

Abstract
Chemoreceptor cells of the carotid bodies (CB) are activated by hypoxia and acidosis, responding with an increase in their rate of neurotransmitter release, which in turn increases the electrical activity in the carotid sinus nerve and evokes a homeostatic hyperventilation. Studies in isolated chemoreceptor cells have shown that moderate hypoxias (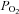 ≈ 46 mmHg) produces smaller depolarisations and comparable Ca2+ transients but a much higher catecholamine (CA) release response in intact CBs than intense acidic/hypercapnic stimuli (20% CO2, pH 6.6). Similarly, intense hypoxia (
≈ 46 mmHg) produces smaller depolarisations and comparable Ca2+ transients but a much higher catecholamine (CA) release response in intact CBs than intense acidic/hypercapnic stimuli (20% CO2, pH 6.6). Similarly, intense hypoxia (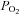 ≈ 20 mmHg) produces smaller depolarizations and Ca2+ transients in isolated chemoreceptor cells but a higher CA release response in intact CBs than a pure depolarizing stimulus (30–35 mm external K+). Studying the mechanisms responsible for these differences we have found the following. (1) Acidic hypercapnia inhibited ICa (∼60%; whole cell) and CA release (∼45%; intact CB) elicited by ionomycin and high K+. (2) Adenylate cyclase inhibition (SQ-22536; 80 μm) inhibited the hypoxic release response (>50%) and did not affect acidic/hypercapnic release, evidencing that the high gain of hypoxia to elicit neurotransmitter release is cAMP dependent. (3) The last effect was independent of PKA activation, as three kinase inhibitors (H-89, KT 5720 and Rp-cAMP; ≥ 10 × IC50) did not alter the hypoxic release response. (4) The Epac (exchange protein activated by cAMP) activator (8-pCPT-2′-O-Me-cAMP, 100 μm) reversed the effects of the cyclase inhibitor. (5) The Epac inhibitor brefeldin A (100 μm) inhibited (54%) hypoxic induced release. Our findings show for the first time that an Epac-mediated pathway mediates O2 sensing/transduction in chemoreceptor cells.
≈ 20 mmHg) produces smaller depolarizations and Ca2+ transients in isolated chemoreceptor cells but a higher CA release response in intact CBs than a pure depolarizing stimulus (30–35 mm external K+). Studying the mechanisms responsible for these differences we have found the following. (1) Acidic hypercapnia inhibited ICa (∼60%; whole cell) and CA release (∼45%; intact CB) elicited by ionomycin and high K+. (2) Adenylate cyclase inhibition (SQ-22536; 80 μm) inhibited the hypoxic release response (>50%) and did not affect acidic/hypercapnic release, evidencing that the high gain of hypoxia to elicit neurotransmitter release is cAMP dependent. (3) The last effect was independent of PKA activation, as three kinase inhibitors (H-89, KT 5720 and Rp-cAMP; ≥ 10 × IC50) did not alter the hypoxic release response. (4) The Epac (exchange protein activated by cAMP) activator (8-pCPT-2′-O-Me-cAMP, 100 μm) reversed the effects of the cyclase inhibitor. (5) The Epac inhibitor brefeldin A (100 μm) inhibited (54%) hypoxic induced release. Our findings show for the first time that an Epac-mediated pathway mediates O2 sensing/transduction in chemoreceptor cells.
Carotid bodies (CB) are small paired sensory organs located in the vicinity of the carotid artery bifurcation. These chemoreceptor organs are formed by clusters of parenchymatous cells (chemoreceptor and sustentacular) surrounded by a dense net of capillaries. Sensory nerve fibres from the carotid sinus nerve (CSN) form synaptic contacts with chemoreceptor cells. Functionally, CBs represent the main arterial chemoreceptor being responsible for the entire hyperventilation that occurs in hypoxic hypoxia (Lahiri, 1976; Honda, 1985) and contributing (∼25–40%) to the compensatory hyperventilation that occurs in situations of acidosis, both respiratory and metabolic in origin (Berkenbosch et al. 1979; Nattie, 1999). These reflex ventilatory responses are initiated at the level of the chemoreceptor cells, which represent the 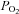 and [H+] sensing elements in this chemoreceptor organ (Fidone & Gonzalez 1986; Gonzalez et al. 1994). Chemoreceptor cells in response to hypoxia and acidosis increase their resting rate of release of neurotransmitters, which in turn augment the action potential frequency in the CSN; central projection of the CSN into the brainstem drives ventilation. Acidic stimuli might directly stimulate the sensory nerve endings of the CSN, this direct effect also contributing to increase the CSN frequency in situations of acidosis (Rigual et al. 1984).
and [H+] sensing elements in this chemoreceptor organ (Fidone & Gonzalez 1986; Gonzalez et al. 1994). Chemoreceptor cells in response to hypoxia and acidosis increase their resting rate of release of neurotransmitters, which in turn augment the action potential frequency in the CSN; central projection of the CSN into the brainstem drives ventilation. Acidic stimuli might directly stimulate the sensory nerve endings of the CSN, this direct effect also contributing to increase the CSN frequency in situations of acidosis (Rigual et al. 1984).
Acidic/hypercapnic stimuli, even the most intense stimuli tested (considered supramaximal) in normoxia and hyperoxia, elicit a level of activity in the CSN that oscillates between 25 and 40% of the maximal hypoxic activity (Fitzgerald & Parks, 1971; Lahiri & Delaney, 1975; Fitzgerald, 1976; Pepper et al. 1995). When the compound output of the CB chemoreceptor cells is measured as the rate of neurotransmitter release (e.g. CA release) from the intact organ, the difference is even more striking, as for example a supramaximal acidic/hypercapnic stimulus (20% CO2-equilibrated solution/pH 6.6) causes a release response 3–4 times smaller than that elicited by a hypoxic stimulus of moderate intensity (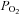 ≈ 46 mmHg) (Rigual et al. 1986vs. 1991; Vicario et al. 2000b; Rocher et al. 2005). In contrast to the integrated responses of the CB, i.e. CSN activity and release of CA, chemoreceptor cell depolarizations produced by intense hypoxia (
≈ 46 mmHg) (Rigual et al. 1986vs. 1991; Vicario et al. 2000b; Rocher et al. 2005). In contrast to the integrated responses of the CB, i.e. CSN activity and release of CA, chemoreceptor cell depolarizations produced by intense hypoxia (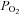 ≈ 20–30 mmHg) are smaller (6.8 ± 0.5 mV) than those produced by acidic/hypercapnic stimulus (20% CO2, pH 6.6; 11.8 ± 0.7 mV) in isolated rabbit chemoreceptor cells (Rocher et al. 2005; see Buckler & Vaughan-Jones, 1994a,b; for data in the rat). Yet, the increases in intracellular Ca2+ produced by intense hypoxia and hypercapnic/acidic stimuli are comparable (Buckler & Vaughan-Jones, 1994a,b; Dasso et al. 2000). Mechanisms causing these variable relationships between cell depolarization and intracellular Ca2+ levels on the one hand and the intensity of exocytosis on the other are unknown. In addition, a moderate pure depolarizing stimulus (30–35 mm external K+) elicits a CA release response comparable to that elicited by moderate hypoxia (
≈ 20–30 mmHg) are smaller (6.8 ± 0.5 mV) than those produced by acidic/hypercapnic stimulus (20% CO2, pH 6.6; 11.8 ± 0.7 mV) in isolated rabbit chemoreceptor cells (Rocher et al. 2005; see Buckler & Vaughan-Jones, 1994a,b; for data in the rat). Yet, the increases in intracellular Ca2+ produced by intense hypoxia and hypercapnic/acidic stimuli are comparable (Buckler & Vaughan-Jones, 1994a,b; Dasso et al. 2000). Mechanisms causing these variable relationships between cell depolarization and intracellular Ca2+ levels on the one hand and the intensity of exocytosis on the other are unknown. In addition, a moderate pure depolarizing stimulus (30–35 mm external K+) elicits a CA release response comparable to that elicited by moderate hypoxia (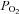 ∼ 46 mmHg; Rocher et al. 2005) but an intracellular Ca2+ rise several times higher than that produced by hypoxia even of high intensity (Buckler & Vaughan-Jones, 1994a,b; Sanchez et al. 2002). Again, mechanisms for the large gain of hypoxic stimulus to elicit neurotransmitter release are unknown. In sum the level of chemoreceptor cell depolarization elicited by hypoxia is smaller than that elicited by acidic/hypercapnic stimuli and both stimuli generate comparable Ca2+ transients; however, more integrated responses elicited by hypoxia, i.e. release of neurotransmitters or neural activity in the CSN, are much greater than those elicited by acidic/hypercapnic stimuli. The general aim of present study has been to find the mechanisms involved in these disparate responses.
∼ 46 mmHg; Rocher et al. 2005) but an intracellular Ca2+ rise several times higher than that produced by hypoxia even of high intensity (Buckler & Vaughan-Jones, 1994a,b; Sanchez et al. 2002). Again, mechanisms for the large gain of hypoxic stimulus to elicit neurotransmitter release are unknown. In sum the level of chemoreceptor cell depolarization elicited by hypoxia is smaller than that elicited by acidic/hypercapnic stimuli and both stimuli generate comparable Ca2+ transients; however, more integrated responses elicited by hypoxia, i.e. release of neurotransmitters or neural activity in the CSN, are much greater than those elicited by acidic/hypercapnic stimuli. The general aim of present study has been to find the mechanisms involved in these disparate responses.
In the present study we have performed electrophysiological and neurochemical experiments aimed at providing answers to the preceding questions. Among others, specific questions addressed in our study include: why do acidic/hypercapnic stimuli which produce larger depolarizations than intense hypoxia cause comparable Ca2+ transients? Conversely, why do hypoxic stimuli elicit a stronger release response than intense acidic stimuli for the same Ca2+ signal? Why do moderate hypoxic stimuli elicit a comparable release response to 30 mm K+ if they generate a weaker intracellular Ca2+ signal? Our data indicate that the weak response to the acidic stimuli is due mainly to a direct inhibition of the exocytotic machinery by the high H+ concentrations. The higher capacity of hypoxia to elicit neurotransmitter release is due to the activation of production of cAMP, which activates exocytosis by an Epac-mediated PKA-independent mechanism.
Methods
Animals and surgery
Adult New Zealand White rabbits (1.5–2.5 kg) were anaesthetized with sodium pentobarbital (40 mg kg−1; i.v., lateral vein of the ear). Animals were tracheostomized and, after adequate dissections, bilateral blocks of tissue containing the carotid bifurcations were removed and placed in a lucite chamber filled with ice cold O2-saturated Tyrode solution (in mm: NaCl, 140; KCl, 5; CaCl2, 2; MgCl2, 1.1; Hepes, 10; glucose, 5.5; pH 7.40). The CBs were identified and cleaned of surrounding tissues under a dissecting microscope. Cleaned CBs were saved in independent vials containing fresh Tyrode solution at 0–4°C until the 8–12 CBs used in a given experiment were collected. Animals were killed with an intracardiac overdose (150 mg (kg weight)−1) of sodium pentobarbital. Experimental procedures were approved by the Institutional Animal Care and Use Committee of the Universities of Valladolid and Miguel Hernandez.
[3H]Catecholamine release experiments and analytical procedures
Groups of six CBs were incubated during 2 h in small glass vials placed in a metabolic shaker (37°C) containing 0.5 ml of Tyrode solution supplemented with 20 μm[3H]tyrosine ([3–5-3H]tyrosine; 30 Ci mmol−1; Amersham Iberica, Madrid, Spain), 1 mm ascorbic acid and 100 μm 6-methyl-tetrahydropterine (Sigma, Madrid, Spain), cofactors of dopamine-β-hydroxylase and tyrosine hydroxylase, respectively (Fidone & Gonzalez, 1982).
At the end of this period of incubation to label CA stores with 3H tyrosine, organs were transferred to individual vials containing 4 ml of [3H]tyrosine-free Tyrode bicarbonate solution (composition as above except for equimolar replacement of 24 mm NaCl with NaHCO3), which, under continuous bubbling with water vapour saturated 20% O2–5% CO2, balance N2, yields a pH of 7.4. For a period of 2 h incubating solutions were renewed every 30 min and discarded, eliminating in this manner the labelled precursor and the readily releasable pool of [3H]CA (Almaraz et al. 1986). Thereafter, incubations continued and the incubating solutions were collected every 10 min for analysis of their [3H]CA content. For most of the experiments, half of the CBs used in a given experiment were used as controls, being subjected to two consecutive identical stimuli (S1 and S2, respectively), consisting of the incubation of the organs in solutions with a low 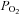 (equilibrated with 7% O2–5% CO2–88% N2 or 2% O2–5% CO2–93 N2). In K+-rich solutions, equimolar amounts of Na+ were removed to maintain the osmolarity, and in solutions with high
(equilibrated with 7% O2–5% CO2–88% N2 or 2% O2–5% CO2–93 N2). In K+-rich solutions, equimolar amounts of Na+ were removed to maintain the osmolarity, and in solutions with high 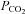 and low pH (equilibrated with 20% O2–20% CO2–60% N2), the desired pH of 7.00, 6.80 or 6.60 was attained by varying the NaHCO3 concentration and adjusting the NaCl accordingly to maintain osmolarity. To study the interactions between hypoxic and acidic/hypercapnic stimuli, or between high K+ and acidic/hypercapnic stimuli, both pairs of stimuli were combined correspondingly (see Results). The other half of the CBs in any given experiment (experimental) were similarly incubated and stimulated except for the presence in the S2 incubation of a variable with respect to the corresponding controls. The use of specific drugs, as well as their protocols of application, will also be described in Results.
and low pH (equilibrated with 20% O2–20% CO2–60% N2), the desired pH of 7.00, 6.80 or 6.60 was attained by varying the NaHCO3 concentration and adjusting the NaCl accordingly to maintain osmolarity. To study the interactions between hypoxic and acidic/hypercapnic stimuli, or between high K+ and acidic/hypercapnic stimuli, both pairs of stimuli were combined correspondingly (see Results). The other half of the CBs in any given experiment (experimental) were similarly incubated and stimulated except for the presence in the S2 incubation of a variable with respect to the corresponding controls. The use of specific drugs, as well as their protocols of application, will also be described in Results.
Immediately upon collection, incubating solutions were acidified to pH 3–3.5 with glacial acetic acid and saved at 4°C to prevent degradation of the released [3H]CA. The analysis of incubating solutions for their [3H]CA content consisted of specific adsorption into alumina of the labelled catechols at pH 8.6, their elution with 1 m HCl and quantification by liquid scintillation spectrometry (Almaraz et al. 1986; Obeso et al. 1992). At the end of the experiments, the CBs were homogenized in 300 μl 0.4 m perchloric acid, centrifuged for 5 min in a microfuge (Beckman) and the [3H]CA present in the supernatants was analysed as incubating solutions.
The stimulus-evoked release of [3H]CA in S1 and S2 was calculated as counts per minute (c.p.m.) above basal release, as shown graphically in Figs 1B and 2, and expressed as a percentage of the [3H]CA present in the organ immediately before the application of the stimulus. Since there is a time-dependent decay in the absolute amount of [3H]CA released, the ratios of the evoked [3H]CA release in S2 to that in S1 (S2/S1) were calculated, and the S2/S1 ratios obtained in control and experimental CBs were compared. Statistical significance of the observed differences was assessed using Student's two-tailed t test for unpaired data; the significance level was established at P < 0.05. There were no statistically significant differences in the magnitude of S1 evoked release between control and experimental CB in any experimental condition. Results are expressed as means ±s.e.m.
Figure 1. Effect of hypoxia and hypercapnic acidosis on chemoreceptor cell membrane potential and on the [3H]CA release from in vitro CB.

A, a sample record (perforated-patch) of membrane potential changes elicited by moderate hypoxia (7% O2, 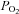 ≈ 46 mmHg) and intense hypercapnic acidosis (20% CO2, pH 6.6) in a dissociated chemoreceptor cell; the remaining time the cell was superfused with control solution (20% O2-5% CO2–75% N2; pH 7.40). B, mean depolarizations produced by moderate hypoxia and intense hypercapnic acidosis (n= 7 and 12, respectively). C, the time course of stimulus-elicited [3H]CA release from two groups of 6 CBs. Stimulus (7% O2 or 20% CO2, pH 6.6) was applied for 10 min as indicated in the drawing. D, total release response evoked (equivalent to c.p.m. above dotted line in C) by moderate hypoxia, intense acidic stimuli and 30 mm K+. Data represent percentage of tissue CA content and are means ±s.e.m. for 6 CBs.
≈ 46 mmHg) and intense hypercapnic acidosis (20% CO2, pH 6.6) in a dissociated chemoreceptor cell; the remaining time the cell was superfused with control solution (20% O2-5% CO2–75% N2; pH 7.40). B, mean depolarizations produced by moderate hypoxia and intense hypercapnic acidosis (n= 7 and 12, respectively). C, the time course of stimulus-elicited [3H]CA release from two groups of 6 CBs. Stimulus (7% O2 or 20% CO2, pH 6.6) was applied for 10 min as indicated in the drawing. D, total release response evoked (equivalent to c.p.m. above dotted line in C) by moderate hypoxia, intense acidic stimuli and 30 mm K+. Data represent percentage of tissue CA content and are means ±s.e.m. for 6 CBs.
Figure 2. Effects of PKA inhibition on the release of [3H]CA elicited by moderate hypoxia.

A and B represent mean time courses of the release obtained in 6 control CBs challenged twice with a hypoxic stimulus (7% O2-equilibrated solutions), and in 6 experimental CBs similarly stimulated except that 30 min prior to and during the second hypoxic challenge, 1 μm H-89 was present in the incubating solution. C, results of similar experiments using the three indicated PKA inhibitors; bars represent ratios of total evoked release obtained in the second challenge to that obtained in the first one (S2/S1) for control (open bars) and experimental CBs (grey bars). Values are means ±s.e.m. for 6–8 CBs.
Cell preparation and electrophysiological recordings
Chemoreceptor cells were obtained from rabbit CBs dissected as in the release experiments. The CBs were incubated (30 min) in nominally Ca2+- and Mg2+-free Tyrode solution (pH 7.2) containing collagenase (2.5 mg ml−1, type IV, Sigma) and bovine serum albumin (6 mg ml−1, Fraction V, Sigma). After incubation tissues were centrifuged (800 g, 5 min) and the pellet was resuspended in a new solution containing collagenase (1 mg ml−1), trypsin (1 mg ml−1, type II, Sigma) and bovine serum albumin (6 mg ml−1) and incubated for an additional 15 min period. Tissues were subjected to mechanical disruption every 10 min by repeated aspiration through a fire-polished Pasteur pipette during both incubation periods. Finally, in the second incubation, dissociated cells were pelleted by centrifugation (800 g, 8 min), washed in an enzyme-free Tyrode solution and resuspended in 100 μl of culture medium (Dulbecco's modified Eagle's medium (DMEM)–F-12; Sigma), supplemented with 5% fetal bovine serum, 2 mm l-glutamine, 100 μg ml−1 streptomicin and 40 μg ml−1 gentamicin. Drops of cell suspension of 20 μl were plated on small poly-l-lysine-coated coverslips kept in 3.5 cm diameter Petri dishes in a humidified incubator (37°C; 5% CO2 in air). Once the cells attached (around 60 min), 2 ml of culture medium was added to the Petri dish to maintain the cells until use (3–36 h later). Coverslips were transferred to the recording chamber (0.15 ml volume; Warner Instruments LLC, Hamden, CT, USA) on the stage of an inverted microscope (Nikon Diaphot-TMD) and superfused by gravity (1.5–3 ml min−1).
The recordings of membrane potential in chemoreceptor cells were performed using the perforated-patch configuration and at 33–35°C. Bath solutions were bicarbonate-buffered and identical to those used in the [3H]CA release experiments (see above). Pipette (borosilicate glass; 1.5 mm o.d.; Clark Electromedical Instruments) solution was (in mm): KCl, 35; potassium gluconate, 95; MgCl2, 3; EGTA, 5; Hepes, 10; pH was adjusted to 7.2 by addition of NaOH (final sodium concentration 14 mm). Nystatin was added at a final concentration of 100–150 μg ml−1 (Albillos et al. 2000). Resistance of pipettes filled with internal solution was 2.0–3.5 MΩ. Voltage signals were recorded with an EPC-7 amplifier (List Medical, Darmstadt, Germany) or an RK 300 amplifier (BioLogic, Claix, France). Pulse generation, data acquisition and analysis were made through an A/D converter (CED 1401, Cambridge Electronic Design, Cambridge, UK) or through a Digidata 1322A (Axon Instruments, Union City, CA, USA) commanded by the software package ‘Strathclyde Electrophysiology Software’ (kindly provided by J. Dempster, Strathclyde University) or pCLAMP software (Axon Instruments). Voltage recordings were filtered at 2 kHz and sampled at 10–16 kHz.
Recordings of calcium currents in chemoreceptor cells were performed at room temperature (20–23°C) using the patch-clamp technique in the whole-cell configuration. When filled with intracellular solution (in mm: CsCl, 130; MgCl2, 2; Hepes, 10; ethylene glycol-bis(-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 10; ATP, 4; and GTP 2; pH 7.2 with CsOH), pipette resistance was 5–6 MΩ. Bath extracellular solutions containing 2 mm Ca2+ were identical to those used in the CA release experiments, but contain 5 × 10−7m tetrodotoxin to block voltage-dependent sodium channels (TTX, Alomone Laboratories, Jerusalem, Israel). Calcium currents were elicited by depolarizing cells to −10 mV from a holding potential of −80 mV every 10 s (pulse duration 20 ms). The effect of hypercapnic acidosis on voltage-dependent calcium currents was tested in standard conditions as well as during superfusion of the cells in the presence of 10−6m Bay K 8644 (Sigma), an activator of L-type calcium channels. Amplitudes of leak-subtracted Ca2+ currents were measured at the end of the voltage pulse and are expressed as means ±s.e.m.
Chemicals
[3–5-3H]Tyrosine was from Amersham Iberica (Madrid, Spain); 6-methyl-tetrahydropterine, collagenase (type IV), trypsin (type II), nystatin, ionomycin, Bay K 8644, SQ-22536, H-89, KT 5720, Rp-cAMP, 8-pCPT-2′-O-Me-cAMP and brefeldin A were all from Sigma (St Louis, MO, USA).
Results
Effect of hypoxia and hypercapnic acidosis on chemoreceptor cell membrane potential. Comparison with the [3H]CA release response
The magnitude of rabbit chemoreceptor cell depolarization induced by moderate hypoxia (superfusion with 7% O2-equilibrated solution) and intense hypercapnic acidosis (superfusion with 20% CO2 solution; pH 6.6) was studied in perforated-patch recorded cells. Some cells discharged spontaneous fast action potentials of 50–80 mV in amplitude in resting normoxic conditions (perfusing solution equilibrated with 20% O2–5% CO2; 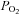 ≈ 136–140 mmHg; Fig. 1A), and after switching the perfusion from normoxia to a moderate hypoxia (equilibrated with 7% O2;
≈ 136–140 mmHg; Fig. 1A), and after switching the perfusion from normoxia to a moderate hypoxia (equilibrated with 7% O2; 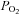 ≈ 46 mmHg) there was a small depolarization accompanied in some cells by an increase in the action potential frequency that did not follow a uniform pattern. On perfusing the cells with a hypercapnic/acidic solution the magnitude of depolarization as well as the spiking frequency was greater than with moderate hypoxia. Mean depolarizations observed were, respectively, 2.5 ± 0.5 and 12.0 ± 0.7 mV in moderate hypoxia and intense hypercapnic acidosis (n= 7 and 12 respectively; Fig. 1B), with resting membrane potential in the recorded cells oscillating between −40 and −55 mV. It has been reported previously that hypoxia of higher intensity (
≈ 46 mmHg) there was a small depolarization accompanied in some cells by an increase in the action potential frequency that did not follow a uniform pattern. On perfusing the cells with a hypercapnic/acidic solution the magnitude of depolarization as well as the spiking frequency was greater than with moderate hypoxia. Mean depolarizations observed were, respectively, 2.5 ± 0.5 and 12.0 ± 0.7 mV in moderate hypoxia and intense hypercapnic acidosis (n= 7 and 12 respectively; Fig. 1B), with resting membrane potential in the recorded cells oscillating between −40 and −55 mV. It has been reported previously that hypoxia of higher intensity (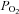 ≈ 15–30 mmHg) produced more intense depolarizations of rabbit chemoreceptor cells (7–13 mV; see Perez-García et al. 2000; Rocher et al. 2005). In rabbit chemoreceptor cells as well as in rat chemoreceptor cells (e.g. Buckler & Vaughan-Jones, 1994a,b; Zhong et al. 1997), their spiking behaviour in response to these stimuli was variable, with some cells firing at relatively high frequencies and some others not spiking at all (Perez-García et al. 2000; Rocher et al. 2005).
≈ 15–30 mmHg) produced more intense depolarizations of rabbit chemoreceptor cells (7–13 mV; see Perez-García et al. 2000; Rocher et al. 2005). In rabbit chemoreceptor cells as well as in rat chemoreceptor cells (e.g. Buckler & Vaughan-Jones, 1994a,b; Zhong et al. 1997), their spiking behaviour in response to these stimuli was variable, with some cells firing at relatively high frequencies and some others not spiking at all (Perez-García et al. 2000; Rocher et al. 2005).
Figure 1C shows the mean time course of the release of [3H]CA elicited by the moderate hypoxic and the intense hypercapnic/acidic stimulus. Both stimuli caused a peak [3H]CA release during the period of the stimulus application that was higher with the hypoxic stimulus, and for both stimuli, the [3H]catechols present in the incubating solutions decreased in the post-stimulus periods to basal levels. Figure 1D shows the magnitude of the evoked response, it being evident that moderate hypoxia elicited a release response considerably more intense (3.10 ± 0.23% of total [3H]CA present in the tissue) than hypercapnic acidosis (0.75 ± 0.08% of total [3H]CA present in the tissue). Thus, moderate hypoxia which produced a smaller depolarization and a smaller intracellular Ca2+ rise than intense hypercapnic acidosis (Buckler & Vaughan-Jones, 1994a,b;) generated a more intense [3H]CA release response. Figure 1D also shows that depolarising chemoreceptor cells with 30 mm K+ elicited a release response comparable to that elicited by 7% O2 and less than half of that elicited by intense hypoxia (∼22 mmHg; Rocher et al. 2005).
Exocytosis of [3H]CA elicited by hypoxia in chemoreceptor cells is enhanced by an adenylate cyclase-dependent and protein kinase A (PKA)-independent mechanism
Hypoxias of moderate intensities producing intracellular Ca2+ levels smaller than those generated by intense hypercapnic acidosis caused a much greater [3H]CA release response. Since hypoxia increases cAMP levels in the CB and agents elevating cAMP levels in the CB increase the release of [3H]CA elicited by hypoxia (Perez-García et al. 1990, 1991), we tested the possibility of this effect being mediated by a PKA-dependent mechanism. Figure 2A shows the mean time course of the release of [3H]CA induced by two consecutive applications of a moderate hypoxic stimulus in control CBs, and Fig. 2B shows identical experiments in which prior to and during the second hypoxic stimulation CBs were incubated in the presence of 1 μm of H-89, a highly specific inhibitor of PKA, at concentration ∼20 times IC50 (IC50= 50 nm). There were no evident alterations either in the time course of the release caused by the PKA inhibitor or in the intensity of the release response (Fig. 2C) since the ratios of the evoked release in the second to the first stimulus (S2/S1 ratios) were not different in control and drug treated organs. Similarly, the widely used PKA inhibitors KT 5720 (IC50= 56 nm) and Rp-cAMP (IC50= 15 μm) at concentrations over 20 times the IC50 lacked significant effects on the [3H]CA release response elicited by hypoxia (Fig. 2C). These and previously published observations (Perez-García et al. 1991) led to the next group of experiments in which we tested the effects of an adenylate cyclase inhibitor.
Figure 3A and B compares the time courses of the release of [3H]CA elicited by hypoxia in the absence and in the presence of the adenylate cyclase inhibitor SQ-22536 (80 μm) evidencing that the drug caused a decrease in the intensity of the [3H]CA release response, without altering the time course. The magnitude of the inhibition of the response is best appreciated by comparing the S2/S1 ratios for control and SQ-22536-treated CBs; the adenylate cyclase inhibitor reduced by nearly 50% the response elicited by the hypoxic stimulus (P < 0.001; Fig. 3D). These findings and those presented in Fig. 2 indicated that cAMP was producing its potentiating effects by a mechanism independent of the classical PKA signalling cascade, and this led us to search for the possible involvement of the recently described cascade mediated by exchange proteins directly activated by cAMP (Epacs; Seino & Shibasaki, 2005; Holz et al. 2006). Then, in a new group of experiments we compared the effects of the cyclase inhibitor (SQ-22536, 80 μm; IC50= 20 μm) with the effects of the cyclase inhibitor plus the Epac activator (8-pCPT-2′-O-Me-cAMP, 0.1 mm; IC50= 2.2 μm) applied simultaneously (Fig. 3C and D). This showed that the Epac activator more than reversed the effect of the adenylate cyclase inhibitor (P < 0.05 when compared with controls). In an additional group of experiments with moderate hypoxia as stimulus, we tested the effect of the 8-pCPT-2′-O-Me-cAMP alone and found that it produced a non-significant tendency to increase the response elicited by the hypoxic stimulus (Fig. 3D). The non-significant effect of the Epac activator was expected, because at this moderate level of hypoxic stimulation the increase in endogenous cAMP is maximal (Perez-García et al. 1990) and therefore the endogenous activation of the Epac pathway should be maximal or near maximal. Finally, using identical protocols we also tested the cyclase inhibitor (SQ-22536, 80 μm) on the release induced by the acidic/hypercapnic stimulus, and found that it did not cause modifications in the time course of the release (not shown) and nor did it alter the magnitude of the response (Fig. 3D), a finding that is consistent with the observation that acidic stimuli per se do not increase the cAMP levels (Perez-García et al. 1990). Thus, this group of experiments indicates the existence in chemoreceptor cells of Epac-mediated mechanism(s) acting as enhancer(s) of the release response to hypoxic stimuli.
Figure 3. Effect of the adenylate cyclase inhibitor SQ-22536, the Epac activator 8-pCPT-2′-O-Me-cAMP and the Epac inhibitor brefeldin A on the release of [3H]CA induced by low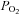 and acidic/hypercapnic stimuli.
and acidic/hypercapnic stimuli.

A and B, the mean time course of release obtained in 6 control CBs challenged twice with a hypoxic stimulus (7% O2-equilibrated solutions; black bars) as drawn, and in 6 experimental CBs similarly stimulated except that 20 min prior to and during the second hypoxic challenge and first post-stimulus, 80 μm SQ-22536 was present in the incubating solution. C, the mean time course of release obtained in 6 control CBs challenged twice with a hypoxic stimulus (7% O2-equilibrated solutions) as drawn. Incubation solutions in control CBs contained 80 μm SQ-22536 during the period marked for drugs, and experimental CB incubation solution contained 80 μm SQ-22536 + 0.1 mm 8-pCPT-2′-O-Me-cAMP during the same period. D, bars represent the release evoked by hypoxia (calculated as in Fig. 2) in control (open bars) and experimental CB as labelled in the drawing (filled bars) calculated from CB in A–C. The figure also shows mean data obtained in eight individual experiments using the Epac inhibitor brefeldin A (100 μm) using protocols comparable to those in B. D also presents mean data obtained for hypercapnic acidosis with protocols identical to those described in A and B. Values are means ±s.e.m. for 6–10 CBs (*P < 0.05; ***P < 0.001).
A confirmation of the participation of Epacs was obtained with the use of brefeldin A, an Epac-signalling inhibitor (Huang & Hsu, 2006; Ster et al. 2009). As shown in Fig. 3D, brefeldin A inhibited the release induced by a moderate hypoxic stimulus by about the same percentage (54%) as the inhibition of adenylate cyclase.
Hypercapnic acidosis inhibits exocytosis in chemoreceptor cells
As shown in the previous figures, even after inhibiting adenylate cyclase the release response elicited by a hypoxic stimulus of moderate intensity is higher than that elicited by a acidic/hypercapnic stimulus of supramaximal intensity. This observation lead us to suspect that hypercapnic–acidosis might have a dual effect: as a stimulus to chemoreceptor cells it would tend to promote a Ca2+-dependent release of CA (Rigual et al. 1991; Vicario et al. 2000b; Rocher et al. 2005), but at the same time, it would tend to inhibit the exocytotic machinery, with the net result of a limited release response. To directly test the hypothesis we performed the experiments shown in Fig. 4 using ionomycin (20 μm) and high external K+ (80 mm) as releasing stimuli. Ionomycin is a Ca2+ ionophore which promotes Ca2+-dependent transmitter release without the need of depolarization and participation of voltage operated Ca2+ channels (Congar et al. 2002), while 80 mm K+ causes release by activating all subtypes of voltage-dependent Ca+ channels (Rocher et al. 2005). In Fig. 4A and B are shown, respectively, the results obtained in a group of 10 control and 10 experimental ionomycin-treated CBs. In control organs the ionophore promoted a prompt release response that subsided very slowly due to the long wash-out time of the ionophore (Sanz-Alfayate et al. 2001), while in experimental organs, in which the incubation with ionomycin was carried out in acidic/hypercapnic solution, the release was greatly inhibited, appearing a frank release response only during the wash-out period after returning to normal pH solution. The percentage of inhibition if evaluated during the period of ionomycin application reached 98.3 ± 12.7% (P < 0.001) and if evaluated during the period of ionomycin application and wash-out periods it represented an inhibition of 45 ± 7% (P < 0.001). Figure 4C and D shows the data obtained with 80 mm external K+. Note (Fig. 4C) that the acidic/hypercapnic solution did not alter the time course of the release response elicited by 80 mm K+, but markedly reduced the intensity of the response. In fact, as shown in Fig. 4D, the inhibition reached 42% (P < 0.01).
Figure 4. Effect of hypercapnic acidosis on the release of [3H]CA by chemoreceptor cells evoked by a calcium ionophore and by 80 mm external K+.

A and B, the mean time courses (n= 10) of [3H]CA release induced by ionomycin (20 μm, 20 min) in control and acidic incubation media. C, mean time courses (n= 6) of the release of [3H]CA elicited by two consecutive applications of 80 mm K+ in control CBs (continuous line) and in experimental CBs (dashed line); in this last group the pH of the incubation solution during the second presentation of 80 mm K+ was 6.60 with 20% CO2. D, the S2/S1 ratios for the 80 mm K+ evoked release in control (open bars) and experimental (filled bar) CBs. Data are means ±s.e.m. (n= 6 CB; **P < 0.01).
Effects of hypercapnic acidosis on Ca2+ currents in isolated chemoreceptor cells
The specific effects of the Epac signalling pathways for hypoxic stimuli and the potent and direct inhibition of the exocytotic machinery exerted by acidic/hypercapnic stimuli evidenced in the experiments just described would satisfactorily explain why acidosis promotes a much weaker release response than hypoxia, for comparable levels of Ca2+. However, there is an additional aspect requiring experimental exploration, namely why acidic stimuli cause Ca2+ transients comparable to intense hypoxia when they generate larger depolarizations. Two hints have directed our experiments to explore the possibility that acidic stimuli are inhibiting Ca2+ currents. First, cAMP does not activate Ca2+ currents in chemoreceptor cells (Lopez-Lopez et al. 1993), so that a specific superactivation of Ca2+ currents by hypoxic stimuli does not appear to be involved. Secondly, Ca2+ entry into chemoreceptor cells during hypoxic and acidic/hypercapnic stimulation occurs mostly via L-type Ca2+ channels (Buckler & Vaughan-Jones, 1994a,b; Rocher et al. 2005), it being well known that these channels are inhibited by low external and internal pH (Takahashi et al. 1993; Klöckner & Isenberg, 1994). Therefore, in the following experiments we tested the hypothesis that the acidic stimuli in spite of producing larger depolarizations than hypoxia cause comparable Ca2+ transients because low pH inhibits Ca2+ currents.
Figure 5A shows a record of the Ca2+ current amplitude obtained at −10 mV in one cell in control, intense acidic/hypercapnic and again in control solution. It is evident that the acidic solution inhibited Ca2+ current in a reversible manner (see inset). In a total of 12 chemoreceptor cells recorded with this protocol we found that mean control Ca2+ current amplitude at −10 mV was 135.6 ± 9.5 pA and that hypercapnic acidosis produced a reversible decrease in the current to 48.0 ± 3.1 pA, representing an inhibition of 63.0 ± 3.4% (P < 0.001) (Fig. 5B). In an additional group of five cells, mean Ca2+ current was 119.0 ± 15.4 pA and Bay-K increased it to 168.0 ± 25.1 pA (41.4% increase; P < 0.01); in the same five cells hypercapnic acidosis reduced the current to 51.8 ± 9.0 pA (57% inhibition) and to 70.8 ± 8.7 (58% inhibition; P < 0.01), respectively, in untreated and Bay-K treated conditions. Thus, the percentage of current increase produced by Bay-K was nearly identical in normal pH and hypercapnic acid solutions (41.4 vs. 44.4%), and the percentage inhibition in both cases also was nearly identical, ∼57% (Fig. 5B), and very similar to that observed in the previous group of cells. The findings indicate that hypercapnic acidosis inhibits in a comparable extent L-type and the rest of Ca2+ channel subtypes because at the recording potential all Ca2+ channels are recruited (Rocher et al. 2005).
Figure 5. Effect of hypercapnic acidosis on chemoreceptor cell calcium currents.

A, time course of calcium current amplitude evoked in a chemoreceptor cell by application of successive voltage pulses to −10 mV (pulse duration 20 ms; pulse frequency 0.1 Hz; Vhold=−80 mV) during superfusion with control and acidic/hypercapnic solutions (20% O2–5% CO2 and 20% O2–20% CO2 equilibrated media, respectively). The inset shows sample current traces at the times indicated (1, 2 and 3). B, mean calcium current amplitude found in 12 chemoreceptor cells recorded with the protocol shown in A (left two bars). Right four bars show mean current amplitudes obtained in 5 chemoreceptor cells recorded with an identical protocol in the absence and presence of 1 μm Bay K 8644. Data are means ±s.e.m.; ***P < 0.001.
Discussion
In perforated-patch recordings we have confirmed the higher efficacy of hypercapnic acidosis (20% CO2, pH 6.6) compared to moderate hypoxia (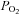 ≈ 46 mmHg) in depolarizing isolated rabbit chemoreceptor cells, and in neurochemical experiments we demonstrate that the acidic/hypercapnic stimulus is far less efficient than moderate hypoxia in promoting exocytotic release of [3H]CA (Obeso et al. 1992). Finding that a cAMP-mediated mechanism potentiates the hypoxic release and not the acidic/hypercapnic release, and that hypercapnic acidosis inhibits exocytosis can satisfactorily explain these opposite relationships, i.e. high membrane depolarization/low exocytosis of [3H]CA for the acidic/hypercapnic stimulus and vice versa for the hypoxic stimulus. The finding that hypercapnic acidosis inhibits Ca2+ currents provides an explanation for previous findings from other laboratories showing that the levels of intracellular Ca2+ produced by hypoxia and hypercapnic acidosis are comparable, even though the acidic stimulus causes a much higher depolarization. The spiking behaviour of chemoreceptor cells will be omitted in the discussion due to its variability from cell to cell. However, in spiking cells, the frequency of spikes parallels the level of depolarization (see Fig. 1A; see also Rocher et al. 2005), and therefore that omission will not detract from our considerations. As a final consideration in this summary, we want to state that our finding indicate that hypoxia as a depolarizing stimulus triggers release of CA, but as a result of its activation of adenylate cyclase and the Epac pathway, releases an amount of CA much larger than expected solely from depolarization.
≈ 46 mmHg) in depolarizing isolated rabbit chemoreceptor cells, and in neurochemical experiments we demonstrate that the acidic/hypercapnic stimulus is far less efficient than moderate hypoxia in promoting exocytotic release of [3H]CA (Obeso et al. 1992). Finding that a cAMP-mediated mechanism potentiates the hypoxic release and not the acidic/hypercapnic release, and that hypercapnic acidosis inhibits exocytosis can satisfactorily explain these opposite relationships, i.e. high membrane depolarization/low exocytosis of [3H]CA for the acidic/hypercapnic stimulus and vice versa for the hypoxic stimulus. The finding that hypercapnic acidosis inhibits Ca2+ currents provides an explanation for previous findings from other laboratories showing that the levels of intracellular Ca2+ produced by hypoxia and hypercapnic acidosis are comparable, even though the acidic stimulus causes a much higher depolarization. The spiking behaviour of chemoreceptor cells will be omitted in the discussion due to its variability from cell to cell. However, in spiking cells, the frequency of spikes parallels the level of depolarization (see Fig. 1A; see also Rocher et al. 2005), and therefore that omission will not detract from our considerations. As a final consideration in this summary, we want to state that our finding indicate that hypoxia as a depolarizing stimulus triggers release of CA, but as a result of its activation of adenylate cyclase and the Epac pathway, releases an amount of CA much larger than expected solely from depolarization.
A statement that we explicitly want to make at the outset of the discussion of the present findings is that the release of CA has been used as an index of the chemoreceptor cell activity with no further assumptions on the significance of CA on the genesis of the activity in the CSN. The validity of the release of CA as an index of chemoreceptor cell function is sanctioned for all of the stimuli tested since there is a clear parallelism between the Ca2+-dependent release of CA (mostly dopamine) and the electrical activity recorded in the CSN, both being highly correlated with the intensity of the stimulus, although the ratios Δrelease/Δelectrical activity is stimulus specific, i.e., varies among different stimuli (Fidone et al. 1982; Obeso et al. 1986; Rigual et al. 1991; Gonzalez et al. 1992; Montoro et al. 1996). It should be acknowledged that according to the current status of our knowledge, it is most likely that several neurotransmitters (e.g. ATP, adenosine, acetylcholine and catecholamine) are involved in the genesis of electrical activity in the CSN (see Conde et al. 2009).
When comparing the release potency of moderate hypoxia and 30 mm K+, it is evident that they produce comparable responses (Fig. 1D) in spite of the consistent observation that a high K+ (depolarizing) stimulus causes a much larger depolarization (30 mV; Rocher et al. 2005) than moderate hypoxia (Fig. 1) and intracellular Ca2+ rises several times higher than moderate hypoxia (Buckler & Vaughan-Jones, 1994a,b;). We believe that the higher gain of hypoxia to elicit neurotransmitter release is mediated by a cAMP-dependent mechanism because hypoxia, and not high K+, produced an increase in cAMP levels in the CB (Perez-García et al. 1990) and because forskolin (an activator of adenylate cyclase) potentiated the release induced by hypoxia and did not affect the release elicited by high K+ (Perez-García et al. 1991). These prior findings obviated testing of cyclase inhibitor and Epac agents under stimulation with high external K+. Our finding of an endogenous cAMP-dependent mechanism enhancing the gain of the coupling of the hypoxic (and not of hypercapnic acidosis) stimulus to the exocytosis was expected, because hypoxia (and not hypercapnic acidosis) increases cAMP levels in the CB independently of the secondary actions of the released neurotransmitters (Perez-García et al. 1990; Cachero et al. 1996; see Perez-García & Gonzalez, 1997), and because forskolin augmented maximally (×5) the release of CA elicited by moderate hypoxia, very moderately the release response elicited by acidic/hypercapnic stimulus and intense hypoxia (×1.5–1.8) and did not affect the release of CA elicited by 30 mm external K+ (Perez-García et al. 1991). Our present findings showing that PKA inhibitors are ineffective in modifying the hypoxic release response, while the inhibitor of adenylate cyclase reduces the response by nearly 50%, go a step further to indicate that cAMP-potentiating effects of the hypoxic Ca2+-dependent release of CA is PKA independent. The regulation of cell processes, particularly Ca2+-dependent exocytosis of transmitters and hormones by cAMP-dependent/PKA-independent mechanisms, has only been recognized in recent years (see Seino & Shibasaki, 2005; Holz et al. 2006). Experimental data indicate that the mechanisms involved in cAMP potentiation of the hypoxic release are mediated by Epacs (or cAMP regulated guanine nucleotide exchange factor; cAMP-GEF), which are known to have many cell targets including the exocytotic machinery and K+ channels. On cAMP binding, the GEF domains of Epacs catalyse the exchange of GDP for GTP in small GTP-binding proteins leading to their activation. The facilitation of exocytosis is mediated by interactions of Epacs with Rab-3 (a member of the small GTP-binding proteins) with the intermediation of Rim-2 (a Rab-3 interacting molecule); Rab-3, which is a key modulator of exocytotic machinery (Sudhof, 2004), would be the ultimate effector of the facilitation of the release process. Overall, at a given level of intracellular calcium this mechanism increases the release probability of secretory granules already in the readily releasable pool and accelerates the refilling of this pool (Renstrom et al. 1997). At the same time, via a different small GTP-binding protein, Rap-1, Epac can lead to downstream activation of the extracellular-signal regulated kinase (ERK) and mitogen-activated protein kinase (MAPK) and modulation of K+ channels (Yuan & Chen, 2006). Then, we hypothesize that exocytotic machinery and K+ channels are the targets for the PKA-independent regulation mediated by cAMP during hypoxic stimulation. In fact, we have previously shown that cAMP analogues (dibutyryl-cAMP) mimic hypoxia in inhibiting K+ currents in isolated rabbit chemoreceptor cells (Lopez-Lopez et al. 1993). Other potential targets of Epac, such as Ca2+ channels and intracellular Ca2+ stores (Seino & Shibasaki, 2005), could be excluded in the case of chemoreceptor cells because cAMP analogues do not modify Ca2+ currents in chemoreceptor cells (Lopez-Lopez et al. 1993) and because intracellular Ca2+ stores appear to be of minimal significance in controlling levels of free Ca2+ (Vicario et al. 2000a; Conde et al. 2006). Anionic channels (Carpenter & Peers, 1997) and gap junctions (Abudara & Eyzaguirre, 1998) in chemoreceptor cells also are regulated by cAMP, but it is not known if this regulation is PKA dependent or independent. Finally, we should mention that preliminary data of experiments in course indicate that moderate hypoxia and the acidic/hypercapnic stimulus interact in such a manner that the release of both stimuli applied simultaneously is higher than the sum of the release of both stimuli applied independently. This behaviour of the release response is analogous to that described by many authors (Fitzgerald & Parks, 1971; Pepper et al. 1995) at the level of CSN discharges, but the more important aspect of our preliminary observations is that the interaction of the stimuli at the level of chemoreceptor cells and their release of DA is totally dependent on cAMP, as it is abolished by SQ-22536, the inhibitor of adenylate cyclase.
Another important finding in this study is the inhibitory effect of hypercapnic acidosis on the release of [3H]CA elicited by ionomycin (Fig. 4), indicating a direct effect of this stimulus on the exocytotic machinery. This finding can satisfactorily explain prior observations indicating that acidic/hypercapnic acidosis, which produces an increase in intracellular Ca2+ comparable to that of moderate hypoxia (Buckler & Vaughan-Jones, 1994a,b; Dasso et al. 2000), produces a much smaller Ca2+-dependent exocytosis of CA (Vicario et al. 2000b; Rocher et al. 2005). Interestingly enough, Krüger et al. (1995) have described a nearly identical inhibition of acidosis for the nicotine-induced Ca2+-dependent release of noradrenaline from guinea pig (inhibition reached 70% at pH 6.8 and 87.8% at pH 6.0), human atrium (76% inhibition at pH 6.0) and bovine adrenal chromaffin cells (inhibition was 75% at pH 6.0). Acidosis also inhibits insulin secretion in β cells (Bigner et al. 1996; Mack, 1998) and vasopressin and oxytocin release in neurohypophyseal synaptosomes (Cazalis et al. 1987); however, the pH sensitivity of the release machinery appears to be different in different systems. Quite recently, Ahdut-Hacohen et al. (2004) have described a mechanism that can explain the acid-mediated inhibition of exocytosis. Using the attached configuration of the patch clamp technique on synaptic vesicles obtained from the electric organ of Torpedo, these authors found that acidification of the recording medium produced a dramatic reduction of the opening frequency (>90% at pH 6.5 vs. the control pH 7.25) of the non-specific ion channels present in the vesicles without modifying their voltage sensitivity, implying that the overall decrease in the vesicle conductance would dramatically decrease the exocytotic vesicle emptying (Ahdut-Hacohen et al. 2004).
The inhibition of voltage-operated Ca2+ channels by the acidic/hypercapnic stimulus (Fig. 5) in chemoreceptor cells conforms with the pH sensitivity of several subtypes of Ca2+ channels in other cell types (Krafte & Kass, 1988; Tytgat et al. 1990; Chen et al. 1996; Kiss & Korn, 1999). It should also be noticed that in the aforementioned study of Krüger et al. (1995) there was also observed a decrease in the intracellular Ca2+ rise evoked by nicotine in bovine chromaffin cells at acidic pH, likely to be related to the pH effect on Ca2+ channels. This finding provides an explanation for prior observations from other laboratories showing that an acidic stimulus, in spite of producing a much higher depolarization than moderate hypoxia, raises intracellular Ca2+ to comparable levels (see Introduction). It must be acknowledged, however, that the correlation between the level of stable depolarization and intracellular Ca2+ levels might be an oversimplification for at least two reasons: first, chemoreceptor cells produce action potentials both during hypoxic and during hypercapnic acidosis stimulation but the variability of the spiking behaviour with both stimuli (Buckler & Vaughan-Jones, 1994a,b; Zhong et al. 1997; Perez-García et al. 2000; Rocher et al. 2005) precludes any further consideration; and second, intracellular acidification would produce a decrease of intracellular Ca2+ buffering-extrusion with the result that for a given amount of Ca2+ entering the cells the actual increase in intracellular Ca2+ measured is higher the lower the intracellular pH (Kohmoto et al. 1990; Cairns et al. 1993).
Findings using 80 mm extracellular K+ as stimulus are comparable to those obtained with ionomycin, i.e. the inhibition observed would result mostly from the inhibition of the exocytosis by acidosis. This interpretation is based on prior observations that the intensity of release of neurotransmitters, including the release of CA in the CB (Almaraz et al. 1986), in response to 60 and 80 mm is maximal and nearly undistinguishable (Vargas & Orrego, 1976; Dismukes et al. 1977). Thus, even if Ca2+ channels are inhibited by acidosis it could be expected that the levels of intracellular Ca2+ needed to produce maximal release are still achieved (see Blaustein, 1975).
In summary, our study demonstrated that moderate hypoxia and strong hypercapnic acidosis depolarized chemoreceptor cells in such a manner that the acidic stimulus more than tripled the depolarizing effect of moderate hypoxia. On the contrary, the acidic/hypercapnic stimulus elicited a neurotransmitter release response that was about a third of the release response elicited by hypoxia. Our data also indicate that the low potency of the acidic/hypercapnic stimulus to elicit neurosecretion is the result of at least two factors: a direct effect of the high proton concentration of the exocytotic machinery and a direct inhibition of voltage-dependent Ca2+ currents. On the contrary, the high gain of the hypoxic stimulus on the exocytosis is mediated by cAMP, via a PKA-independent, Epac-mediated mechanism. This study is the first one implicating Epacs in the control of the O2 chemoreception cascade and opens a completely new path to explore the intimate mechanisms of O2 sensing and its regulation.
Acknowledgments
We want to thank Ma de los Llanos Bravo for technical assistance. The work was supported by grants BFU2007-61848 (DGICYT), CIBER CB06/06/0050 (FISS-ICIII), GR242, VA104A08 and SAN673-VA12/08 (JCyL) and IP052561 (ISCiii).
Glossary
Abbreviations
- CA
catecholamines
- cAMP-GEF
cAMP regulated guanine nucleotide exchange factor
- CB
carotid body
- CSN
carotid sinus nerve
- DA
dopamine
- Epac
exchange protein activated by cAMP
Author contributions
A.R. and A.I.C. performed the neurochemical experiments including the analyses of the data and figure drawing. L.A. performed the electrophysiological experiments including analysis of data and drawings. C.G. was the main designer of the study and prepared the original versions of the manuscript. All authors contributed to successive amelioration of the original version to finally approve of the present version.
References
- Abudara V, Eyzaguirre C. Modulation of junctional conductance between rat carotid body glomus cells by hypoxia and acidity. Brain Res. 1998;792:114–125. doi: 10.1016/s0006-8993(98)00127-9. [DOI] [PubMed] [Google Scholar]
- Ahdut-Hacohen R, Duridanova D, Meiri H, Rahamimoff R. Hydrogen ions control synaptic vesicle ion channel activity in Torpedo electromotor neurones. J Physiol. 2004;556:347–352. doi: 10.1113/jphysiol.2003.058818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albillos A, Neher E, Moser T. R-type Ca2+ channels are coupled to the rapid component of secretion in mouse adrenal slice chromaffin cells. J Neurosci. 2000;20:8323–8330. doi: 10.1523/JNEUROSCI.20-22-08323.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almaraz L, Gonzalez C, Obeso A. Effects of high potassium on the release of [3H]dopamine from the cat carotid body in vitro. J Physiol. 1986;379:293–307. doi: 10.1113/jphysiol.1986.sp016254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkenbosch A, Van Dissel J, Olievier CN, De Goede J, Herringa J. The contribution of the peripheral chemoreceptors to the ventilatory response to CO2 in anaesthetized cats during hyperoxia. Respir Physiol. 1979;37:381–390. doi: 10.1016/0034-5687(79)90083-5. [DOI] [PubMed] [Google Scholar]
- Bigner DR, Goff JP, Faust MA, Burton JL, Tyler HD, Horst RL. Acidosis effects on insulin response during glucose tolerance tests in Jersey cows. J Dairy Sci. 1996;79:2182–2188. doi: 10.3168/jds.S0022-0302(96)76594-3. [DOI] [PubMed] [Google Scholar]
- Blaustein MP. Effects of potassium veratridine ad scorpion venom on calcium accumulation and transmitter release by nerve terminals in vitro. J Physiol. 1975;247:617–655. doi: 10.1113/jphysiol.1975.sp010950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ, Vaughan-Jones RD. Effects of hypercapnia on membrane potential and intracellular calcium in rat carotid body type I cells. J Physiol. 1994a;478:157–171. doi: 10.1113/jphysiol.1994.sp020239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ, Vaughan-Jones RD. Effects of hypoxia on membrane potential and intracellular calcium in rat neonatal carotid body type I cells. J Physiol. 1994b;476:423–428. doi: 10.1113/jphysiol.1994.sp020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cachero TG, Rigual R, Rocher A, Gonzalez C. Cholera and pertussis toxins reveal multiple regulations of cAMP levels in the rabbit carotid body. Eur J Neurosci. 1996;8:2320–2327. doi: 10.1111/j.1460-9568.1996.tb01195.x. [DOI] [PubMed] [Google Scholar]
- Cairns SP, Westerblad H, Allen DG. Changes in myoplasmic pH and calcium concentration during exposure to lactate in isolated rat ventricular myocytes. J Physiol. 1993;464:561–574. doi: 10.1113/jphysiol.1993.sp019651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter E, Peers C. Swelling- and cAMP-activated Cl− currents in isolated rat carotid body type I cells. J Physiol. 1997;503:497–511. doi: 10.1111/j.1469-7793.1997.497bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazalis M, Dayanithi G, Nordmann JJ. Requirements for hormone release from permeabilized nerve endings isolated from the rat neurohypophysis. J Physiol. 1987;390:71–91. doi: 10.1113/jphysiol.1987.sp016687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XH, Bezprozvanny I, Tsien RW. Molecular basis of proton block of L-type Ca2+ channels. J Gen Physiol. 1996;108:363–374. doi: 10.1085/jgp.108.5.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde SV, Caceres AI, Vicario I, Rocher A, Obeso A, Gonzalez C. An overview on the homeostasis of Ca2+ in chemoreceptor cells of the rabbit and rat carotid bodies. Adv Exp Med Biol. 2006;580:215–222. doi: 10.1007/0-387-31311-7_33. [DOI] [PubMed] [Google Scholar]
- Conde SV, Monteiro EC, Obeso A, Gonzalez C. Adenosine in peripheral chemoreception: New insights into a historically overlooked molecule. Adv Exp Med Biol. 2009;648:145–160. doi: 10.1007/978-90-481-2259-2_17. [DOI] [PubMed] [Google Scholar]
- Congar P, Bergevin A, Trudeau LE. D2 receptors inhibit the secretory process downstream from calcium influx in dopaminergic neurons: implication of K+ channels. J Neurophysiol. 2002;87:1046–1056. doi: 10.1152/jn.00459.2001. [DOI] [PubMed] [Google Scholar]
- Dasso LL, Buckler KJ, Vaughan-Jones RD. Interactions between hypoxia and hypercapnic acidosis on calcium signalling in carotid body type I cells. Am J Physiol Lung Cell Mol Physiol. 2000;279:L36–L42. doi: 10.1152/ajplung.2000.279.1.L36. [DOI] [PubMed] [Google Scholar]
- Dismukes K, De Beer AA, Mulder AH. On the mechanism of α-receptor mediated modulation of 3H-noradrenaline release from slices of rat brain neocortex. Naunyn Schmiedebergs Arch Pharmacol. 1977;299:115–122. doi: 10.1007/BF00498553. [DOI] [PubMed] [Google Scholar]
- Fidone S, Gonzalez C. Catecholamine synthesis in rabbit carotid body in vitro. J Physiol. 1982;333:69–79. doi: 10.1113/jphysiol.1982.sp014439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidone S, Gonzalez C. Initiation and control of chemoreceptor activity in the carotid body. In: Fishman AP, Cherniack NS, Widdicombe JG, Geiger SR, editors. Handbook of Physiology, section 3, The Respiratory System, vol II, Control of Breathing. Bethesda: American Physiological Society; 1986. pp. 247–312. [Google Scholar]
- Fidone S, Gonzalez C, Yoshizaki K. Effects of low oxygen on the release of dopamine from the rabbit carotid body in vitro. J Physiol. 1982;333:93–110. doi: 10.1113/jphysiol.1982.sp014441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald RS, Parks DC. Effect of hypoxia on carotid chemoreceptor response to carbon dioxide in cats. Respir Physiol. 1971;12:218–229. doi: 10.1016/0034-5687(71)90054-5. [DOI] [PubMed] [Google Scholar]
- Fitzgerald RS. Single fiber chemoreceptor responses of carotid and aortic bodies. In: Paintal AS, editor. Morphology and Mechanisms of Chemoreceptors. Delhi, India: Vallabhbhai Patel Chest Institute; 1976. pp. 27–35. [Google Scholar]
- Gonzalez C, Almaraz L, Obeso A, Rigual R. Oxygen and acid chemoreception in the carotid body chemoreceptors. Trends Neurosci. 1992;15:146–153. doi: 10.1016/0166-2236(92)90357-e. [DOI] [PubMed] [Google Scholar]
- Gonzalez C, Almaraz L, Obeso A, Rigual R. Carotid body chemoreceptors: from natural stimuli to sensory discharges. Physiol Rev. 1994;74:829–898. doi: 10.1152/physrev.1994.74.4.829. [DOI] [PubMed] [Google Scholar]
- Holz GG, Kang G, Harbeck M, Roe MW, Chepurny OG. Cell physiology of cAMP sensor Epac. J Physiol. 2006;577:5–15. doi: 10.1113/jphysiol.2006.119644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda Y. Role of carotid chemoreceptors in control of breathing at rest and in exercise: studies on human subjects with bilateral carotid body resection. Jpn J Physiol. 1985;35:535–544. doi: 10.2170/jjphysiol.35.535. [DOI] [PubMed] [Google Scholar]
- Huang CC, Hsu KS. Presynaptic mechanism underlying cAMP-induced synaptic potentiation in medial prefrontal cortex pyramidal neurons. Mol Pharmacol. 2006;69:846–856. doi: 10.1124/mol.105.018093. [DOI] [PubMed] [Google Scholar]
- Kiss L, Korn SJ. Modulation of N-type Ca2+ channels by intracellular pH in chick sensory neurons. J Neurophysiol. 1999;81:1839–1847. doi: 10.1152/jn.1999.81.4.1839. [DOI] [PubMed] [Google Scholar]
- Klöckner U, Isenberg G. Calcium channel current of vascular smooth muscle cells: extracellular protons modulate gating and single channel conductance. J Gen Physiol. 1994;103:665–678. doi: 10.1085/jgp.103.4.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohmoto O, Spitzer KW, Movsesian MA, Barry WH. Effects of intracellular acidosis on [Ca2+]i transients, transsarcolemmal Ca2+ fluxes, and contraction in ventricular myocytes. Circ Res. 1990;66:622–632. doi: 10.1161/01.res.66.3.622. [DOI] [PubMed] [Google Scholar]
- Krafte DS, Kass RS. Hydrogen ion modulation of Ca channel current in cardiac ventricular cells. Evidence for multiple mechanisms. J Gen Physiol. 1988;91:641–657. doi: 10.1085/jgp.91.5.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger C, Haunstetter A, Gerber S, Serf C, Kaufmann A, Kubler W, Haass M. Nicotine-induced exocytotic norepinephrine release in guinea-pig heart, human atrium and bovine adrenal chromaffin cells: modulation by single components of ischaemia. J Mol Cell Cardiol. 1995;27:1491–1506. doi: 10.1016/s0022-2828(95)90194-9. [DOI] [PubMed] [Google Scholar]
- Lahiri S. Depressent effect of acute and chronic hypoxia on ventilation. In: Paintal AS, editor. Morphology and Mechanisms of Chemoreceptors. Delhi, India: Vallabhbhai Patel Chest Institute; 1976. pp. 138–146. [Google Scholar]
- Lahiri S, Delaney RG. Stimulus interaction in the responses of carotid body chemoreceptor single afferent fibers. Respir Physiol. 1975;24:249–266. doi: 10.1016/0034-5687(75)90017-1. [DOI] [PubMed] [Google Scholar]
- Lopez-Lopez JR, De Luis A, Gonzalez C. Properties of a transient K+ current in chemoreceptor cells of rabbit carotid body. J Physiol. 1993;460:15–32. doi: 10.1113/jphysiol.1993.sp019456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack RH. Effect of metabolic acidosis on insulin action and secretion in uremia. Kidney Int. 1998;54:603–607. doi: 10.1046/j.1523-1755.1998.00023.x. [DOI] [PubMed] [Google Scholar]
- Montoro RJ, Ureña J, Fernandez-Chacon R, Alvarez DT, López-Barneo J. Oxygen sensing by ion channels and chemotransduction in single glomus cells. J Gen Physiol. 1996;107:133–143. doi: 10.1085/jgp.107.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattie E. CO2, brainstem chemoreceptors and breathing. Prog Neurobiol. 1999;59:299–331. doi: 10.1016/s0301-0082(99)00008-8. [DOI] [PubMed] [Google Scholar]
- Obeso A, Almaraz L, González C. Effects of 2-deoxy D-glucose on in vitro cat carotid body. Brain Res. 1986;371:25–36. doi: 10.1016/0006-8993(86)90806-1. [DOI] [PubMed] [Google Scholar]
- Obeso A, Rocher A, Fidone S, González C. The role of dihydropyridine-sensitive Ca2+ channels in stimulus-evoked catecholamine release from chemoreceptor cells of the carotid body. Neuroscience. 1992;47:463–472. doi: 10.1016/0306-4522(92)90260-9. [DOI] [PubMed] [Google Scholar]
- Pepper DR, Landauer RC, Kumar P. Postnatal development of CO2–O2 interactionin the rat carotid body in vitro. J Physiol. 1995;485:531–541. doi: 10.1113/jphysiol.1995.sp020749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-García MT, Almaraz L, González C. Effects of different types of stimulation on cyclic AMP content in the rabbit carotid body: functional significance. J Neurochem. 1990;55:1287–1293. doi: 10.1111/j.1471-4159.1990.tb03137.x. [DOI] [PubMed] [Google Scholar]
- Perez-García MT, Almaraz L, Gonzalez C. Cyclic AMP modulates differentially the release of dopamine induced by hypoxia and other stimuli and increases dopamine synthesis in the rabbit carotid body. J Neurochem. 1991;57:1992–2000. doi: 10.1111/j.1471-4159.1991.tb06414.x. [DOI] [PubMed] [Google Scholar]
- Perez-García MT, Gonzalez C. Mechanisms of sensory transduction in chemoreceptor cells: the role of second messengers. In: Gonzalez C, editor. The Carotid Body Chemoreceptors. Heidelberg, Germany: Springer Verlag; 1997. pp. 79–96. [Google Scholar]
- Perez-García MT, López-López JR, Riesco AM, Hoppe UC, Marban E, González C, Johns DC. Viral gene transfer of dominant-negative Kv4 construct suppresses an O2-sensitive K+ current in chemoreceptor cells. J Neurosci. 2000;20:5689–5695. doi: 10.1523/JNEUROSCI.20-15-05689.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renstrom E, Eliasson L, Rorsman P. Protein kinase A-dependent and -independent stimulation of exocytosis by cAMP in mouse pancreatic B-cells. J Physiol. 1997;502:105–118. doi: 10.1111/j.1469-7793.1997.105bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigual R, Gonzalez E, Fidone S, Gonzalez C. Effects of low pH on synthesis and release of catecholamines in the cat carotid body in vitro. Brain Res. 1984;309:178–181. doi: 10.1016/0006-8993(84)91026-6. [DOI] [PubMed] [Google Scholar]
- Rigual R, Gonzalez E, Gonzalez C, Fidone S. Synthesis and release of catecholamines by the cat carotid body in vitro: effects of hypoxic stimulation. Brain Res. 1986;374:101–109. doi: 10.1016/0006-8993(86)90398-7. [DOI] [PubMed] [Google Scholar]
- Rigual R, Lopez-Lopez JR, Gonzalez C. Release of dopamine and chemoreceptor discharge induced by low pH and high PCO2 stimulation of the cat carotid body. J Physiol. 1991;433:519–531. doi: 10.1113/jphysiol.1991.sp018441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocher A, Geijo-Barrientos E, Caceres AI, Rigual R, Gonzalez C, Almaraz L. Role of voltage-dependent calcium channels in stimulus-secretion coupling in rabbit carotid body chemoreceptor cells. J Physiol. 2005;562:407–420. doi: 10.1113/jphysiol.2004.075523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez D, Lopez-Lopez JR, Pérez-García MT, Sanz-Alfayate G, Obeso A, Ganfornina MD, González C. Molecular identification of Kv α subunits that contribute to the oxygen-sensitive K+ current of chemoreceptor cells of the rabbit carotid body. J Physiol. 2002;542:369–382. doi: 10.1113/jphysiol.2002.018382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz-Alfayate G, Obeso A, Agapito MT, Gonzalez C. Reduced to oxidized glutathione ratios and oxygen sensing in calf and rabbit carotid body chemoreceptor cells. J Physiol. 2001;537:209–220. doi: 10.1111/j.1469-7793.2001.0209k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seino S, Shibasaki T. PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol Rev. 2005;85:1303–1342. doi: 10.1152/physrev.00001.2005. [DOI] [PubMed] [Google Scholar]
- Ster J, de Bock F, Bertaso F, Abitbol K, Daniel H, Bockaert J, Fagni L. Epac mediates Pacap-dependent long-term depression in the hippocampus. J Physiol. 2009;587:101–113. doi: 10.1113/jphysiol.2008.157461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Dixon DB, Copenhagen DR. Modulation of a sustained calcium current by intracellular pH in horizontal cells of fish retina. J Gen Physiol. 1993;101:695–714. doi: 10.1085/jgp.101.5.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tytgat J, Nilius B, Carmeliet E. Modulation of the T-type cardiac Ca channel by changes in proton concentration. J Gen Physiol. 1990;96:973–990. doi: 10.1085/jgp.96.5.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas O, Orrego F. Elevated extracellular potassium as a stimulus for releasing norephinephrine and 14C α-amino isobutyrate from neocortical slice. Specificity and calcium dependency of the process. J Neurochem. 1976;26:31–34. doi: 10.1111/j.1471-4159.1976.tb04431.x. [DOI] [PubMed] [Google Scholar]
- Vicario I, Obeso A, Rocher A, López-López JR, Gonzalez C. Intracellular Ca2+ stores in chemoreceptor cells of the rabbit carotid body: significance for chemoreception. Am J Physiol Cell Physiol. 2000a;279:C51–C61. doi: 10.1152/ajpcell.2000.279.1.C51. [DOI] [PubMed] [Google Scholar]
- Vicario I, Rigual R, Obeso A, Gonzalez C. Characterization of the synthesis and release of catecholamine in the rat carotid body in vitro. Am J Physiol Cell Physiol. 2000b;278:C490–C499. doi: 10.1152/ajpcell.2000.278.3.C490. [DOI] [PubMed] [Google Scholar]
- Yuan LL, Chen X. Diversity of potassium channels in neuronal dendrites. Prog Neurobiol. 2006;78:374–389. doi: 10.1016/j.pneurobio.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Zhong H, Zhang M, Nurse C. Synapse formation and hypoxic signalling in co-cultures of rat petrosal neurones and carotid body type 1 cells. J Physiol. 1997;503:599–612. doi: 10.1111/j.1469-7793.1997.599bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]