurylase from P. chrysogenum, APS kinase and ATP sulfurylase from T. denitrificans, the bifunctional ATP sulfurylase/APS kinase from A. aeolicus, ATP sulfurylase from S. cerevisiae and ATP sulfurylase from the Riftia symbiont. Black boxes denote the helix–turn–helix, QxRN, HxxH, Walker A and Walker B motifs as well as the two disordered loops in the Thiobacillus APS kinase structure. Black circles mark the locations of residues that are conserved in active ATP sulfurylase sequences but differ in the N-terminal domain of APS kinase from Thiobacillus.
3.2. Subunit structure
The monomer is a 544-residue chain that is comprised of three domains: the two-domain ATP sulfurylase-like motif and an APS kinase domain (Fig. 1 ▶ b). Chains listed above that extend beyond residue 544 include the extra residues in the polyhistidine tag. The N-terminal ATP sulfurylase-like domain comprises residues 1–368. Owing to a genetic deletion, this domain lacks the helix–turn–helix motif seen in active ATP sulfurylase structures (Figs. 3 ▶ b and 4 ▶). In its place is a small hairpin loop stretching from Asp119 to Trp127 (Fig. 4 ▶). The ATP sulfurylase-like domain contains a Rossmann fold as observed in nucleotide-binding proteins (Rao & Rossmann, 1973 ▶), but lacks the residues required to bind ATP or APS (MacRae et al., 2000 ▶; Ullrich & Huber, 2001 ▶; Yu et al., 2007 ▶). The first of the disordered regions (disordered loop 1; Fig. 1 ▶ b) is found in this domain. The C-terminal APS kinase domain displays the typical α/β-fold observed in other APS kinase structures. It consists of five parallel β-strands surrounded by five α-helices. The second disordered region (disordered loop 2; Figs. 1b ▶ and 3b ▶) is found in this domain. The complete monomer displays an architecture similar to that of the previously solved bifunctional ATP sulfurylase/APS kinase from A. aeolicus (Yu et al., 2007 ▶) and ATP sulfurylase from P. chrysogenum (MacRae et al., 2001 ▶, 2002 ▶).
Figure 4.

Ribbon diagram of an overlay of T. denitrificans APS kinase (green) and R-state P. chrysogenum ATP sulfurylase (salmon). The conserved helix–turn–helix motif observed in active ATP sulfurylases (shown here in the P. chrysogenum ATP sulfurylase structure) is highlighted in red. In the inactive sulfurylase domain of T. denitrificans APS kinase, the helix–turn–helix motif is replaced with a truncated β-hairpin loop (indicated by black arrows).
3.3. Quaternary structure
The overall structure of the enzyme is a hexamer with D 3 symmetry, very similar to the P. chrysogenum ATP sulfurylase (MacRae et al., 2001 ▶, 2002 ▶). Superposition of the two noncrystallographic dimers with the one crystallographic dimer resulted in r.m.s.d.s of 0.301 and 0.406 Å (for similar Cα atoms), indicating that the three dimer pairs are very similar in arrangement and intersubunit contacts. While this enzyme adopts an overall hexameric form, all previously solved APS kinase structures have been dimers. Even the bifunctional ATP sulfurylase/APS kinase enzymes from A. aeolicus and mammals assemble into dimers; the dimerization is mediated through the APS kinase domain. However, here the T. denitrificans APS kinase enzyme contains a sulfurylase-like domain that causes trimerization, with the APS kinase domain making contacts across the twofold interface of the two triads to create three kinase dimers within the hexamer.
Each kinase dimer within the hexamer resembles other observed kinase dimer assemblies. However, the T. denitrificans kinase domain dimer adopts a more closed conformation in which the domains are rotated, bringing the two active sites closer together. This closed conformation is similar to that observed in the enzymatically inactive allosteric domain of P. chrysogenum ATP sulfurylase and the A. aeolicus kinase domain, which displays low activity compared with most kinases (Fig. 5 ▶). Conversely, the human and P. chrysogenum APS kinase monomers are in a more open orientation within the dimer (Fig. 5 ▶). Interactions between a semi-conserved hydrophobic patch consisting of Leu404, Leu413, Phe433, Val434 and Ile438 stabilize this dimer interface (black box in Fig. 2 ▶ a, detailed in Fig. 6 ▶ a).
Figure 5.

Overlay of APS kinase dimers showing the ‘open’ and ‘closed’ conformations between monomers. One subunit of the P. chrysogenum APS kinase dimer was aligned with one monomer of the T. denitrificans APS kinase dimer, revealing the two distinct ‘open ’and ‘closed’ orientations between monomers. The overlaid monomers are semitransparent and more distant from the point of view of the figure. ADP and APS are modeled from the P. chrysogenum APS kinase structure (PDB code 1m7g). P. chrysogenum APS kinase is shown in yellow, while T. denitrificans APS kinase (without the sulfurylase-like domain) is shown in green. The P. chrysogenum APS kinase resides in the more open conformation, such that the two active sites are farther apart. This open conformation is also observed in the APS kinase dimer from human PAPS synthetase (not shown for clarity). T. denitrificans APS kinase is in a more ‘closed’ conformation (shown by arrows), which is also observed in the structures of the APS kinase domain from the bifunctional A. aeolicus ATP sulfurylase/APS kinase and of the allosteric domain from P. chrysogenum ATP sulfurylase (not shown for clarity).
Figure 6.

Intersubunit contacts. (a) The hydrophobic interface between the kinase domains that mediates dimerization is shown across the crystallographic twofold, which is slightly rotated down from vertical as shown in Fig. 2 ▶(a) (represented by a black box). The ribbon backbone is semitransparent in the figure. (b) Hydrophilic interactions between the kinase domain (yellow) of one subunit and the sulfurylase domain (aqua) of another within the same triad (represented by the cyan-colored box in Fig. 2 ▶ a). (c) Hydrophilic interactions between the kinase domain (aqua) of one subunit and the sulfurylase domain (orange) of another from the opposite triad (represented by the red-colored box in Fig. 2 ▶ a). Hydrogen bonds are represented by dashed lines.
Each true kinase domain in T. denitrificans also makes contacts with two other sulfurylase-like domains to stabilize the overall hexamer. Within the same triad of the hexamer, each kinase domain makes hydrogen bonds to the nearest sulfurylase-like domain through residues Gln531 to Asn193 and Gln532 to Arg164 (cyan box in Fig. 2 ▶ a, detailed in Fig. 6 ▶ b). There is an additional contact between Gln532 and Glu191 for the kinase domain of monomer C and the sulfurylase-like domain of monomer B. Across the hexamer to the other triad, each kinase domain contacts an N-terminal domain through contacts involving the main-chain O atom of Met394 and Arg215, Glu395 and the main-chain N atom of Phe144 and the main-chain O atom of Glu395 and Arg215 (red box in Fig. 2 ▶ a, detailed in Fig. 6 ▶ c). The interactions between sulfurylase-like domains are similar to those reported for the fungal sulfurylase enzyme (MacRae et al., 2001 ▶, 2002 ▶). The main contacts are created by a helix that stretches from Val145 to Arg161. This helix packs together with the same residues across the triad.
3.4. APS kinase domain
The C-terminal domain of the protein that is encoded by T. denitrificans gene Tbd_0210 is comprised of a single APS kinase domain. The first noticeable feature in this domain is the previously mentioned stretch of disordered residues (478–506; disordered loop 2; Figs. 1 ▶ b and 3 ▶ b). This region corresponds to the active-site lid seen in other kinase structures. Just like in the P. chrysogenum APS kinase ligand-free structure and monomer A of the A. aeolicus bifunctional structure, there was no observable density for this region. Prior to substrate binding this region is mobile, but it becomes ordered upon binding MgATP (Lansdon et al., 2002 ▶; MacRae et al., 2000 ▶). A sequence alignment (Fig. 3 ▶ b) shows that all the conserved residues required for substrate binding are present in this disordered region. One of these residues, Arg485, makes a π-stacking interaction with the adenine ring of ADP in the P. chrysogenum (Lansdon et al., 2002 ▶) and A. aeolicus (Yu et al., 2007 ▶) APS kinase structures as well as in the structure of the truncated human PAPS synthetase structure (Sekulic et al., 2007 ▶). Phe502, which also resides in disordered loop 2, is known to sandwich the adenine ring of APS/PAPS along with Phe419 (which is observed in the electron density). Also part of the disordered region is Lys488, which is seen in other APS kinase structures to reside in a short loop between the two helices of this mobile active-site lid. In these other substrate-bound structures, this residue makes a hydrogen bond with the 2′-hydroxyl of the ribose ring of APS, orienting the 3′-hydroxyl to accept a phosphate from ATP (Lansdon et al., 2002 ▶; Sekulic et al., 2007 ▶). While these three residues (Arg485, Phe502 and Lys488) are disordered in the T. denitrificans structure presented here, we expect similar types of interactions between these strictly conserved residues and the required substrates/products when they bind to the kinase domain.
The APS kinase domain also contains a traditional P-loop or Walker A motif (Walker et al., 1982 ▶; Fig. 3 ▶ b). This sequence, 376GLSGAGKS383, occurs in a short loop between β-strand 12 and α-helix 11 and is known to bind the phosphate moiety of nucleotide substrates. Sitting across from the Walker A motif between β-strand 13 and α-helix 12 is an analog of a Walker B motif (Fig. 3 ▶ b). This 404LDGD407 sequence is highly conserved among known APS kinase enzymes and is in a position to provide a water-mediated hydrogen bond to the magnesium ion coordinated by ATP. This theory was suggested by MacRae et al. (2000 ▶) and confirmed in the MgADP-bound human PAPS synthetase structure (Harjes et al., 2005 ▶).
3.5. ATP sulfurylase-like domain
At first glance, it would seem that residues 1–368 form a functional ATP-sulfurylase domain, which explains the annotation of the original gene (Tbd_0210) as a bifunctional enzyme. However, a number of divergences from typical ATP sulfurylase structures and sequences explain the lack of activity of this domain in the T. denitrificans enzyme. Two important motifs required for substrate binding and catalysis have been affected by changes in the T. denitrificans sequence when compared in an alignment of active ATP sulfurylases (Fig. 3 ▶ b).
Firstly, the conserved QxRN motif, which may stabilize the transition state from ATP to APS (MacRae, 2002 ▶), has been modified to include Gln173 in place of the conserved asparagine (Figs. 3 ▶ b and 7 ▶). Despite having the same functional group, the additional methylene found in glutamine moves the amide N atom an additional 1.5 Å out of its optimal hydrogen-bonding distance to one of the nonbridging O atoms of the phosphosulfate group of APS as observed in the human PAPS synthetase (Harjes et al., 2005 ▶), S. cerevisiae ATP sulfurylase (Ullrich et al., 2001 ▶) and P. chrysogenum ATP sulfurylase structures (MacRae et al., 2001 ▶, 2002 ▶) (or ADP as in the A. aeolicus structure; Yu et al., 2007 ▶). In addition to direct changes to this motif, numerous secondary-layer residues differ in T. denitrificans (Figs. 3 ▶ b and 7 ▶). Ala207, which is typically an aspartate (Fig. 3 ▶ b), lacks the proper functional group to serve as a hydrogen-bonding partner for the conserved arginine from the QxRN motif (172 in T. denitrificans APS kinase). Studies of an Asp-to-Ala mutant of P. chrysogenum ATP sulfurylase at this position have shown the enzyme to be inactive using sulfate as a substrate but that it can use molybdate (Segel et al., unpublished data). In other ATP sulfurylase structures the aspartate hydrogen bonds to the arginine of QxRN to properly orient it for sulfate binding (MacRae et al., 2001 ▶; Ullrich & Huber, 2001 ▶). Glu206, which resides right next to Ala207, could serve as the hydrogen-bonding partner for this arginine. However, Pro208, a known α-helix N-terminal cap that is likely to stabilize the ensuing helix, would disable this glutamate from moving into the proper position for this to occur (Fig. 7 ▶). In active ATP sulfurylases, the amide of glutamine (from the QxRN motif) is oriented to donate a hydrogen bond to sulfate by a conserved arginine on a nearby helix (Beynon et al., 2001 ▶; MacRae et al., 2000 ▶, 2001 ▶; Ullrich et al., 2001 ▶; Ullrich & Huber, 2001 ▶; Yu et al., 2007 ▶); however, this residue is an alanine (Ala253) in the T. denitrificans sequence.
Figure 7.

Ribbon and stick diagram of ATP sulfurylase ‘active site’ overlay of T. denitrificans APS kinase (green) and S. cerevisiae ATP sulfurylase (blue, with the helix–turn–helix shown in red). T. denitrificans APS kinase shows poor orientation of essential ATP sulfurylase active-site residues when compared with ATP sulfurylase from S. cerevisiae. The QxRN-motif residues are in a particularly unfavorable position for ligand binding as a result of an Asn-to-Gln mutation at residue 173 and the mutation of secondary-layer residues (Arg to Ala253 and Asn to Ala207) that help to orient the active-site side chains. Pro208 is likely to stabilize the following helix, preventing it from unwinding to allow Glu206 from acting as a ‘surrogate’ Asp to orient Arg172. Additionally, a His-to-Gln mutation at residue 179 is likely to impede binding of the α- and β-phosphates of ATP. Ligands (APS and PPi) are modeled from the yeast structure (PDB code 1g8h). The helix–turn–helix motif that is missing from Thiobacillus is shown in red for perspective. Residue numbering is based on the Thiobacillus structure.
The second motif affected by sequence changes is a conserved histidine that is part of a HxxH motif which interacts with the β- and γ-phosphates of ATP in functional sulfurylases. In the Thiobacillus gene product, Gln179 replaces the second histidine (Fig. 3 ▶ b) that would normally make contacts with a nonbridging O atom of the ATP substrate (Fig. 7 ▶; Ullrich et al., 2001 ▶; Veitch & Cornell, 1996 ▶; Veitch et al., 1998 ▶). Mutational studies found that a His-to-Ala mutant at this location reduced mouse PAPS synthetase activity by nearly 500-fold (Deyrup et al., 1999 ▶).
Additional sequence and structural changes that can explain the lack of sulfurylase activity also include disordered loop 1 near the defunct active site (Fig. 1 ▶ b). While this loop (residues 266–281) is disordered in the structure reported here, the loop is visible in other crystal structures and has been shown to interact with ATP/APS through multiple side-chain and main-chain interactions (MacRae et al., 2001 ▶; Ullrich et al., 2001 ▶; Ullrich & Huber, 2001 ▶; Yu et al., 2007 ▶). Despite not having electron density to draw conclusions from, simple sequence inspection reveals differences in this area that further explain the lack of ATP sulfurylase activity. Gly265 is an arginine in all other ATP sulfurylase sequences (Fig. 3 ▶ b), which makes a π-stacking interaction with the ATP/APS adenine ring, as seen in the A. aeolicus bifunctional ATP sulfurylase/APS kinase (Yu et al., 2007 ▶) and human PAPS synthetase structures (Harjes et al., 2005 ▶). This arginine also makes a main-chain hydrogen bond to the 2′-hydroxyl group of the ribose ring of ATP or APS (MacRae et al., 2001 ▶, 2002 ▶; Ullrich et al., 2001 ▶). It was previously shown by Deyrup and coworkers that mutating this residue to either alanine or lysine in mouse PAPS synthetase reduced ATP sulfurylase activity by over 80-fold (Deyrup et al., 1999 ▶). Glu266 in the disordered loop is an aspartic acid in all other ATP sulfurylase sequences (Fig. 3 ▶ b). This residue does not appear to make any side-chain contacts with substrates in previously solved structures; however, Deyrup and coworkers also showed that mutating this Asp residue to alanine in mouse PAPS synthetase reduced the ATP sulfurylase activity by over 600-fold. The additional methylene group in glutamate compared with aspartate could create disruptions in substrate binding or activity. Gln268 is an alanine in all other ATP sulfurylase sequences (Fig. 3 ▶ b). This residue is near the sulfate-binding site in other ATP sulfurylase enzymes. The large side chain of this residue could occlude any potential divalent oxyanion substrate from binding to this domain. In active ATP sulfurylases, the loop containing these residues also makes van der Waals contacts with the small helix–turn–helix motif that is genetically deleted in the Thiobacillus protein here (Figs. 3 ▶ b and 4 ▶). The lack of van der Waals contacts helps explains why disordered loop 1 is not observed. With no contacts to hold it in place, the loop is free to move through multiple conformations in the crystal.
4. Discussion
The gene Tbd_0210 from T. denitrificans was originally annotated as a bifunctional ATP sulfurylase/APS kinase based on sequence analysis. However, this gene product only possesses APS kinase activity. This APS kinase from T. denitrificans has been crystallized and its structure has been determined. The protein assembles into a hexamer with three monomers per asymmetric unit, with a crystal twofold generating the biologically active hexamer, which is consistent with size-exclusion chromatography (unpublished data). The hexameric structure is very similar in size and shape to the ATP sulfurylases from both P. chrysogenum (Fig. 2 ▶ b; MacRae et al., 2001 ▶, 2002 ▶) and S. cerevisiae (Ullrich et al., 2001 ▶; Ullrich & Huber, 2001 ▶), both of which contain an inactive C-terminal APS kinase-like domain. Each Thiobacillus APS kinase dimer within the hexamer has a structural arrangement similar to that of the dimeric bifunctional ATP sulfurylase/APS kinase from A. aeolicus (Yu et al., 2007 ▶).
The Thiobacillus C-terminal APS kinase domain forms a dimeric contact within the hexamer that is similar to those of the dimeric APS kinases from other organisms (Lansdon et al., 2002 ▶; MacRae et al., 2000 ▶; Sekulic et al., 2007 ▶; Yu et al., 2007 ▶). Its fold and the orientation of its residues are very similar to previously solved structures. Previous hypotheses suggested that the closed dimer orientation of the A. aeolicus bifunctional enzyme (Yu et al., 2007 ▶) and the allosteric domain of P. chrysogenum ATP sulfurylase (MacRae et al., 2001 ▶, 2002 ▶) were partly responsible for the poor or complete lack of activity observed in these enzymes. However, the T. denitrificans APS kinase adopts a similar closed dimer conformation (Fig. 5 ▶), yet is able to achieve a maximum velocity that is on a par with the most active APS kinase enzymes. This suggests the closed conformation of the APS kinase domain may be the consequence of the presence of an N-terminal sulfurylase domain.
Despite the presence of ligands during crystallization, no electron density for either APS or MgAMP-PNP was observed. Owing to high sequence conservation in the APS kinase domain and the previously determined APS kinase structures, much is already known about the substrate-binding interactions. However, any ligand-induced changes in the overall hexamer conformation or in the inactive N-terminal domain could yield further information as to why the coding sequence for such a large inactive domain remains in the genome.
The structure presented here together with sequence analysis explains the lack of ATP sulfurylase activity of the N-terminal domain. The main differences within the sulfurylase-like domain occur in and around the inoperative active site (Fig. 7 ▶) and the site of the helix–turn–helix that is absent from the N-terminal domain (Figs. 3 ▶ and 4 ▶). While the overall fold of the domain is similar to true ATP sulfurylases from other organisms (Beynon et al., 2001 ▶; MacRae et al., 2001 ▶, 2002 ▶; Ullrich et al., 2001 ▶; Ullrich & Huber, 2001 ▶; Yu et al., 2007 ▶), critical amino-acid replacements and subtle structural changes result in an enzymatically inactive domain. The exact function of the ATP sulfurylase-like domain remains elusive, although kinetic data (Gay et al., unpublished results) suggest that without the inactive N-terminal domain the APS kinase activity suffers from poor efficiency and loses its characteristic substrate inhibition with respect to APS. It is likely that the function of the sulfurylase-like domain is to maintain a hexameric assembly, providing stability for the true kinase domain.
Supplementary Material
PDB reference: APS kinase, 3cr8, r3cr8sf
Acknowledgments
T. denitrificans ATCC 25259 genomic DNA was a kind gift from Sherry Huston and Doug Nelson, University of California, Davis. This work was supported in part by NSF Grant MCB-0515352 to IHS and AJF. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a national user facility operated by Stanford University on behalf of the US Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program and the National Institute of General Medical Sciences.
References
- Adams, P. D., Grosse-Kunstleve, R. W., Hung, L.-W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K. & Terwilliger, T. C. (2002). Acta Cryst. D58, 1948–1954. [DOI] [PubMed]
- Beller, H. R., Chain, P. S. G., Letain, T. E., Chakicherla, A., Larimer, F. W., Richardson, P. M., Coleman, M. A., Wood, A. P. & Kelly, D. P. (2006). J. Bacteriol. 188, 1473–1488. [DOI] [PMC free article] [PubMed]
- Beynon, J. D., MacRae, I. J., Huston, S. L., Nelson, D. C., Segel, I. H. & Fisher, A. J. (2001). Biochemistry, 40, 14509–14517. [DOI] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Cowtan, K. (1994). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr. 31, 34–38.
- DeLano, W. L. (2002). The PyMOL Molecular Graphics System. http://www.pymol.org.
- Deyrup, A., Singh, B., Krishnan, S., Lyle, S. & Schwartz, N. (1999). J. Biol. Chem. 274, 28929–28936. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Hanna, E., MacRae, I. J., Medina, D. C., Fisher, A. J. & Segel, I. H. (2002). Arch. Biochem. Biophys. 406, 275–288. [DOI] [PubMed]
- Harjes, S., Bayer, P. & Scheidig, A. J. (2005). J. Mol. Biol. 347, 623–635. [DOI] [PubMed]
- Hommes, F. A., Moss, L. & Touchton, J. (1987). Biochim. Biophys. Acta, 924, 270–275. [DOI] [PubMed]
- Kissinger, C. R., Gehlhaar, D. K. & Fogel, D. B. (1999). Acta Cryst. D55, 484–491. [DOI] [PubMed]
- Klaassen, C. D. & Boles, J. W. (1997). FASEB J. 11, 404–418. [DOI] [PubMed]
- Kusche, M., Oscarsson, L. G., Reynertson, R., Roden, L. & Lindahl, U. (1991). J. Biol. Chem. 266, 7400–7409. [PubMed]
- Lansdon, E. B., Fisher, A. J. & Segel, I. H. (2004). Biochemistry, 43, 4356–4365. [DOI] [PubMed]
- Lansdon, E. B., Segel, I. H. & Fisher, A. J. (2002). Biochemistry, 41, 13672–13680. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Lillig, C. H., Schiffmann, S., Berndt, C., Berken, A., Tischka, R. & Schwenn, J. D. (2001). Arch. Biochem. Biophys. 392, 303–310. [DOI] [PubMed]
- Lyle, S., Geller, D. H., Ng, K., Stanczak, J., Westley, J. & Schwartz, N. B. (1994). Biochem. J. 301, 355–359. [DOI] [PMC free article] [PubMed]
- MacRae, I. J. (2002). PhD Thesis. University of California, Davis, USA.
- MacRae, I. J. & Segel, I. H. (1997). Arch. Biochem. Biophys. 337, 17–26. [DOI] [PubMed]
- MacRae, I. J., Segel, I. H. & Fisher, A. J. (2000). Biochemistry, 39, 1613–1621. [DOI] [PubMed]
- MacRae, I. J., Segel, I. H. & Fisher, A. J. (2001). Biochemistry, 40, 6795–6804. [DOI] [PubMed]
- MacRae, I. J., Segel, I. H. & Fisher, A. J. (2002). Nature Struct. Biol. 9, 945–949. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Rao, S. T. & Rossmann, M. G. (1973). J. Mol. Biol. 76, 241–250. [DOI] [PubMed]
- Renosto, F., Martin, R. L. & Segel, I. H. (1989). J. Biol. Chem. 264, 9433–9437. [PubMed]
- Renosto, F., Seubert, P. A., Knudson, P. & Segel, I. H. (1985). J. Biol. Chem. 260, 1535–1544. [PubMed]
- Satishchandran, C., Hickman, Y. N. & Markham, G. D. (1992). Biochemistry, 31, 11684–11688. [DOI] [PubMed]
- Satishchandran, C. & Markham, G. D. (1989). J. Biol. Chem. 264, 15012–15021. [PubMed]
- Schriek, U. & Schwenn, J. D. (1986). Arch. Microbiol. 145, 32–38. [DOI] [PubMed]
- Sekulic, N., Dietrich, K., Paarmann, I., Ort, S., Konrad, M. & Lavie, A. (2007). J. Mol. Biol. 367, 488–500. [DOI] [PMC free article] [PubMed]
- Suiko, M., Fernando, P. H., Sakakibara, Y., Nakajima, H., Liu, M. C., Abe, S. & Nakatsu, S. (1992). Nucleic Acids Symp. Ser., pp. 183–184. [PubMed]
- Ullrich, T., Blaesse, M. & Huber, R. (2001). EMBO J. 20, 316–329. [DOI] [PMC free article] [PubMed]
- Ullrich, T. & Huber, R. (2001). J. Mol. Biol. 313, 1117–1125. [DOI] [PubMed]
- Varin, L., DeLuca, V., Ibrahim, R. K. & Brisson, N. (1992). Proc. Natl Acad. Sci. USA, 89, 1286–1290. [DOI] [PMC free article] [PubMed]
- Veitch, D. P. & Cornell, R. B. (1996). Biochemistry, 35, 10743–10750. [DOI] [PubMed]
- Veitch, D. P., Gilham, D. & Cornell, R. B. (1998). Eur. J. Biochem. 255, 227–234. [DOI] [PubMed]
- Walker, J. E., Saraste, M., Runswick, M. J. & Gay, N. J. (1982). EMBO J. 1, 945–951. [DOI] [PMC free article] [PubMed]
- Yu, M., Martin, R. L., Jain, S., Chen, L. J. & Segel, I. H. (1989). Arch. Biochem. Biophys. 269, 156–174. [DOI] [PubMed]
- Yu, Z., Lansdon, E. B., Segel, I. H. & Fisher, A. J. (2007). J. Mol. Biol. 365, 732–743. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: APS kinase, 3cr8, r3cr8sf