

Abstract
Autism is a heterogeneous neurodevelopmental syndrome with a complex genetic etiology. It is still not clear whether autism comprises a vast collection of different disorders akin to intellectual disability or a few disorders sharing common aberrant pathways. Unifying principles among cases of autism are likely to be at the level of brain circuitry in addition to molecular pathways.
Introduction
Autism represents a broadly defined disorder of behavior and cognition with onset prior to age 3 affecting the core domains of language and social development and involving abnormal repetitive and restrictive behaviors. Because autism is characterized by groups of symptoms and signs even in its narrowest conception, it is a highly variable neurodevelopmental syndrome and not a unitary condition. Children diagnosed with autism differ significantly in severity along many cognitive and behavioral dimensions, spawning the term autism spectrum disorder (ASD) to emphasize its full scope.
Basic genetic and neuroscience research in ASD has grown exponentially, reflecting a remarkable trajectory that likely represents many factors, including public awareness and the realization that ASDs are a significant cause of lifetime neuropsychiatric morbidity, affecting nearly 1/150 live births. However, in contrast with many other disorders of the brain, for example neurodegenerative diseases such as Parkinson’s or Alzheimer’s diseases, autism lacks any clear unifying pathology at the molecular, cellular, or systems level. Furthermore, although ASDs appear to be highly heritable overall, their underlying genetic etiology is complex, likely involving many genes, some of which may represent common genetic variation, as well as potential interactions with environmental factors. Thus, ASD research has to contend with not only the complexity and broadness of the phenotype itself, which encompasses the biological basis of human social interactions and language, but genetic and environmental complexity as well. Despite these challenges, measurable progress has been achieved, placing several key questions into relief.
Autism Is Heritable but Genetically Heterogeneous
Three decades of research on autism involving twin and family studies support a significant genetic contribution to its etiology. However, high heritability does not necessarily imply a particular model of genetic transmission or an easily identifiable major gene causing the disorder. On the contrary, the last decade of research in autism genetics reveals significant genetic heterogeneity. For example, several dozen distinct genetic disorders or identified chromosomal abnormalities can result in autism, including Joubert’s syndrome, Rett’s syndrome, tuberous sclerosis, Fragile X syndrome, and maternally inherited duplications of chromosome 15q11-13, the latter two each accounting for 1%–2% of ASD cases (Veenstra-Vanderweele et al., 2004). In all, known rare chromosomal disorders and genetic mental retardation syndromes account for ~10% of ASD, each single cause contributing to no more than 1% of cases on average (Abrahams and Geschwind, 2008).
The existence of considerable genetic heterogeneity is also supported by several dozen genetic linkage studies over the last decade, which have often identified nonoverlapping regions of interest and largely failed to formally replicate autism linkage findings at the level of genome-wide significance. There are a few notable exceptions including regions on chromosome 7q21-35, supported by meta-analysis (Badner and Gershon, 2002), and chromosome 17q, which has been replicated at genome-wide significance (Cantor et al., 2005). A recent large collaborative genome scan by the Autism Genome Project (AGP) of nearly 1200 sibling pairs with ASD (Szatmari et al., 2007) identified several regions of interest, including chromosome 11, but did not identify one region at genome-wide significance, despite a marked increase in sample size over the largest previous studies. Similarly, the homozygosity mapping collaborative for autism (HMCA; Morrow et al., 2008) did not report genome-wide significant loci shared by two or more of the approximately 80 consanguinous families, consistent with the existence of many distinct autism loci in this population as well. However, HMCA investigators were able to identify six independent homozygous deletions segregating with autism in this unique cohort, implicating several new genes in autism susceptibility while again highlighting the genetic heterogeneity of ASD (Morrow et al., 2008).
Whole genome association (WGA) studies using various microarray platforms are beginning to replace linkage studies in the analysis of complex (non-Mendelian) genetic disease including ASD. These genome-wide association analyses test the association of common single-nucleotide variations (SNPs) with disease in a population. If a disease like autism is primarily caused by rare mutations in certain chromosomal regions, WGA is unlikely to be adequately powered to identify most of these, whereas linkage may be powered to identify the chromosomal region where they reside. Subsequent resequencing of genes within a linkage region would then be necessary to identify the actual causal gene. No large WGA studies have yet been published in ASD, but studies of common variant association in linkage regions suggest that analyses performed will be underpowered and that at least a doubling in sample size (many thousands of cases, similar to studies of type I diabetes) will be needed to identify more than a few loci at genome-wide significant association.
These results echo findings in other common diseases with a complex genetic basis, in which early underestimation of heterogeneity and overestimation of the magnitude of risk imparted by any given susceptibility allele led to underestimates of sample sizes needed for adequate power to detect common variant associations. From this perspective, the results of linkage and association studies in autism imply that attaining massive sample sizes through large collaborative efforts and sample sharing—for example, through the Autism Genetic Resource Exchange (AGRE) and the Autism Genome Project (AGP) (Geschwind et al., 2001, Szatmari et al., 2007)—will be necessary to successfully find many common susceptibility genes.
Rare or De Novo Mutations in ASD
Perhaps the most remarkable advance in ASD genetics in the last year was driven by the earlier discovery that regional variations in gene copy number, either heritable or arising de novo (not seen in parents), are a significant source of genetic variation in humans (Sebat et al., 2004). Copy number variation (CNV) is a form of structural variation in the genome in which there is a gain or loss in a chromosomal region greater than 1 kilobase (kb) in size, in contrast to the more common SNPs, which are changes at one base pair of DNA. Recently, Sebat et al. (2007) identified de novo CNVs in 3% of autistic children from multiplex families (having two or more affected members) and in 10% of autistic children from simplex families (having one child with ASD). These findings were presaged by previous studies using lower-resolution methods that identified a number of large chromosomal anomalies associated with ASD and mental retardation (Jacquemont et al., 2006), including a duplicated region on chromosome 15q identified more than a decade ago (Veenstra-Vanderweele et al., 2004). Perhaps because some of these mutations were rare, large, affected other organ systems in addition to the central nervous system, or simply seemed to be special cases, this mechanism was largely unappreciated as a potential cause of idiopathic autism. The CNVs identified by Sebat et al. (2007) were composed of deletions (70%) and duplications (30%) of DNA fragments ranging from 160 kb to several megabases in size, thus containing segments from the size of a single gene to chromosomal regions harboring a dozen or more genes. Such genomic level de novo mutational events were only found in 1% of control individuals and were all duplications rather than the typically more deleterious deletions observed in ASD (Sebat et al., 2007). Remarkably, most of the CNVs were unique, providing an indication that a significant fraction of ASD may be accounted for by rare, essentially private, mutations in simplex (one affected child) autism families. Similarly, the large AGP linkage study identified a handful of rare, likely causal CNVs using a lower-resolution platform (Szatmari et al., 2007).
It is too early to predict with certainty from these data the contribution of de novo CNVs to ASD susceptibility. Larger sample sizes ascertained from independent, clearly defined populations will be necessary to accurately define the role of CNVs. Sample characteristics are important: de novo mutations are observed more frequently in those with more severe intellectual disability or dysmorphology (Jacquemont et al., 2006). The contribution of de novo CNVs is also significantly less in multiplex families having two or more autistic children (Sebat et al., 2007), a finding confirmed in subsequent studies (Weiss et al., 2008; Marshall et al., 2008). Because the number of CNVs detected is clearly related to the resolution of the microarrays used, the contribution of known CNVs to autism is expected to increase beyond 10% as microarray probe density increases. Similarly, single base pair mutations in a few genes encoding the synaptic adhesion proteins neuroligins 3 and 4, the voltage-gated calcium ion channel CaV1.2, the tumor suppressor PTEN, and shank3, a cytoplasmic binding partner of the neuroligins, have been identified in rare cases of ASD. The advent of efficient partial genome sequencing will more fully clarify the contribution of rare single-nucleotide variants to ASD. It should be emphasized that the contribution of inherited CNVs as a source of more common genetic contributions to ASD has not been explored in depth. This will be important because some heritable CNVs may have subtle phenotypic effects and will contribute to common variations in cognition and behavior.
The occurrence of rare de novo mutations in ASD raises important issues regarding mechanisms causing mutations (Lupski, 2007). Paternal age is associated with increasing point mutations in sperm, and complex genetic conditions associated with increasing paternal age may have a higher percentage of new mutations. New mutations may be particularly pronounced in the offspring of older fathers, who may be a reservoir for such de novo events. Notably, advanced paternal age has recently been shown to significantly increase risk for ASD in two distinct patient samples (Cantor et al., 2007; Reichenberg et al., 2006). These data suggest one of many potential mechanisms through which environmental factors could play a role in creating de novo genetic events causing autism, that is, accumulation of mutations in the male germline. Such factors could occur in isolation or in conjunction with genetic susceptibility loci. In the latter case, certain inherited haplotypes, for example, could render specific regions more vulnerable to mutagens, thus increasing the frequency of mutational events. Alternatively, certain regions may be more vulnerable to other environmental factors that could affect chromatin structure or gene expression, leading to epigenetic causes of autism (Jiang et al., 2004).
Multigenic versus Major Gene Contributions
Currently, the predominant genetic model supposes the presence of multigenic inheritance of common polymorphisms contributing to autism risk in multiplex families (Abrahams and Geschwind, 2008). At face value, the paucity of multigenerational pedigrees segregating ASD argues against Mendelian, more specifically, dominant inheritance. However, Wigler and colleagues (Zhao et al., 2007) recently reassessed this notion based on the identification of rare CNVs as significant contributors to autism genetic risk and the strikingly higher incidence in males (4:1 male-female ratio). The model is based on a formal analysis of recurrence risk in multiplex families and is consistent with a significant contribution from two major risk categories: low-risk families in which there is little genetic loading for autism in other family members but in which the proband carries highly penetrant de novo mutations (accounting for about half of the cases) and higher-risk families consistent with dominant inheritance in males (accounting for about one-third of cases). This model fits the family data collected by several groups if the high-risk alleles have lower penetrance with respect to the ASD phenotype in females. Future gene hunting efforts will provide an empirical test of this model. Nevertheless, the current data are consistent with the notion that autism spectrum disorders are caused by a higher proportion of rare mutations than previously anticipated and that the contribution of common variants will mostly consist of alleles with small effect sizes.
Identification of Common Genetic Variants
Moving from genetic linkage studies to identifying the multiple causal variants that likely underlie chromosomal regions with the strongest linkage signals remains a challenge. It may be that rare mutations underlie these signals and identifying these will require large-scale resequencing of the entire region in large numbers of cases and controls. In several cases, the identification of causal common variation has been illusory despite strong overlying linkage signals, supporting the potential role of multiple rare mutations or many common variants, each of small effect size.
Even as the current strategies identify reproducible underlying genetic variants, the question still remains to what aspect of the ASD phenotype are they related? Similarly, the striking phenotypic heterogeneity and clinical variability even among twins suggest that distinct forms of autism may exist or that distinct genetic risk factors may be related to specific phenotypic features. This notion is brought into focus in the recent HMCA study (Morrow et al., 2008), where rare, potentially disease-causing mutations in the gene Slc9a9 were increased in AGRE families with autism and epilepsy, but not in those without epilepsy. Genotype-phenotype correlations will become even more salient as attempts are made to produce relevant animal models, not to mention the needs of families undergoing prognostic counseling in the future.
Moreover, from a neurobiological perspective, different aspects of human cognition and behavior are served by distinct brain regions, which are likely to be patterned and maintained by distinct genetic factors. Thus, specific genetic risk factors may correspond to changes at the level of specific brain structures or neural systems that contribute to autism, such as those serving language or social cognition, rather than the broad syndrome of autism itself (Geschwind and Levitt, 2007; Figure 1). These heritable components or endophenotypes involving language, social responsiveness, or behavioral rigidity are also observed at higher frequency in first-degree relatives of autistic subjects and can be measured as continuous, quantitative variables. Compared with the categorical diagnosis of autism, approaches based on linking quantitative endophenotypes to underlying genetic risk may provide more power, as has been appreciated in other complex genetic conditions. This quantitative trait locus (QTL) approach has the additional benefits of including unaffected relatives and the full range of variation in a particular measured phenotype rather than the arbitrary categorical determination of affected (autistic) and unaffected (not autistic), which has changed over time. We and others have successfully used QTL mapping to identify chromosomal loci related to cognitive endophenotypes, such as language, non-verbal communication, and social cognition (e.g., Alarcón et al., 2008). Moreover, because we postulate that these features involving language, social behaviors, and other behavioral or cognitive traits represent one end of a continuum, normal or otherwise (Figure 1), they are likely to be related to many different neuropsychiatric and neurodevelopmental conditions, in addition to ASD.
Figure 1. Common and Rare Genetic Variations and the Heterogeneity of Autism.
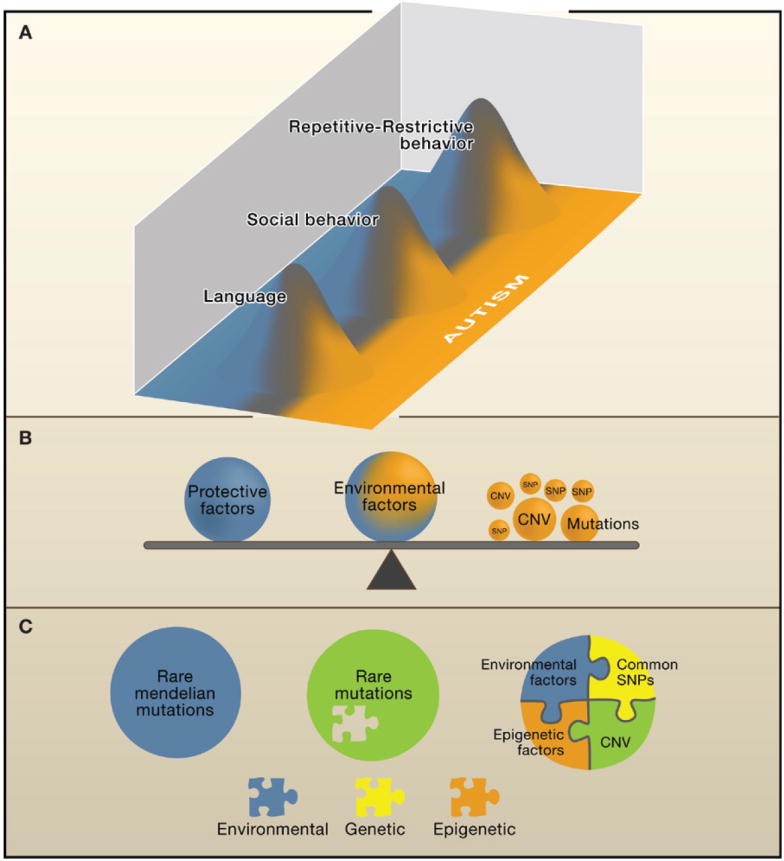
Susceptibility to autism spectrum disorder (ASD) may in some cases reflect the contribution of normal variation in heritable, potentially distinct core components of autism (endophenotypes). This simplified model (A) depicts how a variety of risk variants, large and small, may contribute to ASD in aggregate (B). The notion of hetereogeneous genetic mechanisms acting in different patients is captured by three individual examples depicted by circles (C).
(A) Normal variations in language, social behavior, and repetitive-restrictive behavior are illustrated as overlapping but with distinct functions (blue and orange demarcate the highest level of functioning and progressive abnormality, respectively). In this scheme, moderate abnormality in all three areas is necessary to be diagnosed with autism, but abnormality in one area, for example language, leads to a more circumscribed condition, such as specific language impairment. Patients with Asperger’s syndrome would have normal language but would lie in the orange zone for the other two components. Each of these heritable cognitive-behavioral components (endophenotypes), although genetically complex, is likely to be less heterogeneous than the syndrome of autism. These endophenotypes can be studied separately to increase power to detect common genetic risk variants.
(B) The contribution of genetic or environmental factors is emphasized by size or position of each representative weight. Mendelian mutations, such as de novo CNVs, are depicted by a large ball because they are considered causal in many cases and would tip the balance, placing an individual essentially into the orange zone in all categories shown in (A) (pleiotropy), resulting in autism.
The reality, however, is that there are few mutations that act alone or are fully causal (green circle in C.) Many mutations, including those that are recurrent, are not fully penetrant, and some patients have more than one de novo CNV. Similarly, most mutations, even rare mutations such as those in the gene encoding neuroligin 4, or recurrent mutations such as del (16p), show variable expressivity, resulting in developmental delay or mental retardation in some individuals and autism in others. So, the environment or other common variants that might modulate social behavior or language could be tipping the balance toward autism in a child who would otherwise have had mild or moderate mental retardation (blue circle in C). In other cases, multiple common variants, each with a small effect and related to different components, would push an individual toward the right of the distribution and along with environmental or epigenetic factors would result in an individual with ASD (multicolored circle in C). Each of these scenarios, or a combination of the three, may be acting in a given individual with autism. Defining the extent to which each contributes on a population level to ASD is a goal of current genetic research.
One sign of success comes from a recent high-density SNP association analysis of the chromosome 7q language QTL, in which a common allele of CNTNAP2 was associated with a language endophenotype and a 160 kb deletion in CNTNAP2 was detected in a single proband (Alarcón et al., 2008). Concurrently, another group discovered rare causal de novo chromosomal variation and point mutations in CNTNAP2 (Bakkaloglu et al., 2008), and yet another identified common variation in CNTNAP2 potentially associated with ASD (Arking et al., 2008), providing multiple converging lines of evidence for the involvement of CNTNAP2 in ASD. In addition, Strauss and colleagues had previously discovered a single rare recessive truncating mutation in CNTNAP2 that caused a syndrome of focal epilepsy and neuronal migration abnormalities in affected individuals in an Amish family (Strauss et al., 2006). Remarkably, the majority of affected children were also found to suffer from language delay and ASD, further supporting the role of CNTNAP2 genetic variation related to language systems that are disrupted in ASD. This work on CNTNAP2, in which variation in the same gene may lead to distinct clinical phenotypes, further emphasizes that current notions of disease status based on clinical diagnostic schema can create artificial boundaries between conditions that may share similar genetic underpinnings.
Connecting Genes to Brain and Behavior
Most mutations known to cause autism are de novo mutations often involving multiple genes or identified genetic syndromes. Common variants have been implicated in autism association, but most of these are either in small samples or have not been replicated. Nevertheless, common variations in several genes including EN2 (Benayed et al., 2005), the MET proto-oncogene (Campbell et al., 2006), and others in addition to CNTNAP2 either have been implicated in large samples or independently replicated. But none of these published associations individually account for a large fraction of the genetic risk for ASD.
Considering these common variants and the known rare mutations, autism susceptibility genes appear to have many distinct roles in neural development and neuronal function, ranging from basic metabolism, synaptic transmission, and RNA splicing to neuronal migration. Mutations in genes implicated in these functional categories clearly can cause ASD, but how? Do known mutations converge on a few common molecular pathways or do they represent diverse biological etiologies, and if so, how does disruption of such diverse functions result in the syndrome of autism? The answer to this question goes beyond the boundaries of the current data. Furthermore, several of the known autism genes including NLGN4 clearly cause mental retardation, and others such as the 16p11 CNV are associated with more general forms of developmental delay (Weiss et al., 2008), perhaps more frequently than they cause ASD. So how does disease specificity emerge?
Whatever the known molecular and biological functions of ASD susceptibility genes, they must converge on the disruption of function in brain regions supporting language, social cognition, and behavioral flexibility. This could involve focal gene expression of the specific gene product during development; when the risk allele is expressed, there is disruption of the cortical and subcortical brain networks supporting social responsiveness or language. Remarkably, this appears to be the case for CNTNAP2, which is enriched in highly evolved, anterior regions of the developing human cerebral cortex that overlap with circuitry involved in the development of joint attention (Alarcón et al., 2008), a social precursor to language that is one of the early behaviors disrupted in ASD.
However, most known ASD susceptibility genes do not demonstrate such regionally restricted expression, so other factors must also be operating. The core areas affected in autism involve rapid and coherent integration of information from multiple, higher-level association areas (Geschwind and Levitt, 2007). Such functions could be easily perturbed by minor, but relatively widespread disruptions in neural transmission, for example, due to either subtle mis-wiring or synaptic dysfunction. Circuit mis-wiring could be either local or long distance and could be caused by myriad conditions such as neuronal migration abnormalities, disrupted axon pathfinding, loss or dysfunction of local inhibitory connections, or immature synaptic function, all culminating in what has been referred to as a developmental disconnection (Geschwind and Levitt, 2007). Thus, one would expect to find subtle, widespread differences in many brain systems in subjects with ASD, even those serving primary sensory functions, although these may not be the direct cause of the core features of autism. Such abnormalities, however, may explain the differences in sensory processing, motor function, and sensory-motor integration, in addition to the more global processing differences that have been variably associated with ASD (e.g., Happe and Frith, 2006).
The concepts of focal versus diffuse circuit disruption are not mutually exclusive and both may cause different forms of ASD. Moreover, any unifying framework for understanding autism will necessarily involve testing hypotheses in autism cases with many distinct known etiologies. Now that we possess the tools to continue to identify the genes causing autism, the challenge is to integrate these findings with the study of cellular physiology and brain anatomy and function to bridge the gap between genes and cognition. Given the role of highly adapted language and social cognition systems in autism, we also need to clearly consider the role of human-specific cognitive specializations, carefully integrating model system data with studies in humans. From this perspective, autism is paradigmatic of the challenge facing those who wish to understand diseases affecting higher cognition—the challenge of integrating detailed molecular knowledge with complex circuit function in humans. As this challenge is met and our knowledge increases, leading to etiological understanding of the disorder, our concepts of disease boundaries are likely to change.
Acknowledgments
The author thanks R. Cantor and B. Abrahams for critical reading of the manuscript, G. Coppola for the Figure, L. Hong for editorial assistance, and many colleagues for their collaborations, especially the AGRE consortium and the AGP. We are grateful to the families who participate in AGRE and funding from NIMH (RO1 MH64547, U54 MH68172, R37 MH60233, P50 HD055784), Autism Speaks, and the Simons Foundation.
References
- Abrahams BS, Geschwind DH. Nat Rev Genet. 2008;9:341–355. doi: 10.1038/nrg2346. Erratum: (2008). Nat. Rev. Genet. 9, 493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcón M, Stone JL, Duvall JA, Abrahams BS, Sebat J, Wigler M, Nelson SF, Cantor RM, Geschwind DH. Am J Hum Genet. 2008;82:150–159. doi: 10.1016/j.ajhg.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arking DE, Cutler DJ, Brune CW, Teslovich TM, West K, Ikeda M, Rea A, Guy G, Lin S, Cook EHJ, et al. Am J Hum Genet. 2008;82:160–164. doi: 10.1016/j.ajhg.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badner JA, Gershon ES. Mol Psychiatry. 2002;7:56–66. doi: 10.1038/sj.mp.4000922. [DOI] [PubMed] [Google Scholar]
- Bakkaloglu B, O’Roak BJ, Louvi A, Gupta AR, Abelson JF, Morgan TM, Chawarska K, Klin A, Ercan-Sencicek G, Stillman AA, et al. Am J Hum Genet. 2008;82:165–173. doi: 10.1016/j.ajhg.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benayed R, Gharani N, Rossman I, Mancuso V, Lazar G, Kamdar S, Bruse SE, Tischfield S, Smith BJ, Zimmerman RA, et al. Am J Hum Genet. 2005;77:851–868. doi: 10.1086/497705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DB, Sutcliffe JS, Ebert PJ, Militerni R, Bravaccio C, Trillo S, Elia M, Schneider C, Melmed R, Sacco R, et al. Proc Natl Acad Sci USA. 2006;103:16834–16839. doi: 10.1073/pnas.0605296103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor RM, Kono N, Duvall JA, Alvarez-Retuerto A, Stone JL, Alarcón M, Nelson SF, Geschwind DH. Am J Hum Genet. 2005;76:1050–1056. doi: 10.1086/430278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor RM, Yoon JL, Furr J, Lajonchere CM. Mol Psychiatry. 2007;12:419–421. doi: 10.1038/sj.mp.4001966. [DOI] [PubMed] [Google Scholar]
- Geschwind DH, Levitt P. Curr Opin Neurobiol. 2007;17:103–111. doi: 10.1016/j.conb.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Geschwind DH, Sowinski J, Lord C, Iversen P, Shestack J, Jones P, Ducat L, Spence SJ. Am J Hum Genet. 2001;69:463–466. doi: 10.1086/321292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Happe F, Frith U. J Autism Dev Disord. 2006;36:5–25. doi: 10.1007/s10803-005-0039-0. [DOI] [PubMed] [Google Scholar]
- Jacquemont ML, Sanlaville D, Redon R, Raoul O, Cormier-Daire V, Lyonnet S, Amiel J, Le Merrer M, Heron D, de Blois MC, et al. J Med Genet. 2006;43:843–849. doi: 10.1136/jmg.2006.043166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YH, Sahoo T, Michaelis RC, Bercovich D, Bressler J, Kashork CD, Liu Q, Shaffer LG, Schroer RJ, Stockton DW, et al. Am J Med Genet A. 2004;131:1–10. doi: 10.1002/ajmg.a.30297. [DOI] [PubMed] [Google Scholar]
- Lupski JR. Nat Genet. 2007;39:S43–S47. doi: 10.1038/ng2084. [DOI] [PubMed] [Google Scholar]
- Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y. Am J Hum Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow EM, Yoo SY, Flavell SW, Kim TK, Lin Y, Hill RS, Mukaddes NM, Balkhy S, Gascon G, Hashmi A, et al. Science. 2008;321:218–223. doi: 10.1126/science.1157657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichenberg A, Gross R, Weiser M, Bresnahan M, Silverman J, Harlap S, Rabinowitz J, Shulman C, Malaspina D, Lubin G, et al. Arch Gen Psychiatry. 2006;63:1026–1032. doi: 10.1001/archpsyc.63.9.1026. [DOI] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, et al. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Maner S, Massa H, Walker M, Chi M, et al. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- Strauss KA, Puffenberger EG, Huentelman MJ, Gottlieb S, Dobrin SE, Parod JM, Stephan DA, Morton DH. N Engl J Med. 2006;354:1370–1377. doi: 10.1056/NEJMoa052773. [DOI] [PubMed] [Google Scholar]
- Szatmari P, Paterson AD, Zwaigenbaum L, Roberts W, Brian J, Liu XQ, Vincent JB, Skaug JL, Thompson AP, Senman L, et al. Nat Genet. 2007;39:319–328. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veenstra-Vanderweele J, Christian SL, Cook EH., Jr Annu Rev Genomics Hum Genet. 2004;5:379–405. doi: 10.1146/annurev.genom.5.061903.180050. [DOI] [PubMed] [Google Scholar]
- Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H, Ferreira MAR, Green T, et al. N Engl J Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- Zhao X, Leotta A, Kustanovich V, Lajonchere C, Geschwind DH, Law K, Law P, Qiu S, Lord C, Sebat J, et al. Proc Natl Acad Sci USA. 2007;104:12831–12836. doi: 10.1073/pnas.0705803104. [DOI] [PMC free article] [PubMed] [Google Scholar]