

Abstract
Toll like receptors (TLRs) are essential for host defense. While several TLRs reside on the cell surface, nucleic acid recognizing TLRs are intracellular. For example, the receptor for CpG containing bacterial and viral DNA, TLR9, is retained in the endoplasmic reticulum (ER). Recent evidence suggests that the localization of TLR9 is critical for appropriate ligand recognition. Here we define which structural features of the TLR9 molecule control its intracellular localization. Both the cytoplasmic and ectodomains of TLR9 contain sufficient information while the transmembrane domain plays no role in intracellular localization. We identify a 14 amino acid stretch that directs TLR9 intracellularly and confers intracellular localization to the normally cell surface expressed TLR4. Truncation or mutation of the cytoplasmic tail of TLR9 reveals a vesicle localization motif that targets early endosomes. We propose a model whereby modification of the cytoplasmic tail of TLR9 results in trafficking to early endosomes where it encounters CpG DNA.
Innate recognition of pathogens is mediated by a number of germline-encoded proteins, of which the Toll-like receptors (TLRs1) play a pivotal role. TLRs comprise a family of approximately ten paralogs, depending on species, which initiate inflammatory responses to a diverse array of microbial products (1,2). Examples include bacterial peptidoglycans (PGN), lipopolysaccharide (LPS) and flagellin, which are recognized by TLRs 2, 4, and 5, respectively, and nucleic acids from bacteria and viruses, which are recognized by TLRs 3, 7, 8, and 9 (1,3,4). TLR9, the focus of the current study, responds to bacterial and viral DNA as well as synthetic oligodeoxynucleotides (ODN) that contain unmethylated CpG dinucleotides in specific sequence contexts (5–8). Like all TLRs, TLR9 is a type 1 integral membrane receptor with a ligand-binding ectodomain, and a cytoplasmic portion that belong to the Toll IL-1 Receptor (TIR) family of signaling domains. The TIR domain of TLR9 interacts with the MyD88 adapter protein, initiating a signaling cascade that results in the activation of NF-κB and MAP kinases (9,10).
TLRs recognize their ligands at distinct locations within cells. LPS and PGN are recognized at the cell surface (11–13); however, recognition of flagellin by TLR5 is more compartmentalized and occurs on the basolateral surface of polarized epithelial cells (14). In contrast, nucleic acid recognition occurs intracellularly (13,15–17). TLR9 is retained in the endoplasmic reticulum (ER) prior to CpG DNA exposure (18,19), and it has been proposed that TLR9, localized in the ER, gains access to incoming endosomes containing CpG DNA by direct fusion of the endosome with the ER. Neutralization of lysosomes inhibits signaling through TLR9, suggesting that trafficking to the lysosome is essential for signal transduction (13,15). Furthermore, TLR9, CpG DNA and the adapter protein MyD88 have all been localized to vesicles that can be stained for lysosomal markers (5,13). The endosomal location of TLR9 reflects the requirement for phagocytosis and microbial destruction for the release of DNA prior to signaling.
In the current study, we hypothesize that TLR9 contains specific motifs that control localization and trafficking. Although transmembrane proteins often contain information in their cytoplasmic tails that dictate their cellular localization (20), we found that the internal location of TLR9 was mediated by both the cytoplasmic domain and the ectodomain, but not the transmembrane domain (21). Truncation analysis revealed a region of the cytoplasmic tail required for intracellular retention and a motif that was capable of targeting TLR9 to vesicles. Unexpectedly, the vesicles targeted by this motif were not lysosomes, but instead were early endosomes coincident with endocytosed CpG DNA.
EXPERIMENTAL PROCEDURES
Cell Culture
HeLa, MEF, HEK293 and HEK293T cells were cultured in DMEM containing 2 mM L-glutamine, 50 U/ml penicillin, 50 μg/ml streptomycin, 10 mM HEPES, 1 mM sodium pyruvate, and 10% low endotoxin fetal bovine serum.
Antibodies
Anti-Tac (human CD25, IL-2Rα) mAb (22), used for imaging, was a gift from Dr. Thomas Waldmann, NCI, and was directly labeled with Alexa Fluor 488 using Molecular Probes monoclonal antibody labeling kit. Anti-IL-2Rα (N-19), used for immunoblotting Tac chimeras, was from Santa Cruz. Unconjugated anti-TLR4 (HTA125) and TLR9 (eB72-1665) were from eBioscience. LAMP-1 antibody was from Pharmingen, APC-conjugated secondary antibodies were from Caltag and HRP-conjugated secondary antibodies were from Southern Biotechnology.
Plasmids
TLR9 (Acc# AF259262) and TLR4 (Acc# U93091), tagged at their C-termini with enhanced green fluorescent protein (GFP), were generated by PCR and inserted in frame into pEGFP-N1 (BD Clontech). Constructs containing the extracellular domain of Tac fused to the transmembrane (TM) and TIR domains of TLR9 (818–1032) and TLR4 (635–835) were generated by introducing an EcoRV site directly upstream of the TM domain of the TLR by site directed mutagenesis. The TM, TIR, and stop codon were excised at the EcoRV and a downstream restriction site, and then ligated in frame into compatible sites of pCDNA3 containing the extracellular domain of Tac (1–239) (Acc# NP_000408, a gift from Dr. Remy Bosselut, NCI). The TLR4-9-GFP (TLR41–633TLR9818–1032) and TLR9-4-GFP (TLR91–817TLR4634–835) chimeras were generated by inserting the TLR9 (or TLR4) TIR and TM domains into TLR4 (or TLR9)-GFP in which the TIR and TM domains had been excised. TLR91–810TLR4625–835 was generated by PCR as previously described (26) and put into pFlag-CMV. Truncation mutants were generated by introducing stop codons into the Tac-TIR9 chimera by site directed mutagenesis using the Quick Change Mutagenesis kit (Stratagene) according to the provider’s protocol except that the PCR reactions contained 200 ng of template plasmid and were run for 28 cycles (see Fig. 2). TLR4-9-4 (TLR41–678TLR9875–911TLR4710–839) was generated by stitching PCR. All chimeras were verified by sequencing. Rab5-GFP was a gift from Dr. Juan Bonifacino, NICHD. All plasmids for transfection were prepared using an EndoFree Plasmid Maxi Kit (Qiagen).
Fig. 2.

Diagram of truncation and substitution mutants used in this study. Tac (gray box) was fused to the TM and TIR domains of TLR9 (amino acids 817–1032). Mutant names are indicated to the left. The numbering scheme is according to that used in Genbank, AF259262. Truncations were generated by introducing a stop codon (*) at the indicated amino acid.
Flow Cytometry
HEK293T (3×105) cells were transfected using TransIT (Mirus) in 6 well plates and after 24 hours directly stained for surface expression, or fixed, permeabilized and stained for total expression. Data were collected with a BD FACSCalibur, and analyzed using CellQuest.
Confocal Microscopy
HeLa (3×105) cells were transfected using TransIT on coverslips, then fixed, permeabilized and stained 24 hr later. Cells were visualized using either a Zeiss LSM510 confocal laser-scanning microscope, where images of single 0.8 μm sections were collected, or by a Zeiss AxioImager for non-confocal images. Composite pictures were prepared with Photoshop. Each individual cell image is a representative of at least 5 different cells collected in at least three different experiments. Larger field images were taken with a 40× objective from at least three independent experiments.
Luciferase Assay
HEK293 cells were plated at 2×104 cells per well in 96 well plates. Cells were transfected using TransIT and a total of 200ng DNA per well consisting of NF-κB-luciferase and pSV-β–galactosidase reporter plasmids, and either TLR9, TLR9-4 chimera, MD-2 plus TLR4, or MD-2 plus the TLR4-9 chimera. Cells were stimulated for 24 hours, lysed in Reporter Lysis Buffer (Promega) and assayed for luciferase (Promega) and β-galactosidase (Tropix) activity. NF-κB activity (RLU) was calculated by dividing the luciferase counts by the control β-galactosidase. Salmonella LPS (Sigma) or CpG oligodeoxynucleotide (ODN) 2006 (23) were used as stimulants. Paired t tests were used to determine statistical significance.
Glycosidase Treatment
Transfected HeLa cells were collected in PBS containing 20 mM EDTA with protease inhibitors and sonicated. Sonicates were treated with Endoglycosidase H (EndoH) or Peptide: N-Glycosidase F (PNGaseF), both from New England Biolabs, as described (24). Alternatively, cells were lysed as previously described (18), immunoprecipitated and treated with EndoH (NEB). All treated samples were separated by SDS-PAGE, transferred to nitrocellulose and probed with anti-Tac or anti-GFP antibodies.
RESULTS
The localization of nucleic acid recognizing TLRs is critical for ligand recognition. To determine what molecular mechanisms control TLR9 intracellular localization we constructed chimeric proteins where the ecto- and transmembrane/cytoplasmic (TM-TIR) domains of TLR4 and TLR9 were interchanged. As expected, TLR4, but not TLR9, localized to the cell surface (Fig. 1A). However, no surface expression was detected for TLR9 or the TLR4-9 or the TLR9-4 chimeras, suggesting that both the ecto- and TM-TIR domains of TLR9 contain intracellular localization information. A chimera where the TIR domain of TLR9 was replaced by GFP (TLR9ΔTIR) also failed to localize to the cell surface (Fig. 1A), further supporting the notion that the TM domain and/or the ectodomain of TLR9 contains localization information. Confocal laser scanning microscopy (LSM) revealed that TLR9-4, TLR4-9 and TLR9ΔTIR, like wild type TLR9, were retained in a reticular compartment, which we previously described to be the endoplasmic reticulum (ER) (Fig. 1B) (19). As expected, TLR4 was found both at the cell surface and in a Golgi-like compartment (Fig. 1B) (12,18,19). The TLR4-9 chimera failed to respond to LPS, likely due to redirection of the TLR4 ectodomain from the cell surface to intracellular compartments where it can no longer interact with LPS (Fig. 1C). By contrast, the TLR9-4 chimera, containing the TLR4 TM and cytoplasmic tail, did respond to CpG DNA. In this study we focused on molecular structures contained in the cytoplasmic tail that dictate intracellular localization.
Fig. 1.

Both the ecto and TIR domains of TLR9 localize intracellularly. A, HEK293T cells were transfected with TLR4-GFP, TLR9-GFP, TLR4-9-GFP, TLR9-4-GFP or TLR9ΔTIR-GFP (TLR9-GFP with the TIR domain deleted). After 24 hr, the cells were stained directly (Surface) or fixed, permeabilized, and stained (Total) with anti-TLR4 or anti-TLR9 mAb (black lines). Cells transfected with GFP and stained identically served as negative controls (filled histograms). Cells were gated for GFP expression and only the GFP+ cells are shown. Data are representative of three separate experiments. B, HeLa cells were transfected with the same constructs as in panel A and fixed 24 hours later. GFP fluorescence, differential interference contrast (DIC), and overlays are shown. C, HEK293 cells were transfected with TLR9-4, or TLR9 alone, or with TLR4 plus MD2, or TLR4-9 plus MD2, and stimulated with 100 ng/ml LPS or 5 μg/ml CpG DNA. NF-κB activity was measured 24 hours later. * p<.002, #p <.05. This is one of three replicate experiments.
The studies in Figure 1 suggested that either the TM or the cytoplasmic domain, in addition to the ecto-domain, contained intracellular localization motifs. Other studies have suggested that the transmembrane (TM) domain controls localization of TLR7 and mouse TLR9 (25–27). To determine if the TM domain of human TLR9 also controlled intracellular localization, the ectodomain of CD25 (Tac) was fused to the TM and cytoplasmic tail of TLR9 (Tac-TIR9) (Fig. 2), a strategy that has been used successfully in other systems (24). Tac is processed in the Golgi to a highly glycosylated form that is resistant to EndoH digestion, serving as a sensitive biochemical marker for protein export from the ER. A fusion of Tac and the TLR4 TIR domain (Tac-TIR4) was readily detected on the cell surface by flow cytometry (Supplementary Fig. 1), and contained a high molecular weight EndoH resistant band indicative of Golgi processing. Tac-TIR9 was localized intracellularly, co-localized with WT TLR9 and was completely EndoH sensitive (Supplementary Fig. 1). In contrast, truncation of most or all of the cytoplasmic tail resulted in surface expression of Tac-TIR9 (Fig. 3A and Supplemetary Fig. 2). The Tac-TIR9 truncations contained higher molecular weight, EndoH resistant, glycoforms indicative of trafficking out of the ER and of Golgi processing (Fig. 3B). Since those constructs contained the TLR9 TM domain, we concluded that the human TLR9 transmembrane domain did not mediate intracellular localization. In a previous report, surface expression of mouse FlagTLR91–810TLR4625–835 was detected (26). We generated the same mouse TLR9 chimera, and the analogous untagged human version, and detected no mature glycoform, which would indicate surface expression, in HeLa or mouse embryo fibroblast (MEF) cells (Supplementary Fig. 3 and Supplementary Fig. 4).
Fig. 3.

The TM domain of TLR9 is dispensable for intracellular localization. A, HeLa cells were plated on glass coverslips and transfected with the indicated Tac-fusion plasmids. Twenty-four hours later the cells were fixed and stained with Alexa488 labeled anti-Tac mAb and counterstained with Dapi. Images were collected on an Zeiss AxioImager with a 40× objective at identical settings. Images were handled identically, overlaid and compiled in Photoshop. B, HeLa cells were transfected in 6-well plates with the indicated constructs and 24 hours later harvested for glycosylation analysis. Tac-TIR9 only has an immature glycoform whereas the other chimeras have immature (EndoH sensitive) and mature (EndoH resistant, PNGase partial sensitive) glycoforms. A non-specific band runs at the same molecular weight as the unglycosylated form of TacTIR9Δ888 and TacTIR9Δ870. The TacTIR9Δ902 samples are from the same gel, but were separated from the other samples by the molecular weight marker on the original gel.
To identify motifs that control localization of TLR9, the Tac-TIR9 fusion protein was truncated progressively from the C-terminus (Fig. 2). The cytoplasmic domain of TLR9 contains 196 residues and removal of the last 131 amino acids (i.e. truncation at amino acid 902, TacTIR9Δ902) had little effect on localization since this mutant was not detected on the cell surface and remained EndoH sensitive (Fig. 4). However, an abrupt transition from internal to surface expression was observed when 14 additional residues were deleted (TacTIR9Δ888), as detected by microscopy (Fig. 4A), flow cytometry (Supplementary Fig. 5), and glycoform analysis (Fig. 4B). The 14 residues between TacTIR9Δ902 and TacTIR9Δ888 represented a functional intracellular localization motif since they directed internal localization when grafted into the homologous position in the TLR4 cytoplamsic domain (Tac-TIR4-9-4, Fig. 4 and Supplementary Fig. 5). While there was still surface expression of Tac-TIR4-9-4, the level was significantly reduced compared to Tac-TIR4 (MFI=98 vs MFI=230 respectively). A chimera in which the 14 residues were grafted into the TIR domain of full length TLR4 (TLR4-9-4) had less cell surface expression (data not shown), and contained much less of the EndoH resistant glycoform (Fig. 5A) compared to WT TLR4. TLR4-9-4 also lacked response to LPS; however, both TLR4-9 and TLR4-9-4 demonstrated increased constitutive activity when overexpressed suggesting the cytoplasmic tail retained the ability to transducer signals (Fig. 5B). Together these data indicated that the 14 amino acids between residues 888 and 902 of the TLR9 TIR domain mediated intracellular localization. It is important to note that these results do not rule out other intracellular localization motifs C-terminal to amino acid 902. In fact, a more detailed analysis of cytoplasmic tail truncation mutants revealed several changes in intracellular staining pattern (Fig 6A) and glycosylation pattern (Fig 6B), suggesting there were several, possibly interrelated, localization motifs.
Fig. 4.
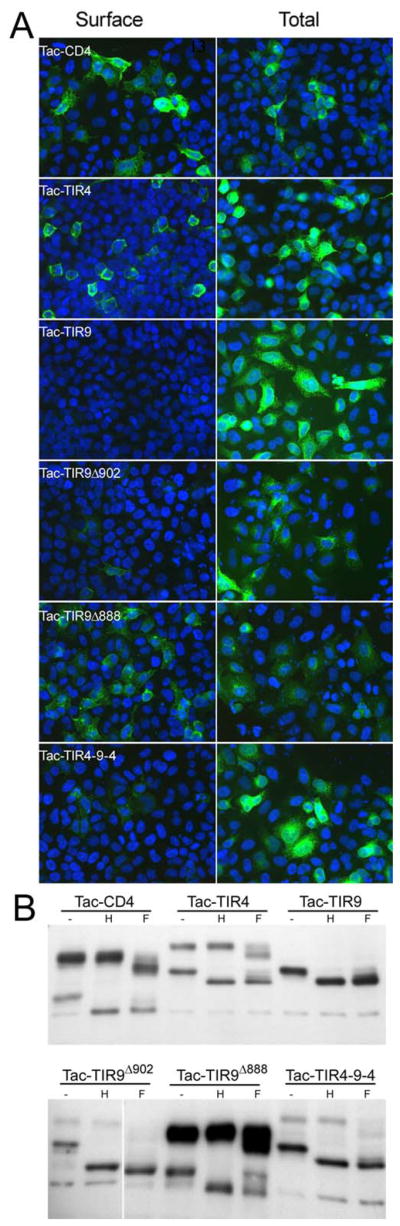
The cytoplasmic tail of TLR9 contains a 14 amino acid localization region. A, HeLa cells were transfected on coverslips with the indicated plasmids. Twenty-four hours later cells were either stained directly with Alexa488 labeled anti-Tac or fixed, permeabilized and stained. Images were taken on a Zeiss AxioImager at the same settings and compiled in Photoshop. B, HeLa cells were transfected in 6 well plates and 24 hours later harvested for glycosylation analysis as in Figure 2. The top and bottom were from two separate gels were developed simultaneously and are shown at the same exposure. The TacTIR9Δ902 samples were separated by the molecular weight marker in the original gel.
Fig. 5.

14 amino acid region confers intracellular localization to TLR4. A, HeLa cells were transfected in 6 well plates with the indicated GFP-tagged chimeras and 24 hours later harvested for GFP immunoprecipitation and glycosylation analysis. Arrowhead indicates mature glycoform. The second and third lanes were separated by the molecular weight marker in the original gel. B, HEK293 cells were transfected with MD2 plus 1.5 or 150 ng/well of TLR4, TLR4-9 or TLR4-9-4. Cells were stimulated with 100 ng/ml LPS and NF-κB activity was measured 24 hours later.
Fig. 6.

Truncation reveal vesicle motif. A, HeLa cells were transfected on coverslips with the indicated plasmids. Twenty-four hours later cells were fixed, permeabilized and stained with Alexa488 labeled anti-Tac. Single slice images (0.8 μM) were taken on a Zeiss LSM510 Meta with a 63x objective and images were compiled in Photoshop. B, HeLa cells were transfected in 6 well plates and 24 hours later harvested for glycosylation analysis as in Figure 2.
TacTIR9Δ888 represented a transition from mostly intracellular to almost exclusive cell surface expression. Since several truncations downstream of amino acid 888 displayed some vesicular staining (Fig. 6), we asked what vesicular compartment was targeted by our mutant and if that compartment was coincident with CpG DNA following its uptake from the medium. TacTIR9Δ898 showed significant targeting to endosomes and some cell surface expression (Fig. 7). Furthermore, co-localization in vesicles with fluorescently labeled CpG DNA was detected when HeLa cells were transfected with TacTIR9Δ898 (Fig. 7A). Unexpectedly, these vesicles were devoid of LAMP-1 (Fig. 7B), but instead contained an early endosome marker, Rab5 (Fig. 7C). Although some studies indicated that TLR9 colocalized with CpG DNA in lysosomes (13), our data are consistent with studies showing that intact TLR9 co-localizes with CpG DNA in early endosomes prior to reaching lysosomes (18). We predicted that there was a motif that targets vesicle localization in the region surrounding amino acid 888. Inspection of the sequence in this region revealed a potential tyrosine-based vesicle localization motif, YXXΦ (residues 888–893, where Φ is a hydrophobic amino acid) (21). Truncation mutants containing the YXXΦ motif were localized intracellularly and in vesicles (TacTIR9Δ898, TacTIR9Δ902). However, truncation mutants lacking the YXXΦ motif were redistributed to the cell surface, supporting our hypothesis that the cytoplasmic tail of TLR9 contained a tyrosine-based vesicle targeting motif.
Fig. 7.

Vesicle motif targets CpG DNA in an early endosome. A, HeLa cells were transfected with TacTIR9Δ898 and incubated with Cy-3 labeled CpG DNA for 1 hour prior to fixation and staining. Tac (green), CpG DNA (red). B, TacTIR9Δ898 transfected HeLa cells were stained for Tac (green) and LAMP-1 (red). C, HeLa cells were co-transfected with TacTIR9Δ898 and Rab5-GFP (green), an early endosome marker, and stained for Tac (red). Single slice images (0.8 μM) were collected on a Zeiss LSM510 Meta confocal microscope with a 63× objective. Sections of overlays were magnified for better visualization of co-localization (insets).
DISCUSSION
Bacterial DNA, a potent stimulus of TLR9, is contained within microorganisms and becomes accessible for recognition only after lysis in phagolysosomes. It has been shown that TLR9 colocalizes with CpG DNA in cytoplasmic vesicles but resides in the ER prior to CpG DNA exposure (5,13,19). Moreover, it has been proposed that TLR9 reaches the endocytic pathway through direct fusion of the ER with the CpG DNA containing endosome (18). What remains unknown is how the localization and movement of TLR9 is orchestrated. Here we show that both the ecto- and TIR domains of TLR9 contain sufficient information for intracellular localization and trafficking, and have identified a specific region within the TIR domain that contributes to intracellular localization and to vesicle targeting. By creating C-terminal truncation mutants in the Tac-TIR9 chimera we identified a 14 amino acid stretch between residues Y888 and W902 (Fig. 2) responsible for intracellular localization. The region contains a tyrosine-based vesicle-targeting motif, YNEL. Truncation of this motif leads to a loss in vesicular localization and redistribution to the cell surface.
Recent data from other laboratories has supported the idea that the transmembrane domain of mouse TLR9 controls intracellular localization (26,27). We have been unable to reproduce these data. Neither Flag-tagged mouse nor untagged human TLR9-4 chimera (TLR91–817TLR4634–835) was detected on the cell surface in HEK293 cells, HEK293T, MEF or HeLa cells (Supplementary Fig. 3, Supplementary Fig. 4 and data not shown). We failed to detect surface expression or mature glycoforms even if the chimera junction, like the previously described chimera, was 7 amino acids upstream of the TM domain (TLR91–810TLR4625–835). Futhermore, we generated a soluble TLR9 ectodomain and found it was also ER retained (C.A.L. personal observation), suggesting that sequences in the ectodomain are associating with chaperones or other luminal proteins to effect TLR9 ER localization. In our hands, a chimera containing the TM and cytoplasmic tail of TLR9 was localized like wild type; however, when most or all of the cytoplasmic tail was truncated from this mutant, the chimera was redistributed to the cell surface (Fig. 3) ruling out any role for the transmembrane domain in TLR9 localization. Lastly, the region that we identified in the cytoplasmic tail could confer, at least partial, intracellular retention to the normally surface expressed TLR4 in the context of either the Tac fusion or the full length WT TLR4. The redistribution of the TLR4-9-4 chimera to a mostly intracellular form eliminated the ability of TLR4 to recognize LPS.
The three-dimensional structure of the TLR9 TIR domain can be inferred by homology with the known structures of the TLR1, TLR2, and IL-1RAPL TIR domains (28, 29). Figure 8 highlights the 888–901 region (blue) on the predicted TIR domain structure of TLR9. This region lies on the surface in the first α helix and the loop that connects the first helix with the second β strand. Contiguous with this region is the BB loop, which contains a proline residue that is essential for signal transduction in most TLRs. In TLR2 this conserved proline is required for the binding of the TLR2-TIR domain to MyD88 (28). However, the proline is dispensable for TLR4 binding to MyD88 and MAL (30). If the TLR9 BB loop interacts with MyD88, as in TLR2, then the recruitment of MyD88 to TLR9 might influence the intracellular localization of TLR9 by interfering with its ability to interact with cytoplasmic localization adapter molecules. If the BB loop is not involved in TLR9-TIR binding to MyD88, as for TLR4, other adapter protein binding or receptor oligomerization may change the structure of the cytoplasmic tail in the region containing the BB loop and thus influence the ability of cytoplasmic localization adapter molecules to bind.
Fig. 8.

Sequence of the region of interest, amino acids 888–901 (top). Model showing location of the region of interest (blue) and the BB loop (red, putative MyD88 binding region) based on the TLR2 TIR structure (bottom) (28). The proposed YNEL vesicle motif is indicated with “A”. The position of the conserved proline residue that is mutated to histidine in TLR4 of C3H/HeJ mice is indicated in yellow.
Although TLR9 is in the ER prior to stimulation and found in lysosomes with CpG DNA at later time points, how TLR9 traffics is not clear. One mechanism by which this might occur is the direct fusion of TLR9-containing ER with early endosomes that contain CpG DNA (18). Alternatively, a CpG DNA-dependent signal upstream may induce the trafficking of TLR9 to the endosomal compartment. Previous results suggest that the TLR9 TIR domain may be tyrosine-phosphorylated in response to CpG stimulation (31) but we have been unable to detect phosphorylation of tyrosine 888 (CAL unpublished observation). Tyrosine 888 of TLR9 is not conserved in other intracellularly localized TLRs (TLRs 3, 7 and 8). Therefore it is not clear whether tyrosine 888 plays a structural role in the formation of the localization site, or serves a recognition function specific for TLR9. Although the surface receptor that would induce TLR-independent CpG DNA signaling events is unknown, surface recognition of CpG DNA by plasmacytoid DC has recently been shown to involve CXCL16 (32).
In summary, our data support a model where a tyrosine-based localization motif in the cytoplasmic tail of TLR9 is involved in trafficking of TLR9 to early endosomes where CpG DNA signaling initiates through TLR9. Further studies are needed to identify what cellular proteins interact with the tyrosine motif and control TLR9 localization and trafficking. How this trafficking relates to the release of DNA following uptake of pathogens remains to be determined.
Supplementary Material
Acknowledgments
We thank Drs. Remy Bosselut and Juan Bonifacino for plasmids used in this study. This work was supported in part by the Intramural Research Program of NIH (NCI) and by NCI/NIH K22 CA113705 (C.A.L.). J.C.B. was supported by an Immunology training grant T32 AI007643.
Footnotes
Abbreviations used in this paper: Toll-like receptor, TLR; oligodeoxynucleotide, ODN; Toll-IL-1 receptor domain, TIR; lipopolysaccharide, LPS; peptidoglycan, PGN; endoplasmic reticulum (ER); green-fluorescent protein, GFP; laser scanning microscopy, LSM.
References
- 1.Takeda K, Kaisho T, Akira S. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 2.Zhang D, Zhang G, Hayden MS, Greenblatt MB, Bussey C, Flavell RA, Ghosh S. Science. 2004;303(5663):1522–1526. doi: 10.1126/science.1094351. [DOI] [PubMed] [Google Scholar]
- 3.Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S. Science. 2004;303(5663):1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 4.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Science. 2004;303(5663):1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 5.Takeshita F, Leifer CA, Gursel I, Ishii KJ, Takeshita S, Gursel M, Klinman DM. J Immunol. 2001;167(7):3555–3558. doi: 10.4049/jimmunol.167.7.3555. [DOI] [PubMed] [Google Scholar]
- 6.Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A. J Exp Med. 2003;198(3):513–520. doi: 10.1084/jem.20030162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. Nature. 2000;408(6813):740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 8.Bauer M, Redecke V, Ellwart JW, Scherer B, Kremer JP, Wagner H, Lipford GB. J Immunol. 2001;166(8):5000–5007. doi: 10.4049/jimmunol.166.8.5000. [DOI] [PubMed] [Google Scholar]
- 9.Subramaniam S, Stansberg C, Cunningham C. Dev Comp Immunol. 2004;28(5):415–428. doi: 10.1016/j.dci.2003.09.016. [DOI] [PubMed] [Google Scholar]
- 10.Yamamoto M, Takeda K, Akira S. Mol Immunol. 2004;40(12):861–868. doi: 10.1016/j.molimm.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 11.Underhill DM, Ozinsky A, Hajjar AM, Stevens A, Wilson CB, Bassetti M, Aderem A. Nature. 1999;401(6755):811–815. doi: 10.1038/44605. [DOI] [PubMed] [Google Scholar]
- 12.Latz E, Visintin A, Lien E, Fitzgerald KA, Monks BG, Kurt-Jones EA, Golenbock DT, Espevik T. J Biol Chem. 2002;277(49):47834–47843. doi: 10.1074/jbc.M207873200. [DOI] [PubMed] [Google Scholar]
- 13.Ahmad-Nejad P, Hacker H, Rutz M, Bauer S, Vabulas RM, Wagner H. Eur J Immunol. 2002;32(7):1958–1968. doi: 10.1002/1521-4141(200207)32:7<1958::AID-IMMU1958>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 14.Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL. J Immunol. 2001;167(4):1882–1885. doi: 10.4049/jimmunol.167.4.1882. [DOI] [PubMed] [Google Scholar]
- 15.Heil F, Ahmad-Nejad P, Hemmi H, Hochrein H, Ampenberger F, Gellert T, Dietrich H, Lipford G, Takeda K, Akira S, Wagner H, Bauer S. Eur J Immunol. 2003;33(11):2987–2997. doi: 10.1002/eji.200324238. [DOI] [PubMed] [Google Scholar]
- 16.Matsumoto M, Funami K, Tanabe M, Oshiumi H, Shingai M, Seto Y, Yamamoto A, Seya T. J Immunol. 2003;171(6):3154–3162. doi: 10.4049/jimmunol.171.6.3154. [DOI] [PubMed] [Google Scholar]
- 17.Nishiya T, DeFranco AL. J Biol Chem. 2004;279(18):19008–19017. doi: 10.1074/jbc.M311618200. [DOI] [PubMed] [Google Scholar]
- 18.Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, Lien E, Nilsen NJ, Espevik T, Golenbock DT. Nat Immunol. 2004;5(2):190–198. doi: 10.1038/ni1028. [DOI] [PubMed] [Google Scholar]
- 19.Leifer CA, Kennedy MN, Mazzoni A, Lee C, Kruhlak MJ, Segal DM. J Immunol. 2004;173(2):1179–1183. doi: 10.4049/jimmunol.173.2.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bonifacino JS, Dell’Angelica EC. J Cell Biol. 1999;145(5):923–926. doi: 10.1083/jcb.145.5.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bonifacino JS, Traub LM. Annu Rev Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- 22.Uchiyama T, Nelson DL, Fleisher TA, Waldmann TA. J Immunol. 1981;126(4):1398–1403. [PubMed] [Google Scholar]
- 23.Hartmann G, Krieg AM. J Immunol. 2000;164(2):944–953. doi: 10.4049/jimmunol.164.2.944. [DOI] [PubMed] [Google Scholar]
- 24.Standley S, Roche KW, McCallum J, Sans N, Wenthold RJ. Neuron. 2000;28(3):887–898. doi: 10.1016/s0896-6273(00)00161-6. [DOI] [PubMed] [Google Scholar]
- 25.Nishiya T, Kajita E, Miwa S, Defranco AL. J Biol Chem. 2005;280(44):37107–37117. doi: 10.1074/jbc.M504951200. [DOI] [PubMed] [Google Scholar]
- 26.Barton GM, Kagan JC, Medzhitov R. Nat Immunol. 2006;7(1):49–56. doi: 10.1038/ni1280. [DOI] [PubMed] [Google Scholar]
- 27.Kajita E, Nishiya T, Miwa S. Biochem Biophys Res Commun. 2006;343(2):578–584. doi: 10.1016/j.bbrc.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 28.Xu Y, Tao X, Shen B, Horng T, Medzhitov R, Manley JL, Tong L. Nature. 2000;408(6808):111–115. doi: 10.1038/35040600. [DOI] [PubMed] [Google Scholar]
- 29.Khan JA, Brint EK, O’Neill LA, Tong L. J Biol Chem. 2004;279(30):31664–31670. doi: 10.1074/jbc.M403434200. [DOI] [PubMed] [Google Scholar]
- 30.Dunne A, Ejdeback M, Ludidi PL, O’Neill LA, Gay NJ. J Biol Chem. 2003;278(42):41443–41451. doi: 10.1074/jbc.M301742200. [DOI] [PubMed] [Google Scholar]
- 31.Sanjuan MA, Rao N, Lai KT, Gu Y, Sun S, Fuchs A, Fung-Leung WP, Colonna M, Karlsson L. J Cell Biol. 2006;172(7):1057–1068. doi: 10.1083/jcb.200508058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gursel M, Gursel I, Mostowski HS, Klinman DM. J Immunol. 2006;177(3):1575–1580. doi: 10.4049/jimmunol.177.3.1575. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.