

Abstract
We investigated the genotype-dependent therapeutic potential of targeting the PI3K/Akt pathway for thyroid cancer. Proliferation of TPC1, Hth7, FTC133, OCUT1, K1, and BCPAP cells that harbored PI3K/Akt-activating genetic alterations was potently inhibited by the Akt inhibitor perifosine whereas SW1736, Hth74, WRO, KAT18, and TAD2 cells that harbored no genetic alterations had no or only modest responses. Inhibition of Akt phosphorylation by perifosine was seen in these cells. Genetic-dependent apoptosis was induced by perifosine in cells selectively tested. Similarly, potent inhibition of cell proliferation by the mTOR inhibitor temsirolimus occurred in virtually all the cells harboring genetic alterations whereas modest inhibition was seen in some of the cells not harboring genetic alterations. Temsirolimus inhibited the phosphorylation of p70S6K, a substrate of mTOR. Knockdown of Akt1/2 or mTOR by shRNA approach inhibited the proliferation and colony formation of FTC133 and OCUT1 cells that harbored genetic alterations in the PI3K/Akt pathway but had no effect on SW1736 and KAT18 cells that did not. Transfection with PIK3CA mutants greatly sensitized SW1736 cells to perifosine and temsirolimus. Growth of xenograft tumors derived from FTC133 cells but not SW1736 cells in nude mice was dramatically inhibited by perifosine. Thus, this work for the first time demonstrates that genetic alterations in the PI3K/Akt pathway confer thyroid cancer cells addiction to this pathway and their sensitivity to inhibition by targeting Akt and mTOR. This genotype-based targeting of the PI3K/Akt pathway using Akt and mTOR inhibitors may offer an effective therapeutic strategy for thyroid cancer and warrants further studies.
Keywords: Thyroid cancer, PI3K/Akt pathway, genetic alteration, perifosine, temsirolimus
Introduction
The phosphatidylinositol-3-kinase (PI3K)/Akt signaling pathway plays a fundamental role in cell growth, proliferation and survival and, when altered, tumorigenesis (1-3). In this signaling, the p110 catalytic subunits of class I PI3Ks, particularly PIK3CA, phosphorylate membrane phosphatidylinositols to produce PI(3,4,5)P3, which in turn binds and recruits the serine/threonine kinase Akt to cell membrane for activation by phosphoinositide-dependent protein kinases, particularly PDK1. A novel Akt inhibitor, perifosine, can selectively prevent the recruitment of the Akt pleckstrin homology domain to the membrane and blocks activation of the downstream signaling (4-5). The PI3K/Akt signaling can also be activated by RET/PTC or Ras. Activated Akt is translocated to the nucleus where it phosphorylates a multitude of downstream targets, leading to changes in cellular functions. An important downstream target of Akt is mTOR, which plays a critical role in mediating the proliferative function of the PI3K/Akt pathway (6). This signaling can be inhibited by specific mTOR inhibitors, such as temsirolimus (CCI-779) that is highly clinically applicable for its improved water solubility and stability compared with rapamycin (7). The signaling of the PI3K/Akt pathway is naturally antagonized by the tumor suppressor gene PTEN product, PTEN, which is a phosphatase that terminates the signaling of this pathway by dephosphorylating PI(3,4,5)P3 (8).
Driven by genetic alterations, the PI3K/Akt pathway is frequently over-activated in human cancers, including thyroid cancer (1,9,10). Follicular thyroid cell-derived thyroid cancer is the most common endocrine malignancy. This cancer is classified into differentiated papillary thyroid cancer (PTC) and follicular thyroid cancer (FTC) and the undifferentiated anaplastic thyroid cancer (ATC) (11). PTC and FTC may progress into poorly differentiated thyroid cancer (PDTC). Genetic alterations in the PI3K/Akt pathway are common in thyroid cancer, including the PIK3CA amplification and mutations, Ras mutations, PTEN mutations, and amplifications of some key genes in this pathway (12-18). These genetic alterations are particularly common and important in aggressive thyroid cancers, such as PDTC and ATC (13,14,17,18), which account for most of the incurable and fetal cases of thyroid cancer. Therefore, the PI3K/Akt pathway is a potentially important and effective therapeutic target in thyroid caner. We propose that the activating genetic alterations in the PI3K/Akt pathway may confer special sensitivity of thyroid cancer cells to inhibition by targeting the pathway, which may form a basis for the development of novel genetic-based therapeutic strategies for this cancer. In the present study, we tested this hypothesis using two clinically applicable inhibitors, perifosine and temsirolimus, as well as the shRNA approach in a large panel of thyroid cancer cell lines for which we characterized the genotypes of the PI3K/Akt pathway.
Materials and Methods
Thyroid cancer cell lines
The thyroid cancer cell lines C643, Hth7, Hth74, and SW1736 were originally from Dr. N.E. Heldin (University of Uppsala, Uppsala, Sweden); KAT18 from Dr. Kenneth B. Ain (University of Kentucky Medical Center, Lexington, KY); OCUT1 from Dr. Naoyoshi Onoda (Osaka City University Graduate School of Medicine, Osaka, Japan); BCPAP from Dr. Massimo Santoro (University of Federico II, Naples, Italy); K1 from Dr. David Wynford-Thomas (University of Wales College of Medicine, Cardiff, UK); WRO-82-1 from Dr. G. J. F. Juillard (University of California-Los Angeles School of Medicine, Los Angeles, CA); and FTC133 from Dr. Georg Brabant (University of Manchester, Manchester, UK). The normal thyroid cell-derived cell line TAD2 was from Dr. Mario Vitale (Università Federico II, Naples, Italy). The TPC1 cell line was provided by Dr. Alan P Dackiw (Johns Hopkins University, Maryland). These cancer cells have been recently characterized to be distinct thyroid cancer cell lines (19). They were all grown at 37°C in RPMI 1640 medium with 10% fetal bovine serum (FBS), except for FTC133 that was cultured with DMEM/HAM'S F-12 medium. For some experiments, cells were treated with perifosine or temsirolimus with the indicated concentrations and time, and the medium and agents were replenished every 24 h. Perifosine and temsirolimus were obtained from Cayman Chemical (Ann Arbor, MI, USA), dissolved in DMSO and ethanol respectively, with a stock concentration of 10 mM, and stored at -20°C.
Analysis of genetic alterations in the PI3K/Akt pathway in thyroid cancer cell lines
We analyzed the major genetic alterations in the PI3K/Akt pathway in all the thyroid cancer cell lines in the present study. K-Ras (exons 1 and 2), N-Ras (exons 1 and 2), H-Ras (exons 1 and 2), PIK3CA (exons 9 and 20) and PTEN (exons 5–7) were analyzed for mutations using our previously designed primers (14,18). For genomic DNA amplification of all the genes by PCR, after 4 min initial denaturing at 95 C, the reaction mixture was run for 35 cycles at 94 C, 54 C, and 72 C, each for 30 sec for denaturing, annealing and elongation, respectively, followed by an elongation at 72 C for 7 min. Copy number of five genes involved in this pathway, including PIK3CA, PIK3CB, PDK1, Akt-1 and -2, that could be functionally important if amplified, was analyzed using the primers and quantitative real-time PCR conditions described previously (18).
Western blotting analysis
Cells were lysed in the RIPA buffer (Santa Cruz, CA). Cellular proteins were resolved on denaturing polyacrylamide gels, transferred to PVDF membranes (Amersham Pharmacia Biotech, Piscataway, NJ), and blotted with appropriate primary antibodies. Anti-phospho-Akt (Sc-7985-R) and Anti-phospho-p70 S6 Kinase (p70S6K) (Sc-7985-R), anti-actin (Sc-1616-R) were purchased from Santa Cruz (Santa Cruz, CA). Anti-Akt-1 (#2967), anti-Akt-2 (#2964) and anti-mTOR (#2972) were purchased from Cell Signaling Technologies, Inc. (Beverly, MA). Antibody against V5-tag (#37-7500) was from Invitrogen (Carlsbad, CA). This was followed by incubation with HRP-conjugated anti-rabbit (Sc-2004) or anti-mouse (Sc-2005) IgG antibodies from Santa Cruz, and antigen-antibody complexes were visualized using the chemilucent ECL detection system (Amersham Pharmacia Biotech, Piscataway, NJ).
Lentiviral vector, shRNA plasmid construction, and cell infection with lentivirus
The lentiviral vector pSicoR-PGK-puro (Addgene Inc. Cambridge, MA, USA), originally constructed as describe (20), was used to express the hairpin RNA specifically against Akt 1/2 (Akt -1 and −2) or mTOR. The oligonucleotide sequences cloned into the vector were as follows. Oligonucleotides simultaneously targeting an identical region of Akt-1/2: sense, 5′-TGT GGT CAT GTA CGA GAT GAT TCA AGA GAT CAT CTC GTA CAT GAC CAC TTT TTT C-3′; antisense, 5′- TCG AGA AAA AAG TGG TCA TGT ACG AGA TGA TCT CTT GAA TCA TCT CGT ACA TGA CCA CA-3′. Oligonucleotides targeting mTOR: sense: 5′ - TAG AGC CGG AAT GAG GAA ACT TCA AGA GAG TTT CCT CAT TCC GGC TCT TTT TTT C - 3′; antisense:5′-TCG AGA AAA AAA GAG CCG GAA TGA GGA AAC TCT CTT GAA GTT TCC TCA TTC CGG CTC TA. The annealed double-stranded oligonucleotides were cloned into the Hpa I and Xhol I sites of pSicoR-PGK-puro. To generate lentiviral particles, human embryonic kidey 293 cells (ATCC, Manassas, VA, USA) were co-transfected with the lentiviral vector and compatible packaging plasmid mixture using Lipofectamine 2000 (Invitrogen, Carlsbad, CA), and the supernatant containing lentivirus was collected 48 h after transfection. For virus infection, cells were exposed to lentivirus-containing supernatant for 24 hours in the presence of Polybrene (Sigma). After selection with puromycin for 1-2 weeks, stable cell pools were used for cell proliferation and colony formation studies.
Cell proliferation assay
Cells (800/well) were seeded into 96-well plates and cultured under various indicated conditions. MTT assay was performed daily over a 5-day time course to evaluate cell proliferation. After 5 days of treatments as indicated, cell culture was added with 10 μl of 5 mg/ml MTT agent (Sigma St. Louis, MO) and incubated for 4 h, followed by addition of 100 μl of 10% SDS solution and a further incubation overnight. The plates were then read on a microplate reader using the test wavelength of 570 nm and the reference wavelength of 670 nm. Three duplicates were done to determine each data point. IC50 values were calculated using the Reed-Muench method (21).
Morphological assay for apoptosis
For morphological examination of apoptotic changes, cells were fixed with 4% paraformaldehyde for 15 min, and then stained with 5μg/ml Hoechst 33342 for 20 min. After PBS washing, the stained cells were observed under a fluorescence microscope. For quantitation of the number of apoptotic cells, 500 cells were counted under microscope as described in (22), and cells with chromatin condensation, margination or nuclear fragmentation were regarded as apoptotic cells, which are the characteristic morphology of apoptotic nuclear (23).
Colony formation assay
For soft-agar colony-formation assay, 2×103 cells (for FTC133) or 5×103 (for KAT18) were plated into 12-well plates with a bottom layer of 0.6% agar and a top layer of 0.3% agar. Following the hardening of soft agar, plates were incubated at 37°C with 5% CO2. After 2-3 weeks of culture, colonies were counted and photographed under a microscope. All the experiments in this study were similarly performed 2-4 times.
Xenograft tumor assay in nude mice
FTC133 (2 × 106) or SW1736 (8 × 106) cells were injected subcutaneously into flanks of nude mice at the age of 4 weeks (Harlan Sprague Dawley, Indianapolis, Inc). When tumors grew to about 5 mm in diameter, animals were grouped into 2 groups (4 mice/group) that have similar average tumor size. The 2 groups were treated daily with the vehicle PBS or 25 mg/kg Perifosine through intraperitoneal injection. Mice were weighed and tumor volume measured at the start of the treatment and twice a week during the course of the therapy. Tumor volumes were calculated by the formula (width)2 × length/2 as described previously (24). After treatment for 2 weeks, tumors were harvested and weighed. The two-tailed Independent-sample T test was used for statistical analysis of differences in tumor volumes and weights between groups.
Results
Characterization of genetic alterations in the PI3K/Akt pathway in thyroid cancer cell lines
We analyzed the genetic alterations in the major components of PI3K/Akt pathway in the thyroid cancer cell lines used in this study. As summarized in Table 1, TPC1, C643 and Hth7 cells harbored RET/PTC1 rearrangement, H-Ras mutation, and N-Ras mutation, respectively. OCUT1 and K1 harbored PI3KCA mutations. It was previously demonstrated that the FTC133 had one PTEN allele deleted and the remaining allele harbored a splice variant IVS4–19G->A (25). Here we also found that this cell line harbored a mutation in the remaining allele of PTEN that led to the formation of a premature stop codon in exon 5. Copy number analysis of the PIK3CA, PIK3CB, PDK1, Akt-1 and -2 genes showed only Akt-1 copy gain in Hth7 and BCPAP cells. The remaining five cell lines, SW1736, KAT18, Hth74, WRO and TAD2, did not harbor any of these genetic alterations in the present study. These results on genetic alterations in thyroid cancer cell lines are consistent with our and other's previous findings in tumor tissues that these are the major genetic alterations in the PI3K/Akt pathway in thyroid cancer (12-18). These cell lines constituted a valuable cell model for testing the genetic-dependent inhibition of thyroid cancer cells in the subsequent experiments of this study.
Table 1. Genotypes of thyroid cancer cell lines and their sensitivity to the Akt inhibitor perifosine and the mTOR inhibitor temsirolimus.
| Cell lines | TPC1 | C643 | Hth7 | FTC133 | OCUT1 | K1 | BCPAP | SW1736 | KAT18 | Hth74 | WRO | TAD2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Derived from | PTC | ATC | ATC | FTC | ATC | PTC | PTC | ATC | ATC | ATC | FTC | Normal |
| Genetic alterations | RET/PTC1 Re-arrangement | H-Ras (G13R+/-) | N-Ras (Q61R+/-), Akt11 copy gain | PTEN (allele deletion and R130+) | PIK3CA (H1047R+/+) | PIK3CA (E542K+/+) | Akt1 copy gain | - | - | - | - | - |
| IC50 of Perifosine (μM) | 3.58 | 4.47 | 4.02 | 1.71 | 2.17 | 10.17 | 4.92 | 35.49 | >1,000 | >1,000 | >1,000 | >1,000 |
| IC50 of Temsirolimus (nM) | 0.11 | 0.64 | 3.98 | 0.01 | 7.33 | 0.01 | 110.46 | >1,000 | 44.55 | 18.9 | 0.20 | 458.74 |
Footnotes: Ras, PIK3CA and PTEN were analyzed for mutations, and PIK3CA, PIK3CB, PDK1, Akt-1 and -2 were analyzed for copy number as described in Material and Methods. +/-, heterozygous mutation; +/+, homozygous mutation. The IC50 values were calculated using the Reed-Muench method (21).
Genetic-selective inhibition of thyroid cancer cell proliferation by the Akt inhibitor perifosine
We examined the concentration response and time course of the effects of perifosine on PI3K/Akt signaling in FTC133 cells in which PTEN is deficient. As shown in Fig 1A, inhibition of Akt phosphorylation was remarkable at 0.2 μM and nearly complete at 1 μM. Complete inhibition was observed at 6 h with perifosine at 5 μM and the inhibitory effect remained for at least 24 h (Fig 1A). We also tested the effect of the Akt inhibitor on other cell lines. As shown in Fig 1B, at 5 μM, perifosine remarkably inhibited Akt phosphorylation in the 7 thyroid cancer cell lines that harbored genetic alternations in the PI3K/Akt pathway, including TPC1, C643, Hth7, FTC133 (again), OCUT1, K1 and BCPAP cells, suggesting that the basal activities of the PI3K/Akt signaling was highly dependent on the genetic alterations in these cells. Perifosine had varying inhibitory effects on Akt phosphorylation in the remaining 5 cell lines, SW1736, KAT18, Hth74, WRO, and TAD2 cells, which did not harbor these genetic alterations, suggesting that the dependence of the PI3K/Akt signaling on such genetic alterations in these cells might not be as high.
Figure 1. Effects of the Akt inhibitor perifosine and the mTOR inhibitor temsirolimus on the PI3K/Akt signaling in thyroid cancer cells.

A) Time- and concentration-dependent effects of perifosine and temsirolimus on the PI3K/Akt signaling in the thyroid cancer cell line FTC133. The drug concentrations and time for treatment are as indicated. B) Effects of perifosine and temsirolimus in various other thyroid cancer cell lines. As indicated, cells were treated with 5 μM perifosine (upper penal) or 5 nM temsirolimus (lower panel) for 24 h. The phosphorylation level of Akt or p70S6K was detected by Western blotting using specific anti-phosphorylated Akt (p-Akt) or p70S6K (p-p70S6K) antibodies. Immunoblotting with antibody against β-actin was used for quality control.
We next examined the effect of perifosine on the proliferation of these cell lines. As shown in Fig 2A, we found that perifosine significantly inhibited proliferation of TPC1, C643, Hth7, FTC133, OCUT1, K1 and BCPAP cells in a concentration-dependent manner, with IC50 values ranging from 1.71 – 10.71 μM (Table 1). These 7 cell lines all harbored genetic alternations in the PI3K/Akt pathway (Table 1). The PTEN-deficient cell FTC133 was the most sensitive to perifosine among the 7 cell lines, suggesting that homozygous inactivation of the PTEN gene probably had the most profound activating effect on the PI3K/Akt pathway. In contrast, SW1736, KAT18, Hth74, and WRO cells that did not harbor genetic alternations in the PI3K/Akt pathway were not as sensitive to perifosine (Fig 2A); the IC50 value was 35.49 μM for the SW1736 cell and >1000 μM for KAT18, and Hth74, and WRO cells (Table 1). As expected, the normal thyroid cell-derived TAD2 cell line that lacked PI3K/Akt pathway-associated genetic alterations did not respond well to perifosine (Fig 2A and Table 1).
Figure 2. Genetic-selective inhibition of thyroid cancer cell growth by the Akt inhibitor perifosine and mTOR inhibitor temsirolimus.
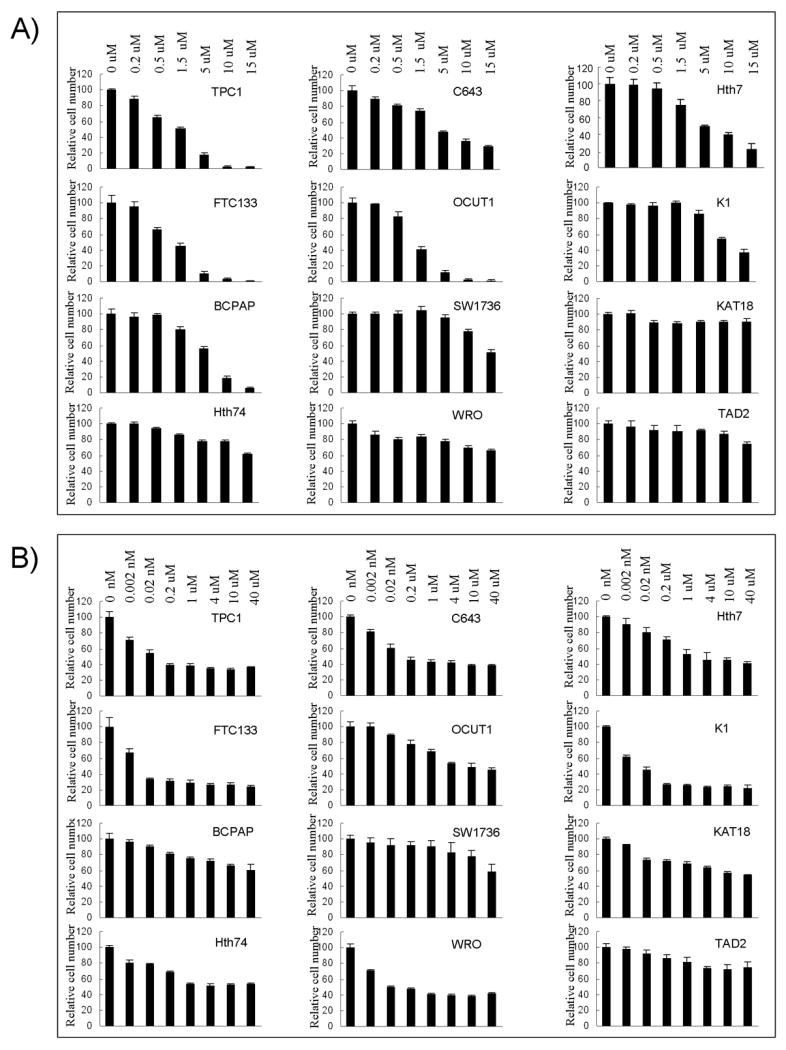
Cells were treated with the indicated concentrations of perifosine (A) or temsirolimus (B) for 5 days with the culture medium and the drug replenished every 24 h, followed by MTT assay to evaluate cell growth. IC50 values of the two drugs for each cell line were calculated according to the Reed-Muench method (21).
Genetic-selective inhibition of thyroid cancer cell proliferation by the mTOR inhibitor temsirolimus
To further explore the important role of genetic alterations in drug sensitivity in the therapeutic targeting of the PI3K/Akt pathway in thyroid cancer cells, we examined the effect of the mTOR inhibitor, temsirolimus, on the proliferation of the 12 thyroid cell lines. The inhibitory effects of temsirolimus on the phosphorylation of p70S6K, a substrate of mTOR that is involved in protein synthesis and cell cycle control (26), were also concentration and time-dependent (Fig 1A). At 5 nM, temsirolimus remarkably suppressed p70S6K phosphorylation in the 12 cell lines (Fig 1B).
We next examined the inhibitory effects of temsirolimus on the proliferation of the 12 cell lines, which, as illustrated in Fig 2B, showed a generally similar inhibitory pattern as perifosine did with few exceptions. Specifically, except for BCPAP cell that showed a modest sensitivity, all the other 6 cells that harbored genetic alterations in the PI3K/Akt pathway were potently sensitive to the inhibition by temsirolimus, with IC50 values ranging from 0.01 – 7.33 nM (Table 1). The lower sensitivity of BCPAP cells to temsirolimus may suggest a weak strength of Akt amplification in this cell in activating the mTOR signaling. Among the cells that did not harbor genetic alterations, except for WRO cell, a resistance or much lower sensitivity to temsirolimus was observed, with IC50 values ranging from 18.9 – 1965.99 nM (Fig 2B, Table 1). As with perifosine, the PTEN-deficient cell line FTC133 was again the most sensitive to temsirolimus (Fig 2B and Table 1).
Genetic-selective inhibition of cell proliferation by siRNA targeting the PI3K/Akt pathway
To confirm the genetic dependency of inhibition of thyroid cancer cells achieved with Akt and mTOR inhibitors, we next used the shRNA approach to explore the inhibitory effects of suppressing the PI3K/Akt pathway by specific knock-down of Akt or mTOR. Four thyroid cancer cell lines, including FTC133, OCUT1, SW1736 and KAT18, were chosen for these experiments. In FTC133 cells, transient knock-down of Akt-1 or -2 individually by siRNA partially suppressed cell proliferation and colony formation, and knock-down of both Akt-1 and -2 resulted in a synergistic and remarkable inhibitory effect (data not shown). As Akt-1 and Akt-2 were the most abundant and important isoforms of Akt in thyroid cancer (27), we chose to knock down the two isofomes (Akt-1/2) simultaneously for stable transfection experiments. With this approach, we were able to knock down Akt-1/2 in all the 4 cell lines with corresponding down-regulation of p-Akt (Fig 3A). We were able to partially knock down mTOR in the 4 cell lines with corresponding down-regulation of p-p70S6K (Fig 3B). Knockdown of Akt-1/2 inhibited proliferation of FTC133 and OCUT1 cells, but not SW1736 and KAT18 cells (Fig 3C). Similarly, partial knockdown of mTOR partially inhibited proliferation of FTC133 and OCUT1 cells but not SW1736 and KAT18 cells (Fig 3C). These results were consistent with the genetic-dependent effects of perifosine and temsirolimus on thyroid cancer cells (Fig 2 and Table 1).
Figure 3. Genetic-selective inhibition of cell proliferation by siRNA targeting the PI3K/Akt pathway.

A) FTC133, OCUT1, SW1736 or KAT18 cells were infected with lentivirus expressing Akt-1/2 siRNA or control lentivirus packaged with empty vector. Stable cell pools, selected with puromycin, were lysed and immunoblotted with corresponding antibodies as described in Materials and Method. The antibody against β-actin was used for quality control of Western blotting. B) Similar procedure was performed using specific siRNA to knock down mTOR. C) Comparison of proliferation rates of FTC133 OCUT1, SW1736 and KAT18 cells with/without stable knock-down of Akt-1/2 or mTOR as indicated. Stable cell pools were seeded into 96-well plates and cell proliferation rate was detected with MTT assay. The OD value (relative cell number) was measured daily over a 5-day time course.
Genetic-selective inhibition of cell colony formation by siRNA targeting PI3K/Akt pathway
We also investigated the effects of stable knockdown of Akt-1/2 or mTOR on colony formation of FTC133, OCUT1, SW1736 and KAT18 cells in soft agar (i.e. anchorage-independent cell growth), a characteristic feature of cellular transformation. As shown in supplemental Fig 1A, colony formation of FTC133 and OCUT1 cells, but not as much SW1736 and KAT18 cells, was inhibited by Akt-1/2 knockdown, in both colony number and size. Similarly, mTOR knock-down significantly inhibited the colony formation of FTC133 and OCUT1 cells but not SW1736 and KAT18 cells (supplemental Fig 1A). Supplemental Fig 1B illustrates more clearly the differential effects of knockdown of Akt-1/2 or mTOR on the colony formation of the 4 cell cells. These results demonstrated the importance of genetic alterations of the PI3K/Akt pathway in maintaining the malignant and transformed state of thyroid caner cells, further supporting the potential effectiveness of genetic-selective therapy of drugs targeting the PI3K/Akt pathway for thyroid cancer.
Expression of mutant PIK3CA increased the sensitivity of SW1736 cells to perifosine and temsirolimus
We next tested whether introduction of mutant PIK3CA into thyroid cancer cells that did not harbor genetic alterations in PI3K/Akt pathway could increase the sensitivity of cells to perifosine and temsirolimus. Two PIK3CA mutants, including H1047R and E545K that are the most common mutations of PIK3CA found in human cancer and showed strong oncogenic effects when transfected in breast cell lines (28), were used to transfect SW1736 cells. Fig 4A and B shows stable expression of exogenous PIK3CA mutants and the corresponding increase in p-Akt level in cells transfected with the PIK3CA mutants. The proliferation of cells transfected with PIK3CA mutants was more potently inhibited by perifosine and temsirolimus than the cells transfected with control vector (Fig 4C). For cells transfected with vector, H1047R and E545K, the IC50 values of perifosine were 30.55 μM, 4.26 μM and 6.14 μM, respectively, and IC50 values of temsirolimus were >1000 nM, 3.57 nM and 10.52 nM, respectively.
Figure 4. Expression of mutant PIK3CA increased the sensitivity of SW1736 cells to perifosine and temsirolimus.

A) Stable expression of PIK3CA mutants H1047R or E545K in SW1736 cells. SW1736 cells were transfected with lentiviral vectors containing mutant PIK3CA or empty vectors and selected with blastomycin for 2 weeks. Stable cell pools were lysed and immunoblotted with antibody against V5 tag or p-Akt to detect the expression of exogenous PIK3CA mutant and p-Akt level. The antibody against β-actin was used for quality control in Western blotting. B) Quantitative illustration of relative p-Akt levels. Densitometry was performed to measure the density of the p-Akt and β-actin bands in Fig 4A. The relative p-Akt levels were obtained by dividing the p-Akt band density by the corresponding β-actin band density. C) Effects of perifosine (left panel) or temsirolimus (right panel) on the proliferation of SW1736 cells transfected with PIK3CA constructs. Cells were treated with perifosine or temsirolimus at the indicated concentrations for 5 days with the culture medium and the drug replenished every 24 h. MTT assay was used to measure cell proliferation and IC50 values were calculated according to the Reed-Muench method (21).
Genetic-selective induction of thyroid cancer cell apoptosis and inhibition of xenograft tumor growth by the Akt inhibitor perifosine
To investigate whether genetic-dependent effects of perifosine and temsirolimus on cell growth involved cell death, we examined pro-apoptotic effects of the two inhibitors on thyroid cancer cell lines using nuclear morphological analysis as described in Material and Methods. After treatment with perifosine, chromatin condensation, margination and nuclear fragmentation, which are characteristic morphologies of apoptotic nuclear (23), were frequently observed in FTC133 and OCUT1 cells that harbored genetic alterations in the PI3K/Akt pathway but not in SW1736 and KAT18 cells that did not (Fig 5A). The apoptotic cell number increased more than 20 folds in the former 2 cells, while it only minimally increased in the latter 2 cells after treatment with perifosine (Fig 5B). We did not see effects of temsirolimus on cell apoptosis in these 4 thyroid cancer cell lines (data not shown).
Figure 5. Genetic-selective induction of thyroid cancer cell apoptosis and inhibition of the xenograft thyroid tumor growth by the Akt inhibitor perifosine.

A) Representative morphology of cell nuclei after treatment with or without perifosine. Cells were treated with 5 μM perifosine for 48 h and apoptotic cells were determined by Hoechst 33342 staining. Cells were observed under a fluorescent microscope and selective apoptotic nuclei are indicated with arrows, showing chromatin margination, condensation and fragmentation, which are characteristics of apoptosis. B) Quantitation of the number of apoptotic cells by examining 500 cells under a fluorescent microscope for each sample. C) Effects of perifosine on the growth of xenograft tumor derived from FTC133 cells. Left panel shows the time course of tumor growth, measured as the average tumor volume in each group at the indicated time of treatment with vehicle (PBS) or perifosine, 25 mg/kg/day peritoneally. Each time point represents the average ± SD of the values obtained from 4 mice in each group. Right panel shows FTC133-derived tumor weights from the 4 individual mice in each group after a 2-week treatment. Animals were finally sacrificed and xenograft tumors were surgically removed and weighted. The average weight of the tumors from each group is indicated with a short horizontal bar. P values were obtained by Independent-sample T test. D) Effects of perifosine on the growth of SW1736-derived xenograft tumor. The experimental procedures were identical as described for FTC133 cell-derived tumors.
Since cell apoptosis was an important mechanism in the genetic-dependent inhibition of thyroid cancer cell growth induced by perifosine, perifosine has a potential to be highly effective in treating thyroid cancer in vivo. We therefore next tested the genetic-dependent effect of perifosine on the growth of xenograft thyroid tumors in nude mice. As shown in Fig 5C, the PTEN-deficient FTC133 cell-derived xenograft tumors progressively grew in the control group while the tumors stopped growing or even shrinked in perifosine-treated group. At the end of the experiment after a 2-week treatment, the tumor volumes and weights were 1.807 ± 0.774 cm3 (control group) vs. 0.177 ± 0.095 cm3 (perifosine group) (p =0.005) and 1.567 ± 0.265 g (control group) vs. 0.198 ± 0.051 g (perifosine group) (p<0.001), respectively (Fig 5C). In contrast, perifosine only had a minimal inhibitory effect on the growth of SW1736-derived xenograft tumors that did not harbor genetic alterations in the PI3K/Akt pathway (Fig 5D). At the end of the experiment after a 2-week treatment, the tumor volumes and weights were 0.449 ± 0.121 cm3 (control group) vs. 0.315 ± 0.160 cm3 (perifosine group) and 0.355 ± 0.115 g (control group) vs. 0.270 ± 0.149 g (perifosine group), respectively (Fig 5D).
Discussion
Thyroid cancer is the most common endocrine malignancy with a rapidly rising incidence in recent decades (29-31), posing an increasing challenge to the treatment of this cancer. Although surgical and radioiodine treatments are generally effective, many thyroid cancer patients have persistent or recurrent disease that is currently incurable and associated with increased morbidity and mortality. PDTC and ATC, which can derive from PTC and FTC or develop de novo, are particularly difficult to treat (32,33). In fact, ATC is one of the most aggressive and deadly human cancers. New therapies are needed for incurable thyroid cancers. The recent several clinical trials on receptor tyrosine kinase inhibitors in thyroid cancers are promising but the results are still limited at this stage (34-36). More effective molecular-targeted therapeutic strategies need to be developed based on molecular mechanisms, particularly the derangements in major signaling pathways, in thyroid cancer.
The PI3K/Akt pathway is a well-explored therapeutic target in human cancers (2,3,37). Given the important role of the PI3K/Akt pathway and its common genetic alterations in thyroid cancer (1,9,10,38), in the present study, we tested the therapeutic potential of targeting this pathway, with a focus on its genotype dependence and the potential of using genetic alterations in this pathway to predict the sensitivity of thyroid cancer cells. Various inhibitors targeting this pathway have been developed for therapeutic testing. Among these are the Akt inhibitors and mTOR inhibitors, which target two key steps, Akt and mTOR, respectively in this pathway. The Akt inhibitor perifosine and mTOR inhibitor temsirolimus are among the most promising inhibitors, which have been or are being actively tested in clinical trials and have shown acceptable toxicity profiles and great promises in some cancers (4,5,39,40). They were therefore specifically tested in the present study.
We demonstrated a striking dependency of perifosine and temsirolimus, particularly the former, on genetic alterations in the PI3K/Akt pathway in the inhibition of thyroid cancer cells. Specific knockdown of Akt and mTOR using shRNA approaches showed identical genetic-dependence patterns in inhibiting the proliferation and anchorage-independent colony formation of thyroid cancer cells. Moreover, transfection of cells with PIK3CA mutants conferred increased sensitivity to perifosine and temsirolimus in cells that did not naturally harbor genetic alterations in the PI3K/Akt pathway. These results suggest that the results obtained with drug inhibitors were indeed genetic-specific. The one exception that showed inconsistent genetic-dependent pattern was the WRO cell, which did not harbor genetic alterations at or upstream of Akt in the PI3K/Akt pathway and, as expected, was highly resistant to the Akt inhibitor perifosine, but was highly sensitive to the mTOR inhibitor temsirolimus. It is likely that an uncharacterized Akt-independent mechanism played a major role in the activation of mTOR signaling in this cell as demonstrated in other cancer cells (41). This mechanism might also occur in the Hth74 cell that did not harbor genetic alterations in the PI3K/Akt pathway and was highly resistant to perifosine but was fairly well sensitive to temsirolimus.
The results in the present study also suggest that thyroid cancer cells are highly addictive to the PI3K/Akt signaling when genetic alterations in this pathway are present, similar to breast cancer cells (42,43) and lung cancer cells (44). It is important to note that the PI3K/Akt-associated genetic alterations tested in the present study, such as PIK3CA, Ras, and PTEN mutations and PIK3CA amplifications, are particularly common in aggressive thyroid cancers, such as PDTC and ATC (13,14,17,18). Therefore, genetic-based targeting of the PI3K/Akt pathway using perifosine or temsirolimus may be a particularly promising therapeutic strategy for these therapeutically challenging thyroid cancers. It is worth noting that the dramatic inhibition of cell proliferation was achieved in cells harboring genetic alterations with perifosine at the concentrations around 5-10 μmol/L, which was within the clinically achievable and safe peak plasma concentration ranges (i.e., 10–15 μmol/L) (45,46). The potency of temsirolimus in the sensitive cells was extremely high, with IC50 in the nanomolar range, consistent with previous findings that temsirolimus at the nanomolar range effectively suppressed the growth of cancer cells (47). Since cell apoptosis was a major mechanism in genetic-dependent inhibition of thyroid cancer cells by perifosine, this inhibitor may have a particularly high clinical potential for effective treatment of thyroid cancer, as supported by its profound genetic-dependent inhibitory effect on the growth of xenograft thyroid tumors.
In summary, we have demonstrated that genetic alterations in the PI3K/Akt pathway confer strong sensitivity of thyroid cancer cells to inhibition by targeting Akt or mTOR. It may thus be possible to use genetic markers in the PI3K/Akt pathway to predict the responses of thyroid cancer to specific inhibitors of this pathway. Given the common genetic alterations in the PI3K/Akt pathway in thyroid cancer, particularly aggressive and incurable types, perifosine and temsirolimus, which have well demonstrated acceptable clinical profiles, are highly promising in genetic-targeted therapies for thyroid cancer.
Supplementary Material
Acknowledgments
We want to thank Drs. Heldin NE, Ain KB, Onoda N, Santoro M, Wynford-Thomas D, Brabant G, Dackiw AP, Juillard GJ, Vitale M, Schweppe RE, and Haugen BR for kindly providing us or facilitating the accessibility to the cell lines used in this study. This work was supported by NIH RO-1 grant CA113507-01 to M Xing.
References
- 1.Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 2002;14:381–95. doi: 10.1016/s0898-6568(01)00271-6. [DOI] [PubMed] [Google Scholar]
- 2.Fresno Vara JA, Casado E, de Castro J, Cejas P, Belda-Iniesta C, Gonzalez-Baron M. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev. 2004;30:193–204. doi: 10.1016/j.ctrv.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 3.Carnero A, Blanco-Aparicio C, Renner O, Link W, Leal JF. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr Cancer Drug Targets. 2008;8:187–98. doi: 10.2174/156800908784293659. [DOI] [PubMed] [Google Scholar]
- 4.van Blitterswijk WJ, Verheij M. Anticancer alkylphospholipids: mechanisms of action, cellular sensitivity and resistance, and clinical prospects. Curr Pharm Des. 2008;14:2061–74. doi: 10.2174/138161208785294636. [DOI] [PubMed] [Google Scholar]
- 5.Gills JJ, Dennis PA. Perifosine: update on a novel Akt inhibitor. Curr Oncol Rep. 2009;11:102–10. doi: 10.1007/s11912-009-0016-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dufner A, Andjelkovic M, Burgering BM, Hemmings BA, Thomas G. Protein kinase B localization and activation differentially affect S6 kinase 1 activity and eukaryotic translation initiation factor 4E-binding protein 1 phosphorylation. Mol Cell Biol. 1999;19:4525–34. doi: 10.1128/mcb.19.6.4525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elit L. CCI-779 Wyeth. Curr Opin Investig Drugs. 2002;3:1249–53. [PubMed] [Google Scholar]
- 8.Sansal I, Sellers WR. The biology and clinical relevance of the PTEN tumor suppressor pathway. J Clin Oncol. 2004;22:2954–63. doi: 10.1200/JCO.2004.02.141. [DOI] [PubMed] [Google Scholar]
- 9.Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xing M. Recent advances in molecular biology of thyroid cancer and their clinical implications. Otolaryngol Clin North Am. 2008;41:1135–46. ix. doi: 10.1016/j.otc.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hundahl SA, Fleming ID, Fremgen AM, Menck HR. A National Cancer Data Base report on 53,856 cases of thyroid carcinoma treated in the U.S., 1985-1995. Cancer. 1998;83:2638–48. doi: 10.1002/(sici)1097-0142(19981215)83:12<2638::aid-cncr31>3.0.co;2-1. see commetns. [DOI] [PubMed] [Google Scholar]
- 12.Wu G, Mambo E, Guo Z, et al. Uncommon mutation, but common amplifications, of the PIK3CA gene in thyroid tumors. J Clin Endocrinol Metab. 2005;90:4688–93. doi: 10.1210/jc.2004-2281. [DOI] [PubMed] [Google Scholar]
- 13.Garcia-Rostan G, Costa AM, Pereira-Castro I, et al. Mutation of the PIK3CA gene in anaplastic thyroid cancer. Cancer Res. 2005;65:10199–207. doi: 10.1158/0008-5472.CAN-04-4259. [DOI] [PubMed] [Google Scholar]
- 14.Hou P, Liu D, Shan Y, et al. Genetic alterations and their relationship in the phosphatidylinositol 3-kinase/Akt pathway in thyroid cancer. Clin Cancer Res. 2007;13:1161–70. doi: 10.1158/1078-0432.CCR-06-1125. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y, Hou P, Yu H, et al. High prevalence and mutual exclusivity of genetic alterations in the phosphatidylinositol-3-kinase/akt pathway in thyroid tumors. J Clin Endocrinol Metab. 2007;92:2387–90. doi: 10.1210/jc.2006-2019. [DOI] [PubMed] [Google Scholar]
- 16.Abubaker J, Jehan Z, Bavi P, et al. Clinicopathological analysis of papillary thyroid cancer with PIK3CA alterations in a Middle Eastern population. J Clin Endocrinol Metab. 2008;93:611–8. doi: 10.1210/jc.2007-1717. [DOI] [PubMed] [Google Scholar]
- 17.Santarpia L, El Naggar AK, Cote GJ, Myers JN, Sherman SI. Phosphatidylinositol 3-kinase/akt and ras/raf-mitogen-activated protein kinase pathway mutations in anaplastic thyroid cancer. J Clin Endocrinol Metab. 2008;93:278–84. doi: 10.1210/jc.2007-1076. [DOI] [PubMed] [Google Scholar]
- 18.Liu Z, Hou P, Ji M, et al. Highly prevalent genetic alterations in receptor tyrosine kinases and phosphatidylinositol 3-kinase/akt and mitogen-activated protein kinase pathways in anaplastic and follicular thyroid cancers. J Clin Endocrinol Metab. 2008;93:3106–16. doi: 10.1210/jc.2008-0273. [DOI] [PubMed] [Google Scholar]
- 19.Schweppe RE, Klopper JP, Korch C, et al. Deoxyribonucleic acid profiling analysis of 40 human thyroid cancer cell lines reveals cross-contamination resulting in cell line redundancy and misidentification. J Clin Endocrinol Metab. 2008;93:4331–41. doi: 10.1210/jc.2008-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ventura A, Meissner A, Dillon CP, et al. Cre-lox-regulated conditional RNA interference from transgenes. Proc Natl Acad Sci U S A. 2004;101:10380–5. doi: 10.1073/pnas.0403954101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Welkos S, O'Brien A. Determination of median lethal and infectious doses in animal model systems. Methods Enzymol. 1994;235:29–39. doi: 10.1016/0076-6879(94)35128-7. [DOI] [PubMed] [Google Scholar]
- 22.Delaney CA, Pavlovic D, Hoorens A, Pipeleers DG, Eizirik DL. Cytokines induce deoxyribonucleic acid strand breaks and apoptosis in human pancreatic islet cells. Endocrinology. 1997;138:2610–4. doi: 10.1210/endo.138.6.5204. [DOI] [PubMed] [Google Scholar]
- 23.Saraste A. Morphologic criteria and detection of apoptosis. Herz. 1999;24:189–95. doi: 10.1007/BF03044961. [DOI] [PubMed] [Google Scholar]
- 24.Gray MJ, Wey JS, Belcheva A, et al. Neuropilin-1 suppresses tumorigenic properties in a human pancreatic adenocarcinoma cell line lacking neuropilin-1 coreceptors. Cancer Res. 2005;65:3664–70. doi: 10.1158/0008-5472.CAN-04-2229. [DOI] [PubMed] [Google Scholar]
- 25.Weng LP, Gimm O, Kum JB, et al. Transient ectopic expression of PTEN in thyroid cancer cell lines induces cell cycle arrest and cell type-dependent cell death. Hum Mol Genet. 2001;10:251–8. doi: 10.1093/hmg/10.3.251. [DOI] [PubMed] [Google Scholar]
- 26.Berven LA, Crouch MF. Cellular function of p70S6K: a role in regulating cell motility. Immunol Cell Biol. 2000;78:447–51. doi: 10.1046/j.1440-1711.2000.00928.x. [DOI] [PubMed] [Google Scholar]
- 27.Ringel MD, Hayre N, Saito J, et al. Overexpression and overactivation of Akt in thyroid carcinoma. Cancer Res. 2001;61:6105–11. [PubMed] [Google Scholar]
- 28.Zhang H, Liu G, Dziubinski M, Yang Z, Ethier SP, Wu G. Comprehensive analysis of oncogenic effects of PIK3CA mutations in human mammary epithelial cells. Breast Cancer Res Treat. 2008;112:217–27. doi: 10.1007/s10549-007-9847-6. [DOI] [PubMed] [Google Scholar]
- 29.Leenhardt L, Grosclaude P, Cherie-Challine L. Increased incidence of thyroid carcinoma in France: a true epidemic or thyroid nodule management effects? Report from the French Thyroid Cancer Committee. Thyroid. 2004;14:1056–60. doi: 10.1089/thy.2004.14.1056. [DOI] [PubMed] [Google Scholar]
- 30.Davies L, Welch HG. Increasing incidence of thyroid cancer in the United States, 1973-2002. JAMA. 2006;295:2164–7. doi: 10.1001/jama.295.18.2164. [DOI] [PubMed] [Google Scholar]
- 31.Enewold L, Zhu K, Ron E, Marrogi AJ, et al. Rising thyroid cancer incidence in the United States by demographic and tumor characteristics, 1980-2005. Cancer Epidemiol Biomarkers Prev. 2009;18:784–91. doi: 10.1158/1055-9965.EPI-08-0960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Volante M, Rapa I, Papotti M. Poorly differentiated thyroid carcinoma: diagnostic features and controversial issues. Endocr Pathol. 2008;19:150–5. doi: 10.1007/s12022-008-9040-4. [DOI] [PubMed] [Google Scholar]
- 33.Neff RL, Farrar WB, Kloos RT, Burman KD. Anaplastic thyroid cancer. Endocrinol Metab Clin North Am. 2008;37:525–38. xi. doi: 10.1016/j.ecl.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 34.Sherman SI, Wirth LJ, Droz JP, et al. Motesanib diphosphate in progressive differentiated thyroid cancer. N Engl J Med. 2008;359:31–42. doi: 10.1056/NEJMoa075853. [DOI] [PubMed] [Google Scholar]
- 35.Cohen EE, Rosen LS, Vokes EE, et al. Axitinib Is an Active Treatment for All Histologic Subtypes of Advanced Thyroid Cancer: Results From a Phase II Study. J Clin Oncol. 2008 doi: 10.1200/JCO.2007.15.9566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gupta-Abramson V, Troxel AB, Nellore A, et al. Phase II Trial of Sorafenib in Advanced Thyroid Cancer. J Clin Oncol. 2008 doi: 10.1200/JCO.2008.16.3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 38.Smallridge RC, Marlow LA, Copland JA. Anaplastic thyroid cancer: molecular pathogenesis and emerging therapies. Endocr Relat Cancer. 2009;16:17–44. doi: 10.1677/ERC-08-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Malizzia LJ, Hsu A. Temsirolimus, an mTOR inhibitor for treatment of patients with advanced renal cell carcinoma. Clin J Oncol Nurs. 2008;12:639–46. doi: 10.1188/08.CJON.639-646. [DOI] [PubMed] [Google Scholar]
- 40.Mills EJ, Rachlis B, O'Regan C, Thabane L, Perri D. Metastatic renal cell cancer treatments: an indirect comparison meta-analysis. BMC Cancer. 2009;9:34. doi: 10.1186/1471-2407-9-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Memmott RM, Dennis PA. Akt-dependent and -independent mechanisms of mTOR regulation in cancer. Cell Signal. 2009;21:656–64. doi: 10.1016/j.cellsig.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hennessy BT, Lu Y, Poradosu E, et al. Pharmacodynamic markers of perifosine efficacy. Clin Cancer Res. 2007;13:7421–31. doi: 10.1158/1078-0432.CCR-07-0760. [DOI] [PubMed] [Google Scholar]
- 43.She QB, Chandarlapaty S, Ye Q, et al. Breast tumor cells with PI3K mutation or HER2 amplification are selectively addicted to Akt signaling. PLoS ONE. 2008;3:e3065. doi: 10.1371/journal.pone.0003065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Engelman JA, Chen L, Tan X, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–6. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crul M, Rosing H, de Klerk GJ, et al. Phase I and pharmacological study of daily oral administration of perifosine (D-21266) in patients with advanced solid tumours. Eur J Cancer. 2002;38:1615–21. doi: 10.1016/s0959-8049(02)00127-2. [DOI] [PubMed] [Google Scholar]
- 46.Van Ummersen L, Binger K, Volkman J, et al. A phase I trial of perifosine (NSC 639966) on a loading dose/maintenance dose schedule in patients with advanced cancer. Clin Cancer Res. 2004;10:7450–6. doi: 10.1158/1078-0432.CCR-03-0406. [DOI] [PubMed] [Google Scholar]
- 47.Shi Y, Gera J, Hu L, et al. Enhanced sensitivity of multiple myeloma cells containing PTEN mutations to CCI-779. Cancer Res. 2002;62:5027–34. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.