

Abstract
The objective of the present study was to evaluate how different ligand interactions of profilin-1 (Pfn1), an actin-binding protein that is upregulated during capillary morphogenesis of vascular endothelial cells (VEC), contribute to migration and capillary forming ability of VEC. We adopted a knockdown-knockin experimental system to stably express either fully-functional or mutants of Pfn1 that are impaired in binding to two of its major ligands, actin (H119E mutant) and proteins containing polyproline domains (H133S mutant), in a human dermal microvascular cell line (HmVEC) against near-null endogenous Pfn1 background. We found that silencing endogenous Pfn1 expression in HmVEC leads to slower random migration, reduced velocity of membrane protrusion and a significant impairment in matrigel-induced cord formation. Only re-expression of fully-functional but not any of the two ligand-binding deficient mutants of Pfn1 rescues the above defects. We further show that loss of Pfn1 expression in VEC inhibits three-dimensional capillary morphogenesis, MMP2 secretion and ECM invasion. VEC invasion through ECM is also inhibited when actin and polyproline interactions of Pfn1 are disrupted. Together, these experimental data demonstrate that Pfn1 regulates VEC migration, invasion and capillary morphogenesis through its interaction with both actin and proline-rich ligands.
Keywords: Profilin-1, ligand interactions, cell motility, invasion, MMP2, capillary morphogenesis, lamellipodial protrusion, vascular endothelial cells
INTRODUCTION
Migration of vascular endothelial cell (VEC) is critical for capillary outgrowth from pre-existing blood vessels during angiogenesis [1]. Reorganization of actin cytoskeleton that occurs during cell migration is a dynamic process that involves both actin polymerization and depolymerization in a precise spatiotemporal fashion. This actin remodeling process is regulated by a wide range of actin-binding proteins including those involved in monomer sequestering, nucleating, elongating, severing, depolymerizing, and capping of actin filaments [2]. Based on changes in the expression profiles in VEC undergoing capillary morphogenesis, some of the important actin-binding proteins including thymosin β4, profilin, gelsolin and VASP (vasodilator stimulated phosphoprotein) have been previously implicated in angiogenesis [3], and among these proteins, at least thymosin β4 has been confirmed as a pro-angiogenic molecule in vivo [4]. In a previous study, we showed that silencing profilin-1 (Pfn1 - the founding member and the ubiquitously expressed member of profilin family of genes) expressions in human umbilical vein endothelial cells (HUVEC) dramatically impairs their ability to form planar cord-like structures on matrigel (a commonly adopted in vitro representation for angiogenesis) [5]. This observation suggested for the first time that Pfn1 might serve an important role in capillary morphogenesis of VEC.
Pfn1 was originally identified as a G-actin sequestering molecule [6]; however, later studies showed that Pfn1 enhances actin polymerization by catalyzing ADP-to-ATP exchange on G-actin and shuttling ATP-G-actin to the barbed ends of actin filaments [7, 8]. Pfn1-depletion leading to reduced F-actin content in various cell types is in clear support of Pfn1’s role as an enhancer of actin polymerization in vivo [5, 9]. Since Pfn1 has an affinity for poly-L-proline sequences, it also binds to several major proline-rich protein families which either nucleate and/or elongate actin filaments in cells (example: VASP, WASP (Wiskott-Aldrich Syndrome Protein) and diaphanous) [10-12]. Pfn1’s interaction with these proline-rich cytoskeletal regulators is thought to be important for generation of actin-based structures in cells. However, abolishing Pfn1’s interaction with proline-rich ligands by overexpression of dominant-negative mutants has been shown to either promote [13] or inhibit [14] the formation of actin-based protrusive structures depending on the cell-type, suggesting complex and context-dependent functions of such interactions. Finally, Pfn1 is also a ligand for phosphatidylinositol-4,5-bisphosphate (PIP2) [15]. The two binding sites of Pfn1 for PIP2 overlap with its actin and polyproline binding regions [16]. Biochemical studies have shown that PIP2 competes with actin and partly, polyproline binding of Pfn1 [15, 17]. Although it remains to be seen, PIP2 could therefore potentially act as a negative regulator of Pfn1’s function in vivo.
Previous studies from our laboratory have shown that the effect of Pfn1 depletion on overall cell motility is context-dependent. While loss of Pfn1 expression inhibits migration of normal VEC [5], breast cancer and normal human mammary epithelial cells show an opposite trend [9, 18] suggesting a complexity of Pfn1’s role in cell migration. While all of those studies demonstrate that cell migration is sensitive to perturbation of Pfn1’s function, how different ligand interactions of Pfn1 contribute to whole cell migration and physiological processes that critically rely on cell migration have never been examined. In the present study, we examine the role of actin and polyproline interactions of Pfn1 in migration, invasion and capillary morphogenesis of VEC.
MATERIALS AND METHODS
Reagents
Polyclonal Pfn1 and monoclonal GAPDH antibodies were purchased from Cytoskeleton Inc (Denver, CO) and Abd Serotec (Raleigh, NC), respectively. Monoclonal actin antibody was obtained from Chemicon (Billerica, MA). Monoclonal vimentin antibody was a product of Pharmingen (San Diego, CA). Rhodamine-phalloidin and DAPI were obtained from Invitrogen (Carlsbad, CA). Collagen type I and growth-factor reduced matrigel are products of BD Biosciences (Bedford, MA).
Cell culture
HUVEC (Human umbilical vein endothelial cells; source: Cambrex Biosciences, Walkersville, MD) were cultured in the complete EBM2 growth media (also commercially available from the same source). HMEC-1 (an immortalized human dermal microvascular cell line - referred to as HmVEC here) was cultured in MCDB 131 media (Gibco, Gaithersburg, MD) supplemented with 10% FBS, 10 mM L-Glutamine (Gibco, Gaithersburg, MD), 1 ng/mL EGF (BD Biosciences, Bedford, MA), and 1 ng/ml hydrocortisone (Sigma, St. Louis, MO).
siRNA transfection, plasmid construction and retroviral infection
For gene silencing of Pfn1, cells were transfected with 100 nM of either our previously described Pfn1-specific siRNA [18] or smart-pool control-siRNA commercially available from Dharmacon Inc (Lafayette, CO) according to the manufacturer’s instructions. Generation of our original plasmids encoding GFP-Pfn1 and its point mutants (GFP-Pfn1-H119E and GFP-Pfn1-H133S) has been previously described [9]. These constructs were further modified by introducing a two base-pair silent mutation (does not change the peptide encoding) in the Pfn1-siRNA targeting region before subcloning into pQCXIP retroviral vector (Clontech, Mountainview, CA) at Not1 and BamH1 restriction sites. Retrovirus packaging and subsequent infection of HMEC-1 cells were carried out according to the manufacturer’s instructions. Infected cells were selected for puromycin resistance (250 ng/ml) and finally, stable cells were sorted based on their GFP-fluorescence before experimental use.
Capillary morphogenesis assay
Matrigel-induced planar cord morphogenesis assay has been previously described. Briefly, two-hundred microliters of matrigel was polymerized in the wells of a 48-well plate at 37°C for 30 minutes prior to seeding 25,000 cells on the top of matrigel. Cord formation was assessed 8 hours after cell-seeding, and was quantified by measuring the total cord length/10X field of observation which was then averaged for three fields per well from a duplicate set of samples for each experimental condition.
Three-dimensional (3D) capillary morphogenesis assay was carried out according to a published protocol [3] with slight modification. Essentially, 150 μl of neutralized collagen-I solution was premixed with cells and plated in duplicate in the wells of a 8-well Lab-Tek chamber slide at the final concentrations of collagen and cells equal to 2.5 mg/ml and 2×106 /ml, respectively. The collagen solution was allowed to polymerize for 30 minutes, and then overlaid with the complete growth medium with 50 ng/ml bFGF, 50 ng/ml VEGF, and 50 ng/ml PMA. At the end of 96 hours of incubation, cells were stained with rhodamine-phalloidin and DAPI. Images of capillaries were taken at 4 random 10X fields of observation per chamber slide and the mean value of the total capillary length/10X field was used for statistical comparison.
Cell migration / Kymograph assay
Speed of cell migration was measured from time-lapse motility assay as previously described [5]. Briefly, cells were sparsely plated on a 35 mm tissue-culture dish and after an overnight incubation, time-lapse imaging of randomly migrating cells was performed simultaneously at 3 locations and at an interval of 2-3 minutes for a total duration of 90 minutes. The acquired images were analyzed using the NIH ImageJ software to compute the total distance traveled by cells during the observation time. Membrane dynamics was studied from additional time-lapse movies which were recorded for shorter time (total duration - 10 minutes) but at a higher temporal resolution (5-sec interval). Kymographs marking the beginning to the end of protrusion were constructed based on 1-pixel wide (0.3 μm) lines drawn at multiple locations (3 to 4) across the protruding membrane. Membrane fluctuation less than 4 pixels (1.2 μm) was disregraded for quantitative analyses. All images were acquired and analyzed using Metamorph and NIH ImageJ softwares, resepectively.
Cell invasion assay
The overall experimental set-up for measuring ECM invasion of VEC was identical to that used for 3D capillary morphogenesis assay except in this case, real-time imaging of cells were performed at an interval of 10 minutes for a total duration of either 72 hours (for HUVEC) or 48 hours (for HmVEC). The average invasion speed was scored by analyzing the stack of time-lapse images by NIH Image J software.
Protein extraction/ Immunoblotting
Total cell lysate was extracted by modified RIPA buffer (50 mM Tris-HCl -pH 7.5, 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholate, 0.3% SDS, 2 mM EDTA) supplemented with 50 mM NaF, 1mM sodium pervanadate, and protease inhibitors. For biochemical fractionation experiments, we used our previously adopted protocol [9]. Briefly, cells were first washed with ice-cold F-actin stabilization buffer (50mM PIPES-pH 6.9, 50mM NaCl, 5% glycerol, 5mM EGTA, 5mM MgCl2, 1mM ATP, 1mM DTT, 0.1% β-mercaptoethanol) and then extracted with buffer A (F-Actin stabilization buffer supplemented with 0.1% triton-X and protease inhibitors) for 10 minutes to remove soluble proteins (contain G-actin). Culture plate was washed with buffer A and was further extracted with modified RIPA buffer, clarified by centrifugation to obtain the triton-insoluble fraction. The purity of the triton-insoluble fraction was confirmed by positive and negative immunoreactivity for vimentin and GAPDH antibodies, respectively. For immunoblotting, antibodies were used at the following concentrations: Pfn1(1:500), GAPDH (1:2000), actin (1:000) and vimentin (1:1000).
Phalloidin staining
Cells were washed with warm PBS, fixed with 4% formaldehyde for 10 minutes, permeabilized with 0.5% Triton X-100 in PBS for 10 minutes before incubating with rhodamine-phallodin (in PBS) with or without DAPI for 30 minutes. Cells were washed 5 times ((3 times with PBS containing 0.02% tween followed by 2 times with PBS) before mounting on slides. All fluorescence images were acquired with Olympus IX-71 inverted microscope.
Gelatin Zymography
Cells were initially plated in 60 mm culture dish (seeding density: 6×105 and 7×105 cells for HUVEC and HmVEC, respectively) in complete growth media for 24 hours before serum-starving for another 21 hours (the seeding density was optimized to ensure sub-confluent culture at the day of experiment). Conditioned media was collected, gently centrifuged at 300 g for 3 minutes to remove cell debris, if any, and then concentrated in centrifugation filter units (molecular weight cut-off: 10 KDa; Millipore, Billerica, MA) at ∼2500 g for 40 min. Samples of equivalent protein content were run on a 10% gelatin gel (Bio-Rad, Hercules, CA). The zymogram gel was subsequently processed completely according to the manufacturer’s instructions before acquiring images for densitometric quantification of MMP bands.
Statistics and data representation
All statistical tests were performed with ANOVA followed by Newman-Keuls post-hoc test for multiple comparisons whenever applicable, and a ‘p’ value less than 0.05 was considered to be statistically significant. Experimental data were represented as box and whisker plots where dot represents the mean, middle lines of box indicates median, top of the box indicates 75th percentile, bottom of the box measures 25th percentile and the two whiskers indicate the 10th and 90th percentiles, respectively.
RESULTS
Generation and characterization of stable VEC lines with perturbed Pfn1 function
Several different point-mutants of Pfn1 that are defective in binding specifically to actin (H119E, R74E) and polyproline (H133S, W3A, W3N) have been reported in the literature [13, 19, 20]. In previous studies, some of these mutants were also overexpressed in cells to assess the effects of perturbing ligand interactions of Pfn1 on formation of actin-based structures including filopodia [20], neurite outgrowth [13] and comet tail induced by bacterial pathogens [14]. In this study, we have adopted a “knock-down/knock-in” approach to study for the first time the effects of abolishing Pfn1’s interaction with actin and proline-rich ligands on whole cell migration and capillary morphogenesis of VEC. Specifically, we engineered HmVEC to stably express either GFP-Pfn1 (fully functional form of Pfn1) or its point-mutants (GFP-Pfn1-H119E (actin-binding deficient) or GFP-Pfn1-H133S (polyproline-binding deficient)) by retroviral transduction. Fusing GFP at the N-terminus of Pfn1, as done here, preserves its biochemical functions and cellular localization of the fusion protein similar to the endogenous Pfn1 [21], and is therefore, a valid approach. Loss of specific ligand-binding function of the two GFP-tagged Pfn1 mutants was biochemically confirmed in our previous study [9]). As a control, HmVEC was transduced with GFP-encoding retrovirus. All of these Pfn1 constructs were rendered Pfn1-siRNA resistant by introducing additional silent mutations in the siRNA-targeting region. This strategy enabled us to express these various constructs in cells in the background of strongly suppressed endogenous Pfn1 expression achieved via Pfn1-siRNA treatment.
Fig 1A shows the fluorescence micrographs of FACS-sorted HmVEC lines expressing GFP-Pfn1 and its various mutants. Statistical analyses of average fluorescence intensity of individual cells in cultures of our different HmVEC lines revealed ∼50% coefficient of variation in GFP-fluorescence (data not shown). In an unsynchronized culture where cells exist at different phases of cell cycle, a 50% coefficient of variation in fluorescence intensity indicates that we did not have a large cell-to-cell variation in the level of expression of GFP-fused protein in any of our stable HmVEC lines (this is also evident from the micrographs presented in Fig 1A). The immunoblot in Fig 1B shows the relative expression levels of exogenous GFP-Pfn1 (or its mutants) vs endogenous Pfn1 in the various HmVEC lines. The endogenous Pfn1 level was found to be similar between the different cell lines. We performed densitometric quantification of GFP-Pfn1 (or its mutants) bands relative to that of respective endogenous Pfn1. The average expression level of exogenous Pfn1 was similar between the different cell lines and within a range of 60-70% of the respective endogenous Pfn1 level. Based on this data taken together statistical distribution of fluorescence of individual cells in culture, we further estimated that the exogenous Pfn1 was either below or equal to the endogenous Pfn1 level in 85-90% of the total cell population. For those cells with strongest GFP-fluorescence (generally excluded from subsequent single cell motility analyses), the level of overexpression was estimated to be equal to 1.5 fold.
Figure 1. Generation of stable Pfn1 mutants of HmVEC.
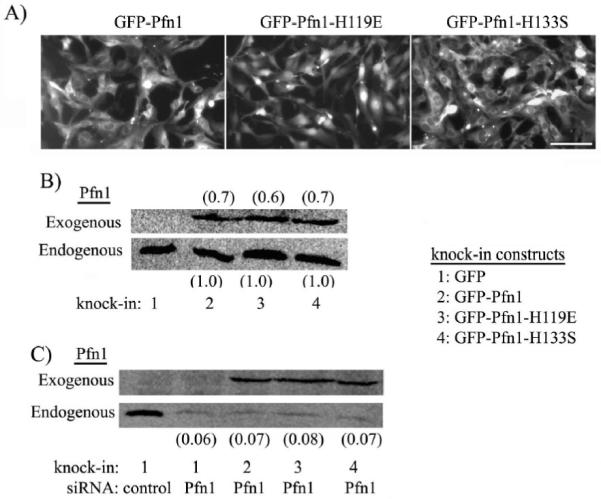
(A) Fluorescence micrographs of HmVEC expressing GFP-Pfn1 and its mutants (GFP-Pfn1-H119E, GFP-Pfn1-H133S). (B) Pfn1 immunoblot showing relative levels of exogenous and endogenous levels of Pfn1 in the various sublines of HmVEC (the ratio of exogenous to endogenous Pfn1 is indicated by the numbers based on densitometric quantification of immunoblot data averaged from 2 different experiments). (C) Expression of exogenous GFP-Pfn1 or its mutants in HmVEC in the silenced endogenous Pfn1 background (the numbers indicate the relative endogenous Pfn1 level for different treatment conditions).
Next, to demonstrate the efficacy of our “knockdown-knockin” system, we transfected all of our stable HmVEC sublines with Pfn1-siRNA. As a control group for all of the experiments, GFP expressers were also transfected with control siRNA (this treatment condition was referred to as “control group” from here on). Fig 1C shows a representative Pfn1-immunoblot confirming the expression of exogenous GFP-Pfn1 or its mutants in various HmVEC sublines against a strongly suppressed endogenous Pfn1 background (silencing efficiency >90%).
Since Pfn1 plays an important role in actin polymerization, we asked whether silencing the overall expression or disrupting specific ligand interactions of Pfn1 has any effect on polymerized actin in HmVEC. Phalloidin staining revealed significantly reduced actin filaments in HmVEC when Pfn1 expression was silenced (Fig 2A). To quantitatively represent this difference, we performed biochemical fractionation experiments which showed ∼56% less actin in the triton-insoluble fraction (contains F-actin) of lysate of Pfn1-silenced HmVEC compared to the same extracted from control siRNA transfected cells (Fig 2B). The total expression level of actin was unchanged after Pfn1 depletion (Fig 2B). This data is consistent with our previous observation in HUVEC and MDA-MB-231 breast cancer cell line [5, 9]. We also performed similar fractionation experiments with the various stable cell lines of HmVEC. However, we did not find any significant difference in the actin content in the triton-insoluble fractions between the different stable cell lines (Fig 2C).
Figure 2. Effects of perturbing Pfn1 on actin cytoskeleton in HmVEC.
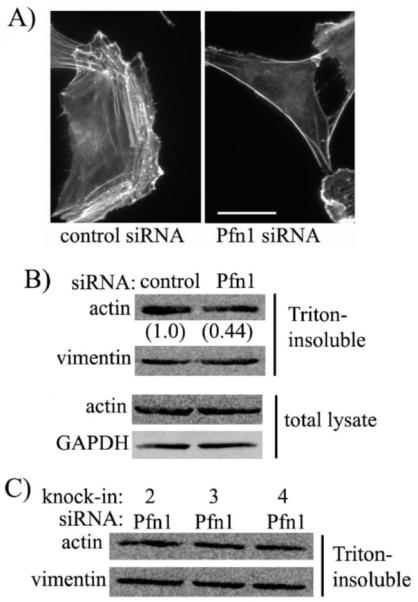
(A) Phalloidin staining of GFP-expressers treated with either control or Pfn1-siRNA (bar - 30 μm). (B) Immunoblots comparing total and triton-insoluble fraction of actin levels between control and Pfn1-siRNA treated HmVEC (the numbers indicate the relative F-actin levels between the two siRNA treated conditions - data summarized from 2 independent experiments). (C) Relative actin content in triton-insoluble fractions of different stable sublines of HmVEC. GAPDH and vimentin immunoblots serve as the loading controls for total lysate and triton-insoluble fraction of lysate, respectively.
Both Actin and Polyproline Interactions of Pfn1 are important for lamellipodial protrusion and migration of VEC
To determine whether Pfn1’s interaction with both actin and polyproline ligands contribute to overall VEC migration, we compared the average speed of migration between the different sublines of HmvEC from time-lapse motility measurements. Because of certain degree of variation in the expression of GFP-tagged proteins in the polyclonal culture of our stable cell lines, cells which were fairly bright for GFP-fluorescence were only chosen for time-lapse measurements (extremely bright cells were also excluded). Also, between the different experimental groups, cells with relatively similar levels of GFP-fluorescence were selected for final data analyses. A box and whisker plot comparing the relative speed of migration between the different groups of cells demonstrates that silencing Pfn1 expression inhibits the average speed of migration of GFP-expressing HmVEC by nearly 37% (Fig 3A). Re-expression of GFP-Pfn1 in a silenced endogenous Pfn1 background resulted in an average migration speed close to 91% of that of control group of cells. In fact, we did not find any statistically significant difference in the average speed of migration between the control group and GFP-Pfn1 re-expressers suggesting that expression of GFP-Pfn1 is able to fully rescue the inhibition of migration resulting from endogenous Pfn1 depletion. However, re-expression of neither of the Pfn1 mutants was able to rescue the motility defect induced by silencing Pfn1 expression since the average speed of migration of GFP-Pfn1-H119E and GFP-Pfn1-H133S expressers were found to be 40% and 37% less than that of control cells, respectively, and these differences were statistically significant (p<0.01). Overall, these results demonstrate that both actin and polyproline interactions of Pfn1 are indispensable for efficient VEC migration.
Figure 3. Role of actin or polyproline interactions of Pfn1 in membrane protrusion and migration of HmVEC.
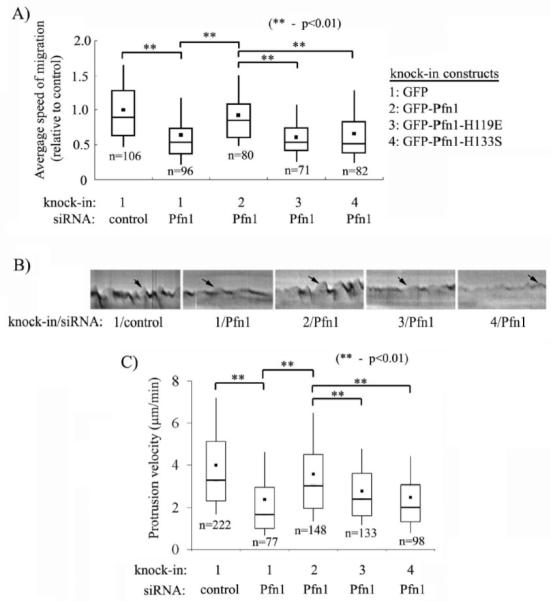
(A) A box and whisker plot showing the average speed of migration of different HmVEC sublines relative to that of control cells (n: number of cells analyzed from a total of 4 independent experiments;). (B) Representative kymographs of different groups of cells (black arrow marking the ascending portion of a saw-tooth waveform indicates membrane protrusion). (C) A box and whisker plot comparing the average protrusion velocity between the different groups (n: number of protrusion events analyzed from a total of 4-5 experiments). Construct and siRNA annotations are same as in panel A.
Lamellipodial protrusion initiates and defines the direction of cell movement,and it is actin polymerization at the cell membrane that translates to effective membrane protrusion. Although Pfn1 binds to a plethora of ligands and therefore it has been implicated in a wide range of cellular functions, its role has been most extensively studied in the context of actin polymerization. We have recently demonstrated that Pfn1 depletion leads to slower membrane protrusion in HUVEC [18]. Therefore, we queried whether perturbing actin or polyproline interactions of Pfn1 alters the lamellipodial dynamics of HmVEC. To address this question, we analyzed the leading edge movement of the different groups of HmVEC from kymographs of 1-pixel (0.3 μm) wide lines that were drawn normal to the leading edge and in the direction of protrusion. Fig 3B depicts a set of representative kymographs of the protrusion events of HmVEC under different experimental conditions where leading edge traces (marked by the arrows) reveal cycles of typical lamellipodial protrusion and withdrawal (resemble saw-tooth waveforms). It is evident from Fig 3B that either silencing the overall expression of Pfn1 or the expression of Pfn1-mutants leads to a much flatter kymograph trace suggesting that membrane dynamics is suppressed by inhibition of Pfn1 function. We performed quantitative analyses of the actual protrusion velocity (equal to the slope of the ascending portion of a saw-tooth waveform) of the different experimental groups and these data are summarized in the form of a box and whisker plot in Fig 3C. The average protrusion velocity of Pfn1-depleted HmVEC (= 2.3 μm/min) was found to be nearly 40% less than that of control siRNA-treated cells (= 4 μm/min), and this data is consistent with our earlier finding with HUVEC [18]. Re-expression of GFP-Pfn1 m/min) was found to be nearly 40% less than that of control siRNA-treated cells (= 4 μm/min), in the silenced endogenous Pfn1 background increased the average protrusion velocity to 3.6 μm/min, and this value was not statistically different from the velocity scored for control cells. The average velocity of protrusion of both GFP-Pfn1-H119E (= 2.7 μm/min) and GFP-Pfn1-H133S (=2.5 μm/ min) expressers were found to be less than that of GFP-Pfn1 expressing cell line within statistical significance. We also noted that both Pfn1-depleted and the mutant cells display reduced frequency of protrusion (data not shown). Overall, these data demonstrate that both actin and polyproline interactions of Pfn1 are required for efficient lamellipodial protrusion of VEC.
Capillary morphogenesis of VEC requires Pfn1’s interactions with actin and proline-rich ligands
We next evaluated the sensitivity of capillary morphogenesis of HmVEC to disruption of ligand interactions of Pfn1 in matrigel-induced planar cord-formation assay. Fig 4A depicts the representative cord formation by the different group of cells. To quantitatively represent the difference in cord forming ability between the various cell lines, we measured the total cord length/10X field of observation for each cell line, and these data are summarized in the form of a box and whisker plot in Fig 4B. Our data shows that the average cord length /field of GFP expressers bearing control siRNA (=5347±681 μm) is significantly higher than the value scored for the same cells in a Pfn1-depleted condition (=3136±972 μm), and this data is consistent with our previous observation with HUVEC [5]. No statistically significant difference in the average cord length was found between GFP-Pfn1 re-expressers (=4941±1327 μm) and control cells thus demonstrating that re-expression of GFP-Pfn1 can rescue cord morphogenesis defect of HmVEC caused by Pfn1 depletion. However, the average cord lengths of both GFP-Pfn1-H119E (=3327±701 μm) and GFP-Pfn1-H133S (=2473±488 μm) were found to significantly less compared to that of GFP-Pfn1 re-expressers. These data show that both actin and polyproline interactions of Pfn1 are indispensable for cord-morphogenesis of VEC.
Figure 4. Cord morphogenesis of HmVEC requires functional interactions of Pfn1 with both actin and proline-rich ligands.

(A) Representative images of matrigel-induced cord formation by different groups of cells at 8 hrs after cell-seeding. (B) A box and whisker plot summarizing the cord morphogenesis data from a total of 2-3 independent experiments. (C) A line graph compares the relative spreading ability of different groups of cells at different time-points after seeding on matrigels (diamond: GFP/control-siRNA, cross: GFP/Pfn1-sRNA, triangle: GFP-Pfn1/Pfn1-siRNA, square: GFP-Pfn1-H119E/Pfn1 siRNA, and open circle: GFP-Pfn1-H133S/ Pfn1-siRNA). Data here are summarized from a total of two independent experiments with a duplicate set of samples for each experimental condition.
VEC initially need to spread and then elongate to form cord-like structures on matrigel. It was apparent from Fig 4A that a significant fraction of HmVEC displayed round morphology (suggesting spreading defect) when Pfn1 function was inhibited by either siRNA treatment or expression of the mutants. Since the images shown in Fig 4A represent end-point assessments, we also examined the spreading behavior of these different groups of cells in a time-course fashion within the first 4 hours after cell-seeding on matrigel, the results of which are shown in Fig 4C. While for all experimental groups, there was a general trend of increase in % of spread cells (identified by elongated morphology) as a function of time, inhibiting Pfn1 function, either through silencing the endogenous expression or disrupting actin/polyproline interactions, clearly led to much reduced spreading efficiency when compared to control cells or GFP-Pfn1 re-expressers at all time-points of evaluation. These data demonstrate that both actin and polyproline interactions of Pfn1 are required for VEC spreading.
Loss of Pfn1 expression inhibits MMP2 secretion, ECM invasion and 3-D capillary morphogenesis of VEC
Finally, capillary formation in vivo occurs within 3-D ECM environment and involves cellular changes that may not be completely represented in a 2D experimental model as in matrigel-based cord formation assay. Therefore, we asked whether 3-D capillary morphogenesis of VEC within an ECM scaffold is also affected by Pfn1 depletion. Specifically, we chose to examine the effect of silencing Pfn1 expression on capillary morphogenesis of VEC seeded inside collagen matrix. We chose HUVEC as the VEC model system for these experiments since we found that HmVEC cell line does not form robust capillary-like structures when embedded inside collagen matrix (note that in almost all of the previous studies, the ability of HmVEC to form capillary-like structures was also assessed using either matrigel or collagen-based 2-D cord-morphogenesis assay [22-24]). Immunoblot data in Fig 5A shows that Pfn1 expression can be strongly suppressed in HUVEC within 96 hours after siRNA transfection. Three-dimensional capillary morphogenesis experiments showed HUVEC transfected with control siRNA form prominent network of capillary-like structures (marked by rhodamine-phallodin staining) as expected (Fig 5B). However, Pfn1 depletion severely inhibits capillary morphogenesis of HUVEC as judged by significant reduction in both number and length of capillary-like structures observed in this culture. This difference is quantitatively represented by a box and whisker plot in Fig 5C which shows that total length of capillary-like structures/ 10X field of observation formed by control siRNA treated cells (=3438 ± 546 μm) was nearly 2-fold greater than the same scored for Pfn1-depleted cells (=1820 ± 602 μm) thereby establishing that Pfn1 plays an indispensable role in 3D capillary morphogenesis of VEC.
Figure 5. Silencing Pfn1 expression inhibits ECM invasion, MMP2 secretion and 3D capillary morphogenesis.

(A) Pfn1-immunoblot of HUVEC extracts prepared 96 hours after siRNA transfection with GAPDH blot serving as the loading control. (B) Capillary formation (arrows) by control and Pfn1-siRNA treated HUVEC within collagen matrix (blue: DAPI, red: rhodamine-phalloidin) (scale bar - 200 μm) (C) A box and whisker plot comparing the mean values of total capillary length per 10X field of observation between the two transfection conditions (n=2 experiments). (D) A box and whisker plot showing the relative invasion speed of control and Pfn1-siRNA treated HUVEC and HmVEC through collagen (‘n’ indicates the number of cells analyzed for each cell type pooled from 2 independent experiments). (E) Gelatin zymogram of conditioned media from HUVEC and HmVEC showing relative levels of MMP2 between control and Pfn1-siRNA treated conditions (the numbers indicate the relative levels of MMP2 between the two transfection conditions summarized from 2 and 4 independent experiments for HUVEC and HmVEC, respectively; * indicates statistical significance with p<0.01).
We previously showed that silencing Pfn1 expression inhibits planar migration of HUVEC on tissue culture substrate. However, it is known that cells assume dramatically different morphologies when invading through 3-D matrices vs. migrating on planar tissue-culture substrata and EC is no exception to this rule. Since ECM invasion by VEC is critical for capillary formation in vivo, we performed time-lapse imaging of control and Pfn1-siRNA treated HUVEC seeded inside collagen matrix for 96 hours and analyzed the average speed of invasion of HUVEC under these two transfection conditions. A box and whisker plot in Fig 5D shows that the loss of Pfn1 expression is associated with a 37% decrease in the average speed of HUVEC invasion through collagen matrix. We performed similar invasion experiments with HmVEC which also resulted in a 50% reduction in the average speed of invasion when Pfn1 expression was suppressed (Fig 5D).
Matrix metalloproteases (MMPs) play key role in proteolytic degradation of ECM proteins during cell invasion and angiogeneis. We therefore examined whether suppressing Pfn1 expression in VEC has any effect on MMP secretion by performing gelatin zymography of conditioned media from subconfluent cultures of both HUVEC and HmVEC following either control or Pfn1 siRNA treatment. For control siRNA treated HUVEC, we were able to detect strong bands representing the characteristic prozymogen (higher molecular weight) and active (lower molecular weight) forms of MMP2 (Fig 5D). These band intensities were significantly weaker when Pfn1 expression was suppressed. We estimated a 70% decrease in total (active plus inactive forms) MMP2 secretion by HUVEC as a result of silencing Pfn1 expression. This trend was reproducible for HmVEC although the % decrease in MMP2 secretion as a result of Pfn1 suppression (equal to 20%) was lower than that observed for HUVEC (note that for HmVEC, we were only able to detect the prozymogen form of MMP2 and we did not see any MMP9 band for either of the VEC type). Overall, these results demonstrate that loss of Pfn1 expression in VEC has an inhibitory effect on MMP2 secretion.
We finally examined the effect of abolishing actin and polyproline interaction of Pfn1 on 3D collagen invasion by HmVEC. Similar to our data obtained from planar migration experiments, the average invasion speed of GFP-Pfn1-H119E and GFP-Pfn1-H133S expressers were found to be 50% and 60% lower than that of GFP-Pfn1 expressers, respectively (Fig 6). This suggests that both actin and polyproline interactions of Pfn1 are important for VEC invasion through ECM.
Figure 6. HmVEC invasion through ECM requires actin and polyproline interactions of Pfn1.
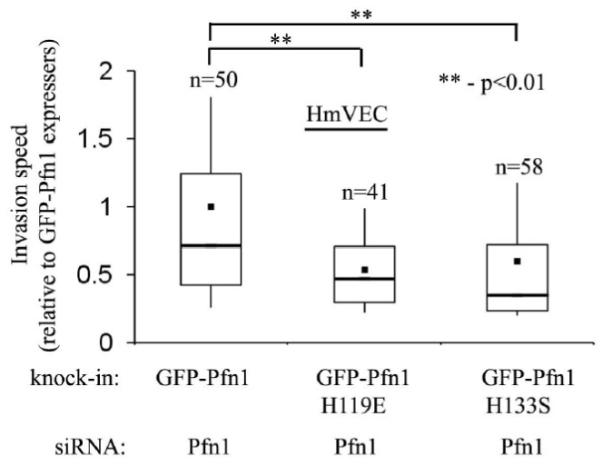
A comparison of the average speed of invasion of different HmVEC lines with perturbed Pfn1 function (data normalized to the speed of invasion of GFP-Pfn1 re-expressers and ‘n’ indicates the number of cells analyzed for each cell type pooled from 2 independent experiments).
DISCUSSION
The role of ligand-binding of Pfn1 has been previously assessed in the context of actin polymerization (reviewed in [25]) and formation of specific actin-based protrusive structures including filopodia [11, 20], neurite outgrowth [13], and actin comet-tail induced by bacterial pathogens (a molecular mimicry of lamellipodial protrusion- [14]). Although actin-based protrusion is an integral part of cell migration, the overall process of cell migration spatiotemporally integrates many different events and is much more complex. Therefore, findings from those studies cannot be extrapolated to determine the contribution of specific ligand interactions of Pfn1 in whole cell migration, a knowledge that is presently lacking in the literature. How different ligand interactions of Pfn1 regulate lamellipodial dynamics has also not been examined so far. Another technical issue with all of the previous studies is the use of overexpression of Pfn1 mutants in a dominant-negative fashion. While this is a commonly used strategy, a general drawback of dominant negative approach is that one has to express a given mutant in large molar excess compared to the endogenous protein in order to assure dominant negative action and this could potentially result in experimental artifacts due to hyper-functionality of other ligand interactions which are not targeted by the mutation. In the present study, we for the first time have used a knockdown/knockin strategy to evaluate the effect of expressing specific ligand-binding deficient mutants of Pfn1 in a strongly suppressed (>90% silencing) endogenous background on lamellipodial dynamics, migration, invasion and capillary morphogenesis of VEC, and these are the novel aspects of this study.
We here demonstrate that either depleting the overall expression or abrogating actin/polyproline interactions of Pfn1 leads to reduced velocity of lamillipodial protrusion and slower VEC migration. We showed that the effect of silencing the overall expression of Pfn1 on cell motility and lamellipodial protrusion was closely mimicked by disrupting either of actin or polyproline interactions of Pfn1thus suggesting that Pfn1 must interact simultaneously with actin and proline-rich ligands to drive membrane protrusion and cell motility. Alteration in lamellipodial dynamics by Pfn1 mutations clearly suggest that actin and polyproline interactions of Pfn1 play key role in regulating actin dynamics at the leading edge. However, we did not find any effect of Pfn1 mutation on the actin content in triton-insoluble fraction of HmVEC. A few points are worth discussing here. First, actin content in the triton-insoluble fraction here may not represent the total F-actin level in cells as demonstrated previously in the case of polymorphonuclear leukocytes [26]. Therefore, subtle differences in either the total and/or localized changes in F-actin content between the different mutant groups of cells, if any, can be undetected in our experiments. Second, it is also not completely surprising that silencing Pfn1 expression but not expression of either of the mutants affected the actin content in the triton-insoluble fraction of HmVEC. This is because particularly the H133S mutant of Pfn1 should, in principle, polymerize actin through barbed-end elongation without involving proline-rich actin regulators. Finally, an alternative possibility is that there may be a complicated downstream effect of Pfn1 mutants altering the expression and/or activity of other actin-binding proteins compensating for F-actin changes. Based on our earlier findings that expression levels of some of major promoters of actin polymerization including VASP, N-WASP and mDia are not sensitive to Pfn1 depletion in HUVEC [5], we, however, think changes in expression of other actin-binding proteins are far less likely.
Our results with H119E mutant of Pfn1 is consistent with previous studies that showed cdc42/N-WASP-induced actin microspike formation, Rac-induced membrane ruffles, intracellular propulsion speed of bacterial pathogens (an indirect assessment of velocity of protrusion), and neurite outgrowth can be suppressed by disrupting Pfn1-actin interaction [11, 13, 14]. Since the H119E substitution does not affect Pfn1’s binding to polyproline sequences, this mutant can lead to slower membrane protrusion and motility by still binding to proline-rich regulators of actin polymerization at the leading edge (example: VASP, WASP) but not being able to increase local G-actin concentration at or near these regulatory proteins to enhance actin polymerization.
Disruption of polyproline interactions of Pfn1, on the other hand, has been shown to have different responses on the generation of actin-based protrusion depending on the context. Overexpression of H133S mutant of Pfn1 dramatically inhibited intracellular movement of bacterial pathogens based on which it was postulated Pfn1’s interaction with proline-rich ligands might be important for actin-based protrusion [14], and our present finding is consistent with this pathogen data. These results are further supported by existing biochemical data which show that the rate of F-actin elongation by some of the major proline-rich actin regulators is enhanced in the presence of Pfn1 [27]. Interestingly, neurite outgrowth, a process that is also driven by actin polymerization, was found to be actually facilitated when polyproline interaction of Pfn1 was abrogated by overexpression of W3A mutant [13]. Although the reason for this apparent discrepancy between the different studies is not clear, a few possibilities should be considered. First, a simple explanation could be that Pfn1’s action on actin cytoskeleton can be cell-specific. This is not completely unlikely since dictyostelium amobae tend to have an increase in their F-actin content upon genetic deletion of Pfn1 while other mammalian cells including VEC show reduced F-actin after Pfn1 depletion. Second, Pfn1’s effect on actin cytoskeleton can be dependent on its level of expression. At a very high cellular concentration, Pfn1 can act as a G-actin sequestering protein if enough barbed ends are not available for elongation or even induce F-actin depolymerziation through modulation of capping proteins as proposed by Bubb and colleagues [28]. Therefore, the resulting effect of H133S mutant on actin polymerization should be a function of the level of overexpression, a factor which is likely to vary between different studies. Third, the two different polyproline-deficient mutants of Pfn1 may have subtle difference in their phospholipid binding, and particularly in an overexpression-based setting this can have a major influence on the functional status of Pfn1 if one considers phospholipid binding to be a critical regulator of Pfn1 function. The last two points further exemplify the necessity of evaluating the effects of Pfn1-mutants at a close-to physiological level of expression which can be achieved in a knock-down/knock-in experimental system, as adopted in the present study. Future studies are needed to identify which of the polyproline interactions of Pfn1 are critical for VEC migration.
Finally, we showed that Pfn1 is required for ECM invasion, MMP2 secretion and capillary morphogenesis of VEC. Very recently, Pfn1-dependent dissociation of thymosin-β4 (Tβ4) from actin resulting in Tβ4-ILK (integrin-linked kinase)-AKT-linked pathway of MMP2 production has been demonstrated in bovine EC [29]. Whether Pfn1 also has a direct effect on MMP2 production remains to be identified. We show that interaction to both actin and proline-rich ligands contribute to VEC invasion and differentiation to capillary-like structures. These in vitro findings pave the way for future in vivo studies to investigate whether suppressing Pfn1 function could be an effective anti-angiogenic strategy. It will be also interesting to examine if augmenting Pfn1 function has a positive influence on the angiogenic response of VEC. These future studies will recognize whether Pfn1 could be a novel angiogenesis target.
ACKNOWLEDEGMENTS
We would like to thank Steve Chirieleison for technical assistance. This work is supported by grants from the National Institute of Health (R01-CA108607-01), American Heart Association (0665414U), and Competitive Medical Research fund from the University of Pittsburgh Medical Center to P. R.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Bauer SM, Bauer RJ, Velazquez OC. Angiogenesis, vasculogenesis, and induction of healing in chronic wounds. Vasc Endovascular Surg. 2005;39:293–306. doi: 10.1177/153857440503900401. [DOI] [PubMed] [Google Scholar]
- [2].Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112:453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- [3].Salazar R, Bell SE, Davis GE. Coordinate induction of the actin cytoskeletal regulatory proteins gelsolin, vasodilator-stimulated phosphoprotein, and profilin during capillary morphogenesis in vitro. Experimental Cell Research. 1999;249:22–32. doi: 10.1006/excr.1999.4460. [DOI] [PubMed] [Google Scholar]
- [4].Philp D, Goldstein AL, Kleinman HK. Thymosin beta4 promotes angiogenesis, wound healing, and hair follicle development. Mech Ageing Dev. 2004;125:113–115. doi: 10.1016/j.mad.2003.11.005. [DOI] [PubMed] [Google Scholar]
- [5].Ding Z, Lambrechts A, Parepally M, Roy P. Silencing profilin-1 inhibits endothelial cell proliferation, migration and cord morphogenesis. J Cell Sci. 2006;119:4127–4137. doi: 10.1242/jcs.03178. [DOI] [PubMed] [Google Scholar]
- [6].Carlsson L, Nystrom LE, Sundkvist I, Markey F, Lindberg U. Actin polymerizability is influenced by profilin, a low molecular weight protein in non-muscle cells. Journal of Molecular Biology. 1977;115:465–483. doi: 10.1016/0022-2836(77)90166-8. [DOI] [PubMed] [Google Scholar]
- [7].Theriot JA, Mitchison TJ. The three faces of profilin. Cell. 1993;75:835–838. doi: 10.1016/0092-8674(93)90527-w. [DOI] [PubMed] [Google Scholar]
- [8].Pantaloni D, Carlier MF. How profilin promotes actin filament assembly in the presence of thymosin beta-4. Cell. 1993;75:1007–1014. doi: 10.1016/0092-8674(93)90544-z. [DOI] [PubMed] [Google Scholar]
- [9].Zou L, Jaramillo M, Whaley D, Wells A, Panchapakesa V, Das T, Roy P. Profilin-1 is a negative regulator of mammary carcinoma aggressiveness. Br J Cancer. 2007;97:1361–1371. doi: 10.1038/sj.bjc.6604038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Reinhard M, Giehl K, Abel K, Haffner C, Jarchau T, Hoppe V, Jockusch BM, Walter U. The proline-rich focal adhesion and microfilament protein VASP is a ligand for profilins. EBMO Journal. 1995;14:1583–1589. doi: 10.1002/j.1460-2075.1995.tb07146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Suetsugu S, miki H, Takenawa T. The essential role of profilin in the assembly of actin for microspike formation. EMBO Journal. 1998;17:6516–6526. doi: 10.1093/emboj/17.22.6516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Watanabe N, Madaule P, Reid T, Ishizaki T, Watanabe G, Kakizuka A, Saito Y, Nakao K, Jockusch BM, Narumiya S. p140mDia, a mammalian homolog of Drosophila diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. Embo J. 1997;16:3044–3056. doi: 10.1093/emboj/16.11.3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lambrechts A, Jonckheere V, Peleman C, Polet D, De Vos W, Vandekerckhove J, Ampe C. Profilin-I-ligand interactions influence various aspects of neuronal differentiation. J Cell Sci. 2006;119:1570–1578. doi: 10.1242/jcs.02884. [DOI] [PubMed] [Google Scholar]
- [14].Mimuro H, Suzuki T, Suetsugu S, Miki H, Takenawa T, Sasakawa C. Profilin is required for sustaining efficeient intra- and intercellular spreading of Shigella Flexneri. Journal of Biological Chemistry. 2000;275:28893–28901. doi: 10.1074/jbc.M003882200. [DOI] [PubMed] [Google Scholar]
- [15].Lassing I, Lindberg U. Specific interaction between phosphatidylinositol 4,5-bisphosphate and profilactin. Nature. 1985;314:472–474. doi: 10.1038/314472a0. [DOI] [PubMed] [Google Scholar]
- [16].Lambrechts A, Jonckheere V, Dewitte D, Vandekerckhove J, Ampe C. Mutational analysis of human profilin I reveals a second PI(4,5)-P2 binding site neighbouring the poly(L-proline) binding site. BMC Biochem. 2002;3:12. doi: 10.1186/1471-2091-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lambrechts A, Verschelde JL, Jonckheere V, Goethals M, Vandekerckhove J, Ampe C. The mammalian profilin isoforms display complementary affinities for PIP2 and proline-rich sequences. Embo J. 1997;16:484–494. doi: 10.1093/emboj/16.3.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bae YH, Ding Z, Zou L, Wells A, Gertler F, Roy P. Loss of profilin-1 expression enhances breast cancer cell motility by Ena/VASP proteins. J Cell Physiol. 2008 doi: 10.1002/jcp.21677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bjorkegren-Sjogren C, Korenbaum E, Nordberg P, Lindberg U, Karlsson R. Isolation and characterization of two mutants of human profilin I that do not bind poly(L-proline) FEBS Lett. 1997;418:258–264. doi: 10.1016/s0014-5793(97)01376-8. [DOI] [PubMed] [Google Scholar]
- [20].Suetsugu S, Miki H, Takenawa T. Distinct roles of profilin in cell morphological changes: mircospikes, membrane ruffles, stress fibers, and cytokinesis. FEBS Letters. 1999;457:470–474. doi: 10.1016/s0014-5793(99)01086-8. [DOI] [PubMed] [Google Scholar]
- [21].Wittenmayer N, Rothkegel M, Jockusch BM, Schluter K. Functional characterization of green fluorescent protein-profilin fusion proteins. European Journal of Biochemistry. 2000;267:5247–5246. doi: 10.1046/j.1432-1327.2000.01600.x. [DOI] [PubMed] [Google Scholar]
- [22].Kellouche S, Mourah S, Bonnefoy A, Schoevaert D, Podgorniak MP, Calvo F, Hoylaerts MF, Legrand C, Dosquet C. Platelets, thrombospondin-1 and human dermal fibroblasts cooperate for stimulation of endothelial cell tubulogenesis through VEGF and PAI-1 regulation. Exp Cell Res. 2007;313:486–499. doi: 10.1016/j.yexcr.2006.10.023. [DOI] [PubMed] [Google Scholar]
- [23].Liu J, Wang XB, Park DS, Lisanti MP. Caveolin-1 expression enhances endothelial capillary tubule formation. The Journal of biological chemistry. 2002;277:10661–10668. doi: 10.1074/jbc.M110354200. [DOI] [PubMed] [Google Scholar]
- [24].Marchetti M, Vignoli A, Russo L, Balducci D, Pagnoncelli M, Barbui T, Falanga A. Endothelial capillary tube formation and cell proliferation induced by tumor cells are affected by low molecular weight heparins and unfractionated heparin. Thromb Res. 2008;121:637–645. doi: 10.1016/j.thromres.2007.06.015. [DOI] [PubMed] [Google Scholar]
- [25].Witke W. The role of profilin complexes in cell motility and other cellular processes. Trends in cell biology. 2004;14:461–469. doi: 10.1016/j.tcb.2004.07.003. [DOI] [PubMed] [Google Scholar]
- [26].Cano ML, Cassimeris L, Fechheimer M, Zigmond SH. Mechanisms responsible for F-actin stabilization after lysis of polymorphonuclear leukocytes. The Journal of cell biology. 1992;116:1123–1134. doi: 10.1083/jcb.116.5.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Barzik M, Kotova TI, Higgs HN, Hazelwood L, Hanein D, Gertler FB, Schafer DA. Ena/VASP proteins enhance actin polymerization in the presence of barbed end capping proteins. The Journal of biological chemistry. 2005;280:28653–28662. doi: 10.1074/jbc.M503957200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bubb MR, Yarmola EG, Gibson BG, Southwick FS. Depolymerization of actin filaments by profilin. Effects of profilin on capping protein function. The Journal of biological chemistry. 2003;278:24629–24635. doi: 10.1074/jbc.M302796200. [DOI] [PubMed] [Google Scholar]
- [29].Fan Y, Gong Y, Ghosh PK, Graham LM, Fox PL. Spatial coordination of actin polymerization and ILK-Akt2 activity during endothelial cell migration. Developmental cell. 2009;16:661–674. doi: 10.1016/j.devcel.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]