

Abstract

Supramolecular complex of a cationic surfactant and oppositely charged disulfide containing polyelectrolyte was found to form micelle type aggregates at concentration much lower than the critical aggregate concentration (CAC) of the surfactant itself. We show that this difference can be utilized to generate stimulus-sensitive disassembly of these structures. This can be achieved either by converting the polyelectrolyte counterions to monovalent counterions in response to a stimulus or by simply weakening the interaction between the polymer and the surfactant in the presence of a stimulus. We have utilized three different stimuli to demonstrate these possibilities.
Keywords: Polyelectrolyte-surfactant aggregate, Disassembly, Glutathione, Redox, pH sensitive, Ionic strength
Introduction
Surfactant assemblies, such as micelles and vesicles, are widely studied due to their container properties and have been shown to be useful in various applications ranging from environmental to medicinal applications.1 Polymeric versions of these surfactants have been extensively explored due to their greater stability and lower critical aggregation concentrations (CAC).2 The utility of the macromolecular micelles in various applications dramatically increases when one considers the possibility of these assemblies being responsive to an external stimulus such as pH, temperature, ionic strength, or redox environment.3 For example, redox sensitive supramolecular aggregates are interesting, because of the possibility of them being susceptible towards disassembly in specific disease locations where the redox environment is significantly different from that of healthy cells. One such example of this is the reducing environment near cancer cells due to the over-expression of a tripeptide called glutathione (GSH).4 It is well established that disulfide bonds can be cleaved by GSH5 and thus there have been several attempts in designing amphiphilic systems containing this particular redox sensitive functionality.6 We have recently demonstrated a small-molecule surfactant system wherein the hydrophobic tail was connected to the ionic head group with a disulfide bond and the corresponding micelles were found to be capable of releasing encapsulated guest molecules in the presence of glutathione due to the disassembly of the micelles.7 However, in this system the CMC of the surfactant was rather high and the cleavage of the disulfide bond produced insoluble alkyl chains which are not desirable for biological applications. Recently we have reported the formation of amphiphilic homopolymer8 like structure, where supramolecular assemblies were achieved using electrostatic interactions among suitably chosen polyelectrolytes and surfactants.9 We demonstrated that the original CMC of the surfactant is reduced significantly due to the polymeric effect and such systems could be used for protein sensing.9 We envisaged that these systems would also be useful for stimuli-sensitive disassembly with two complementary approaches by: (i) changing from a polyvalent interaction to a monovalent one in the presence of an external stimulus; (ii) weakening the interaction between the polyelectrolyte and the surfactant in the presence of an external stimulus. In this paper, we show examples of both of these approaches. A redox-sensitive polymer assembly was used to demonstrate the former approach, while pH and ionic strength dependencies were exploited for the latter.
Experimental
Materials and Methods
All chemicals and solvents were purchased from commercial sources and were used as such, unless otherwise mentioned. Synthesis of polymer P2 was described earlier.9 1H NMR spectra were recorded on a 400 MHz Bruker NMR spectrometer using the residual proton resonance of the solvent as the internal standard. Chemical shifts are reported in parts per million (ppm). When peak multiplicities are given, following abbreviations are used: s, singlet; d, doublet; t, triplet m, multiplet. 13C-NMR spectra were proton decoupled and recorded on a 100 MHz Bruker spectrometer using carbon signal of the deuterated solvent as the internal standard. Emission spectra were recorded in JASCO FP-6500 spectrofluorimeter. Molecular weights of the polymers were estimated by gel permeation chromatography (GPC) using a PMMA standard and RI detector. The samples were eluted using 0.1 M LiCl solution of DMF. Dynamic light scattering studies were carried out in Zetasizer nano series Malvern instrument. TEM measurements were carried out using a JEOL 100CX 100KV TEM.
Synthesis of P1
50 µL of glacial acetic acid was added to a solution of polymer P1a10 (Mn = 5560, PDI =1.2, 200 mg, 0.78 mmol) in DMF (0.5 mL). Thiopropionic acid (91 mg, 0.86 mmol) was added to the solution and it was stirred at room temperature for 6 h. The stirring was stopped and the polymer was precipitated in diethylether, filtered, and dried under vacuum. Yield: 84%. 1H NMR (DMSO): δ(ppm): 4.2-4.09 (broad peak, 2H), 2.88-2.86 (broad peak, 4H), 2.62 (broad peak, 2H), 2.05-1.65 (m, 2H), 0.97-0.71(m, 3H). 13C NMR (DMSO): δ(ppm): 177.4, 176.0, 63.9, 44.9, 37.0, 34.3, 33.1, 29.9, 25.0.
Synthesis of P2
26.8 mg (0.187 mmol) of Cu(I)Br was added to a 10 mL round bottom flask equipped with a septa and gas inlet/outlet. This flask was purged with a flow of argon for 5 minutes. Then, 64.5 mg (0.374 mmol) of N,N,N’,N’,N’’-pentamethyl diethylenetriamine (PMDETA) was added to this flask and the resultant solution was stirred for 5 more minutes. Following this, a solution of the t-buty1 methacrylate (2.00 mL, 14.1 mmol) in degassed DMF (1.5 mL) was added and stirred for another 5 minutes. To this mixture, the initiator ethy1-2-bromoisobutyrate (EBIB) (36.5 mg, 0.187 mmol) was added and the flask was transferred to a preheated oil bath at 60 °C. The polymerization reaction was carried out at this temperature under argon atmosphere for 12 h. The reaction was stopped and the polymer was precipitated from 3:1 (v/v) methanol/water mixture and dried under vacuum for 5 h. Yield: 56%. Molecular weight: Mn= 8200 (PDI-1.5). 1H NMR (CDCl3): δ(ppm): 2.1-1.80 (m, 2H), 1.43 (broad s, 9H), 1.12-1.02 (m, 3H). 13C NMR (CDCl3): δ(ppm): 175.4, 83.0, 44.3, 34.8, 28.7, 24.6.
To a solution of 500 mg (3.53 mmol) of this polymer in dichloromethane (1 mL) was added 2 mL of trifluoroacetic acid. The reaction mixture was stirred at room temperature under inert atmosphere for 12 h. The product polymer precipitated out of solution. The reaction mixture was concentrated in vacuo and diethylether was added to precipitate the polymer as a white powder. The product was filtered and dried under vacuum to get the desired polymer, P2. Yield: 83%. 1H NMR (DMSO): δ(ppm): 1.92-1.60 (m, 2H), 1.00-0.90 (m, 3H). 13C NMR (DMSO): δ(ppm): 179.4, 46.1, 34.3, 24.3
Preparation of polymer-surfactant complex
Measured amount of polymer was added to appropriate volume of 5.0 mM aqueous NaOH solution so that the ratio of NaOH: polymer repeat unit = 1:1 and the solid polymer was dissolved by sonication over 10−15 minutes. To this solution (2.0 mL), a solution of DTAB (2.0 mL, 5.0 mM) was added and the mixture was allowed to equilibrate for 1 h.
Determination of CAC
A stock solution (concentration 10−4 M) of Nile red in dichloromethane was made from which 0.2 mL was transferred to another vial and the solvent was removed. To this, 2 mL of the stock solution of surfactant /polymer-surfactant complex was added. The mixture was sonicated for 30 minutes and filtered into a cuvette and emission spectrum was recorded (λex = 550 nm). Fluorescence emission intensity of Nile red was monitored at different concentrations. The emission intensity was then plotted against the concentration of the surfactant/ polymer-surfactant complex and the CAC was determined to be the inflection point observed in such a plot.
Glutathione induced dye release study
Nile red (1 × 10−5M) was encapsulated in the assembly formed by 1:1 mixture of DTAB and sodium salt of polymer P1 (1.18 mM). To this solution (2 mL), one mole equivalent of glutathione (50 µL, 4.72 × 10−2 M aqueous solution) was added. The intense pink color started fading away immediately and the emission from Nile red almost completely disappeared suggesting complete release of the encapsulated dye.
Salt induced dye release study
Nile red encapsulated in a 1:1 mixture of DTAB and P2 was prepared following the procedure described above. The concentration of the Nile red was 1 × 10−5 M whereas the concentration of the polymer-surfactant complex was 11.4 mM. 2 mL of this solution was taken in a cuvette and to this solution, a measured amount of 5.65 M NaCl solution was added and the emission intensity of Nile red was monitored as a function of salt concentration. Total volume of NaCl solution that was added in the end of the experiment was 50 µL, which is insignificant compared to the total volume of the solution (2 mL). Thus the change in emission intensity due to dilution effect was considered negligible. In the control experiment, Nile red was encapsulated inside the micelle formed by only DTAB instead of DTAB-P2 complex. The emission intensity of Nile red was then plotted against the salt concentration of the surfactant/ polymer-surfactant complex and the salt concentration required for complete disassembly was determined to be the inflection point observed in such a plot.
pH dependent dye release study
pH induced dye (Nile red) release study was also carried out following similar procedure as explained for the salt study. In this case, instead of salt solution, HCl solution (0.1 M, 639 µL) was added in steps into 9 mL solution of (DTAB+P2) and the emission intensity of the Nile red was monitored as a function of solution pH. For checking the reversibility, measured amounts of NaOH solution were added. Before each spectral measurement the pH of the solution was checked using a pH meter.
TEM studies
TEM images were taken using a JEOL 100CX 100KV TEM. Samples were prepared by dipping copper EM grids in aqueous solution of 0.1 mmol P2-DTAB complex with and without HCl (1 mM), the excess of solvent was removed by placing the tissue in the bottom of the EM grid.
Results and Discussions
To test the hypothesis of whether converting a polyvalent counterion for the surfactant to monovalent counterions would result in disassembly, we designed and synthesized polyelectrolyte P1 (Figure 1). In this polymer, each ionic carboxylate unit is connected to the polymer backbone by a labile disulfide bond. Complexation of P1 with the surfactant decyltrimethylammonium bromide (DTAB) reduces the CAC of the surfactant, as we have shown in our previous report,9 and thus the resulting complex can encapsulate guest molecules at much lower concentrations compared to the DTAB alone. We envisaged that cleavage of the disulfide bond in the presence of glutathione (GSH) would disrupt the polyvalent interactions and the ensuing disassembly of the supramolecular aggregate will result in the release of the encapsulated guest molecules, as shown in Figure 2.
Figure 1.

Structures of polymers and surfactant.
Figure 2.
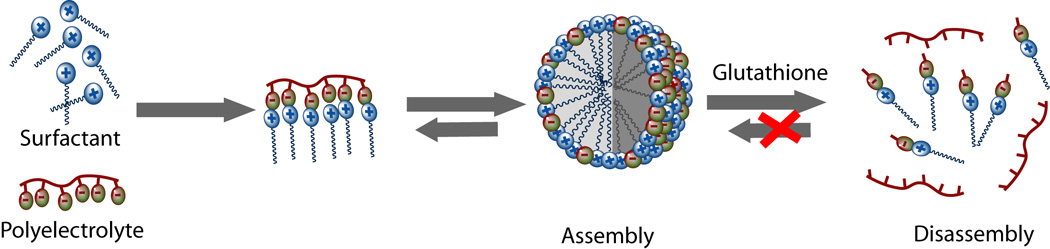
Schematic representation for the redox-sensitive assembly/disassembly process
Polymer P1 was synthesized in 84% yield from the parent polymer, P1a,10 containing pyridy1 disulfide moiety by reaction with thiopropionic acid under mild acidic conditions (Scheme 1). The product polymer was characterized by 1H and 13C NMR; the absence of any aromatic proton signal in the 1H NMR spectra confirmed the complete substitution of the pyridy1 disulfide moiety. The polymer P1 was dissolved in H2O in the presence of 1 mole equivalent (with respect to the repeat unit of the polymer) of NaOH. To this polyanionic polymer, one equivalent of DTAB per carboxylate functionality was added. To assess the CAC of the polymer-surfactant complex, we used fluorescence behavior of Nile red as the probe.11 Nile red is not soluble in water and thus does not show any emission in water. However, when encapsulated in the hydrophobic pocket of a supramolecular aggregate, the intensity of the emission band enhances drastically. The emission spectra of the encapsulated nile red in 1:1 complex of sodium salt of P1 and DTAB, was monitored as a function of the surfactant-polymer complex concentration. The spectral variations are shown in Figure 3. It can be noticed that unless the concentration of the polymer-surfactant complex reaches a threshold value, the emission intensity of the Nile red is close to zero, beyond which the intensity of Nile red emission starts increasing in linear fashion. The critical aggregation concentration (CAC) of the polymer-surfactant complex was determined from the inflection point of this plot and was found to be 7.2 × 10−4 M. This value is nearly two orders of magnitude lesser than the CAC of DTAB (6.6 × 10−2 M) itself. Such a drastic reduction in CAC is certainly reflective of the polyvalent effect.
Scheme 1.

Synthesis of polymer P1
Figure 3.
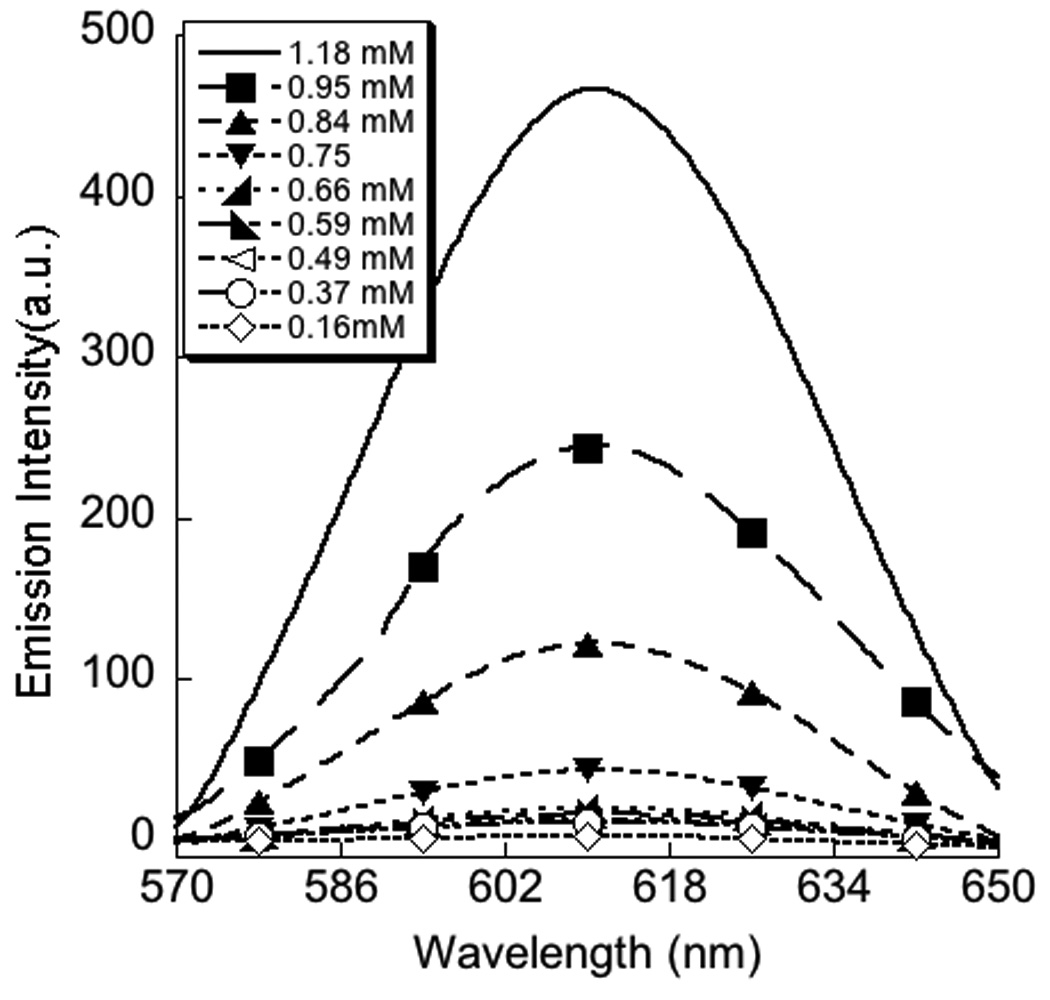
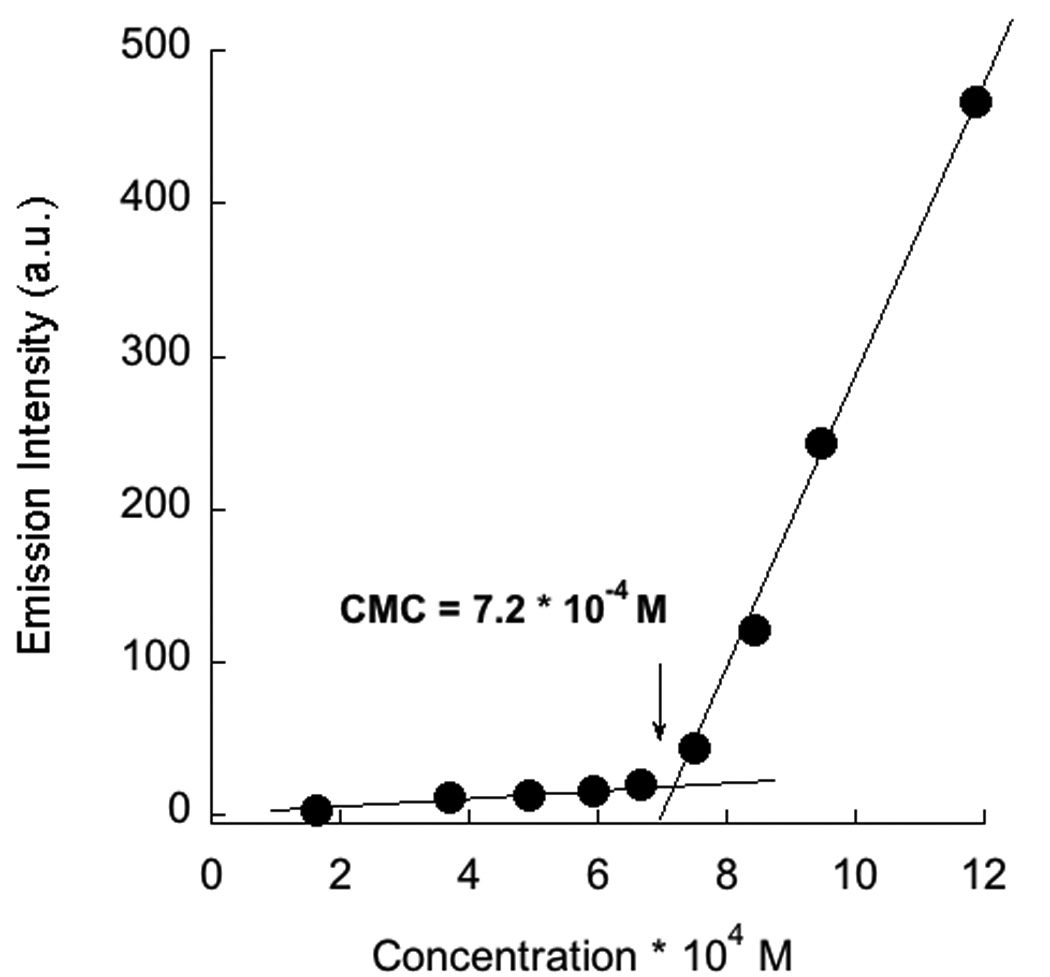
Determination of CAC of 1:1 mixture of DTAB+P1 by nile red emission spectra; left-change in emission spectra of nile red as a function of concentration of (DTAB+P1) complex; right- plot of emission intensity at emission maxima (615 nm) as a function of concentration of (DTAB+P1) complex
The difference in CAC between the surfactant and the polymer-surfactant complex suggests that there is a possibility of disassembling the polymeric aggregate, if an appropriate redox stimulus is applied in the concentration range in-between the two CACs. To test this possibility, we investigated the release of the hydrophobic dye molecule (Nile red) in the presence of glutathione (GSH). Nile red was encapsulated within the hydrophobic interiors of a P1:DTAB complex (1.18 mM). Upon addition of GSH, the pink color of the solution due to the presence of Nile red disappeared almost spontaneously and the emission intensity of Nile red was reduced to almost zero when monitored after 20 minutes (Figure 4). This result shows that the reductive cleavage of the disulfide does cause the disassembly of micelle. We have carried out two control experiments to further insure that this is indeed the case. In the first experiment, we checked whether GSH by itself could cause any change in the emission intensity of the fluorescent probe (Nile red). Addition of GSH to a solution of Nile red does not cause any change in the fluorescence behavior of the solution, which shows that the observed reduction is not the inherent response of nile red to the tripeptide. In another control experiment, we studied the effect of GSH on the nile red encapsulated complex of poly(methacrylic acid) (P2):DTAB under identical experimental condition and found no effect on the emission property of the probe, suggesting that it is indeed the cleavage of disulfide bonds that is responsible for the disassembly of the P1:DTAB complex.
Figure 4.
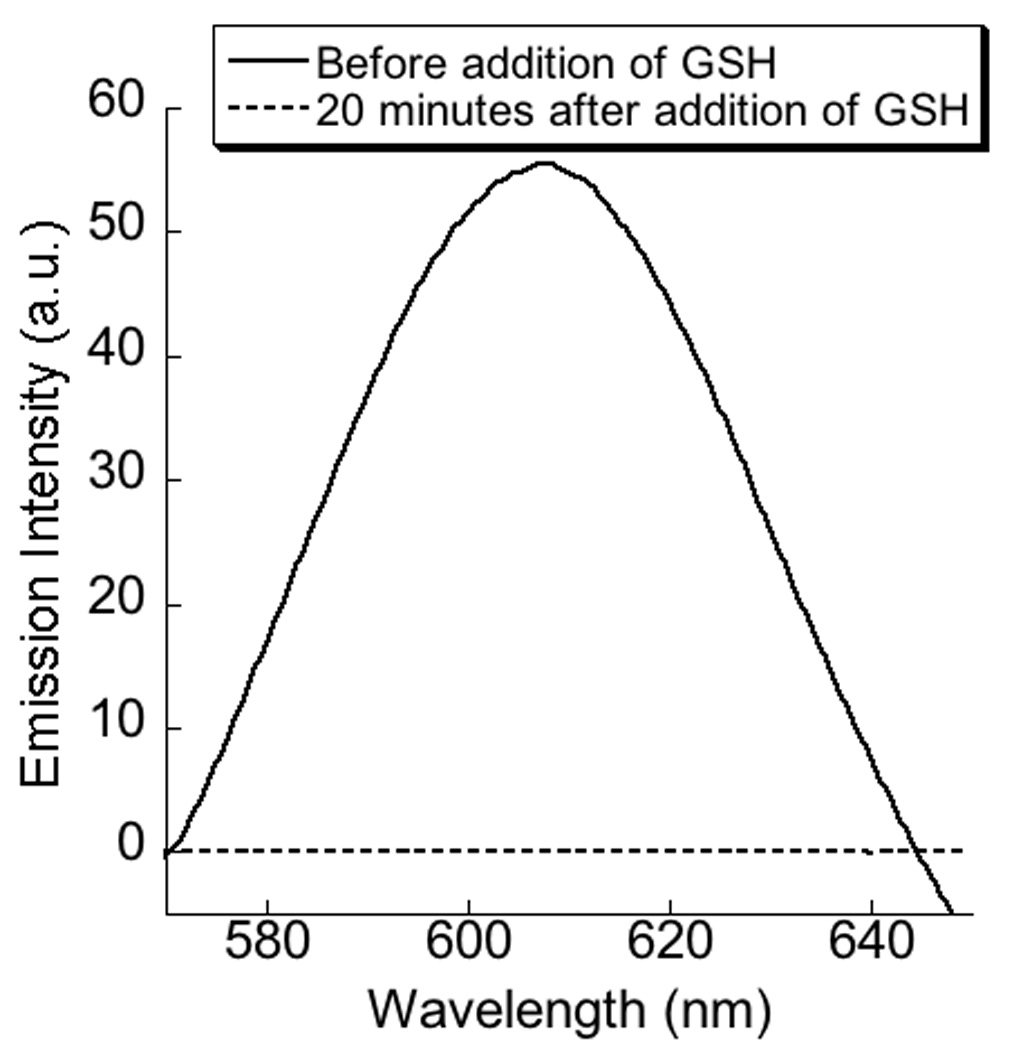
Top- Scheme showing glutathione induced cleavage reaction; bottom- effect of Glutathione on the emission spectra of encapsulated nile red in 1:1 complex of (P1 + DTAB).
A complementary approach to disassemble the micelle in the presence of the stimulus is to use it to weaken the interaction between the polyelectrolyte and the surfactant. For this purpose, the polymer P2 was used as the polyelectrolyte. The CAC of the complex P2:DTAB was found to be 8 × 10−4 M, 9 quite similar to that observed in the P1:DTAB complex. Since the complexation is based on electrostatics, it is possible to lower the strength of the interaction by increasing the ionic strength of the solution due to charge screening effects. To test this possibility, we probed the encapsulation of Nile red within a 1:1 P2:DTAB complex (11.4 mM) at different ionic strengths. A sharp decrease in the emission intensity of the dye molecule was observed between 8 and 10 mM sodium chloride concentration (Figure 5), which suggests that a disassembly indeed occurs at high ionic strengths. We have independently confirmed that the micellar assembly of DTAB itself at this ionic strength is intact, when the concentration of the surfactant is above its CAC. Therefore, the observed guest molecule release is most likely due to the electrostatic screening between the polymer and the surfactant molecules at high ionic strengths.
Figure 5.
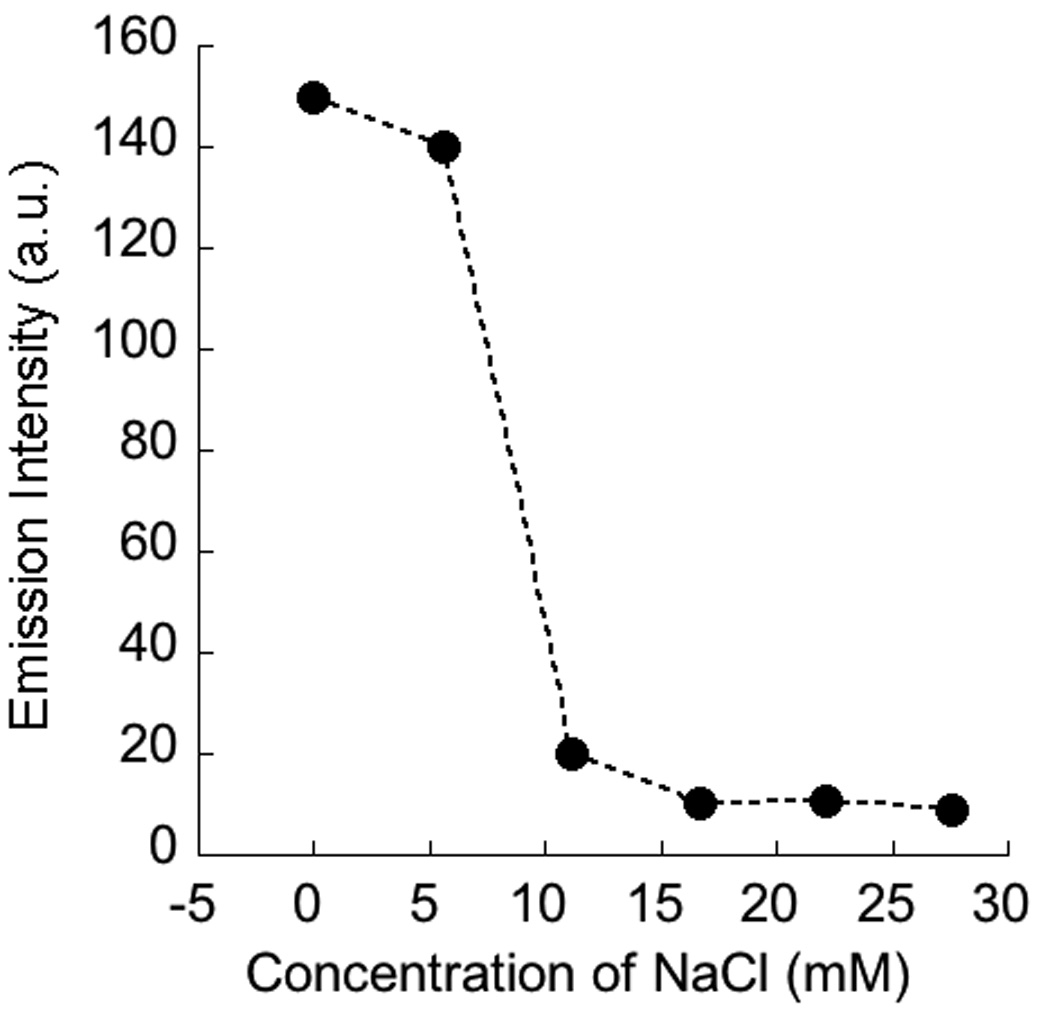
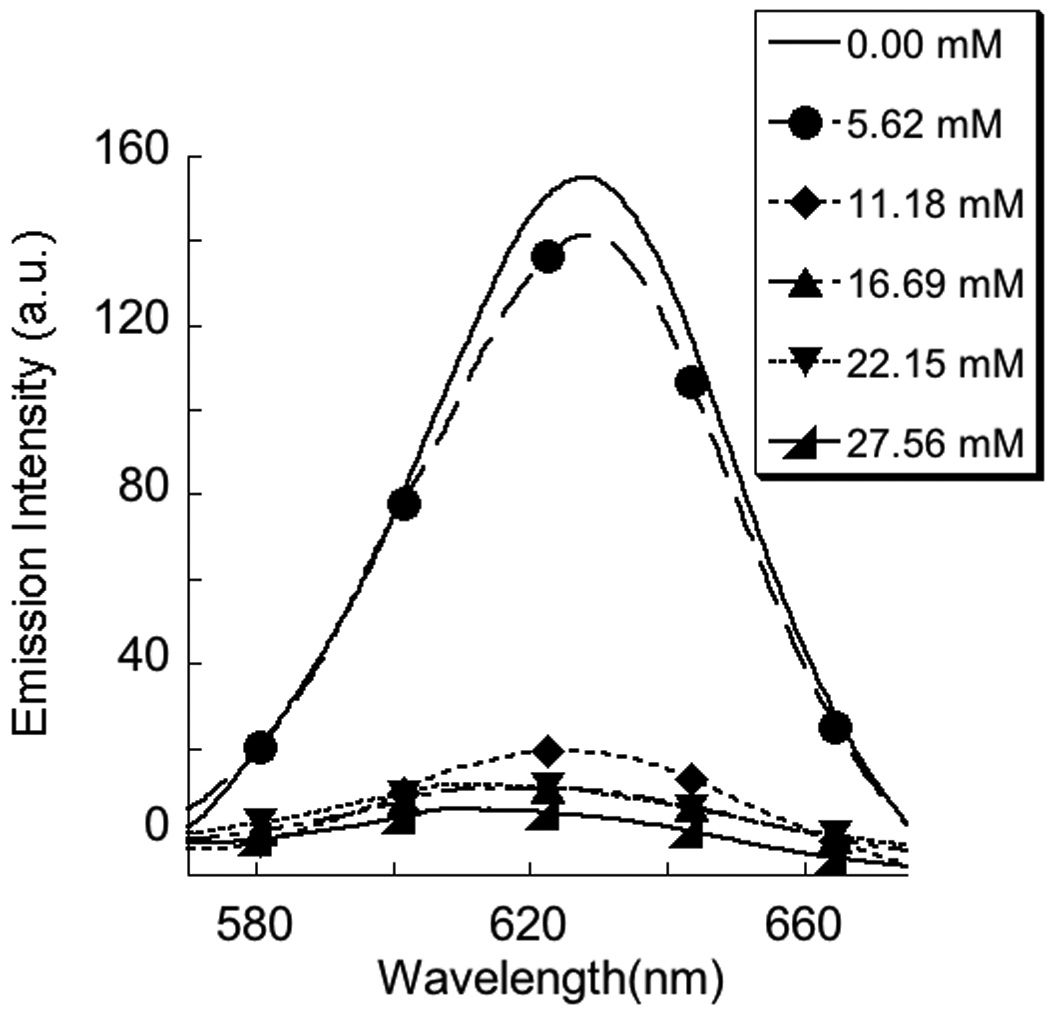
Change in the emission intensity of nile red (monitored at 625 nm) encapsulated in 1:1 complex of P2 + DTAB (a) as a function of NaCl concentration; (b) the emission spectra represented in (a).
The fact that the anionic polymer contains carboxylic acid groups and the cationic counterpart is based on a tetraalkylammonium moiety also provides an opportunity to induce a similar disassembly using pH change as the stimulus. This is because the change in pH affects the charged nature of carboxylic acid (anionic to neutral with decreasing pH), but should have no effect on the charge of the DTAB, since this is based on the non-protonatable quaternary ammonium cation. Nile red fluorescence was probed at different pHs in a 1:1 P2:DTAB complex, and it was observed that the emission intensity indeed reduced with decreasing pH (Figure 6), indicating the disassembly of the micelle between the pHs of 5.5 and 4.5. This is expected considering that the average pKa of polymethacrylic acid is 5.0.12
Figure 6.
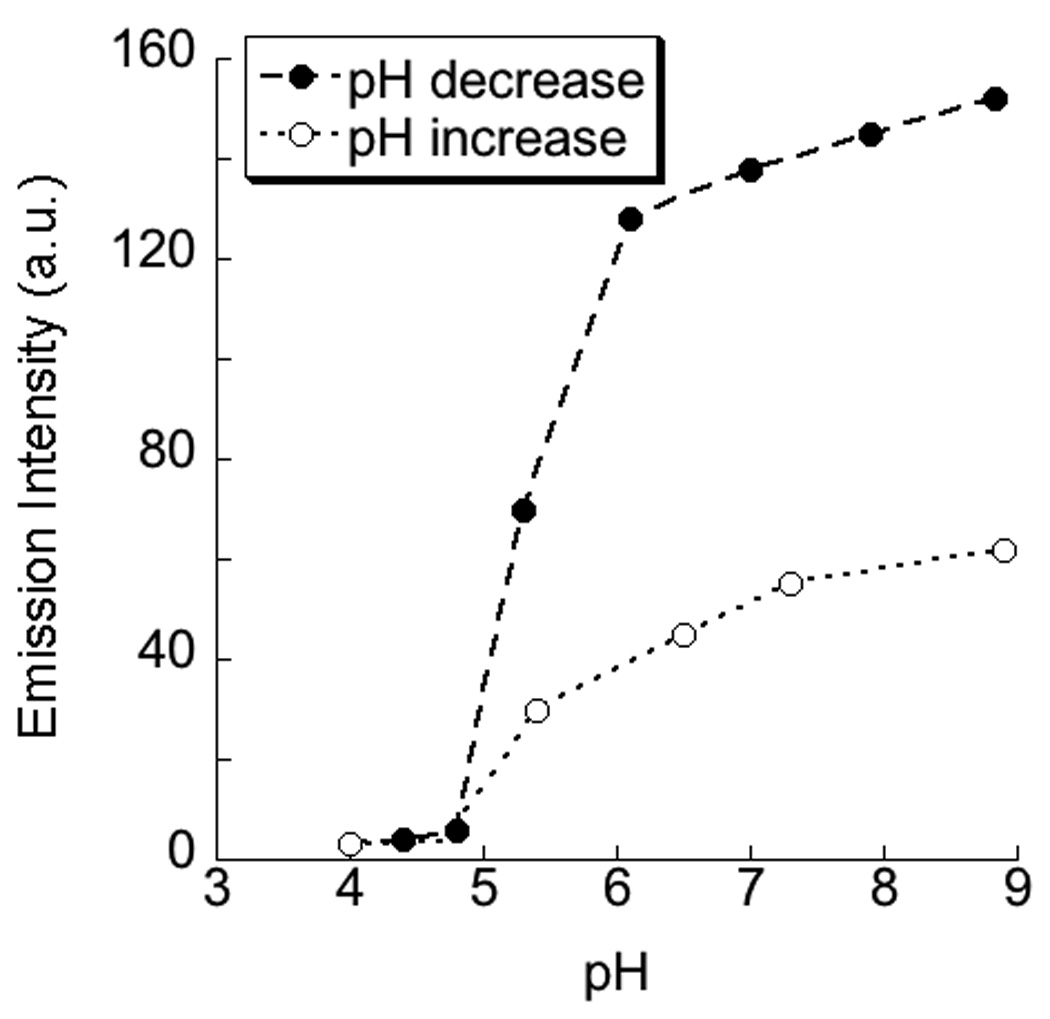
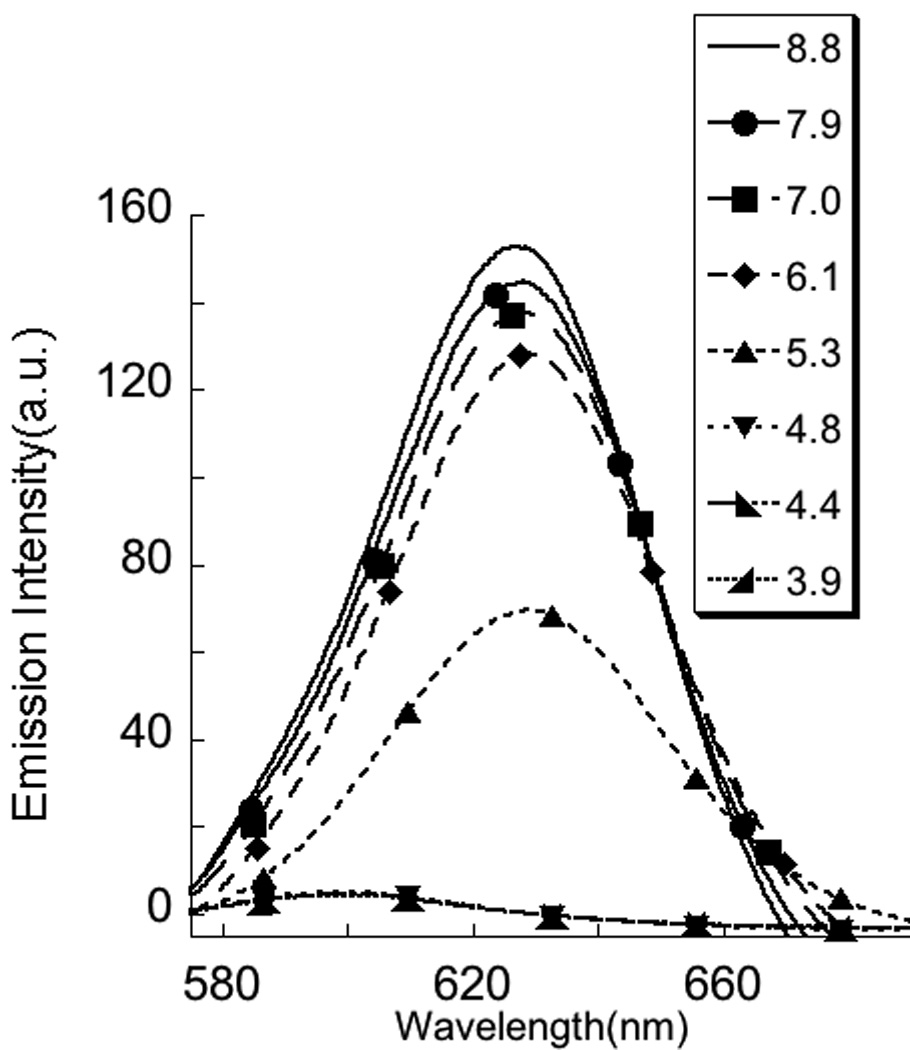
Change in the emission intensity of nile red (monitored at 625 nm) encapsulated in 1:1 complex of P2 + DTAB (a) as a function of pH; (b) the emission spectra represented in (a).
The pH-based disassembly is also interesting because of the possible reversibility in the assembly/disassembly process. Accordingly, when the previous mixture is subjected to a pH-increase, the emission intensity of the Nile red could be recovered to 40% of its original value (Figure 6). One could conclude that the results suggest that the process is only partially reversible. However, note that this data does show full reversibility with respect to pH changes, if one accounts for the ionic strength change that accompanies the pH change cycle. In our case, generation of NaCl (7.1 mM) during the pH changes could have caused the partial disruption of the micelle, which accounts for the incomplete recovery of nile red emission. If we fit this pH-based data into the ionic strength data in Figure 5, the results are indeed consistent with this supposition.
Having tested the utility of these polymer-surfactant complexes for stimuli-sensitive dye release, we were interested in obtaining further supporting evidence for disassembly. Thus, we studied the nature of these aggregates using transmission electron microscopy (TEM) and dynamic light scattering (DLS) studies. TEM images of 1:1 complex of DTAB: P2 at pH 8 are shown in Figure 7. Near spherical micelle-type aggregates could be observed by TEM with average diameter of approximately 70 nm. In the DLS studies, the average diameter of these particles was found to be of 67 nm which is comparable to that observed in TEM images. As with the dye encapsulation studies, the disassembly process could be observed in both TEM and DLS studies. The spherical particles, which were present at pH 8 (Figure 7a), disappeared completely at a lower pH (~4.0) (Figure 7b). Similarly in DLS studies, the correlation function obtained was very poor for both lower pH and the high salt concentration conditions. These results suggest the absence of large enough particles in the solution for scattering. Combined, these results provide clear supporting evidences for the disassembly-based dye release observed in the dye-encapsulation studies.
Figure 7.

TEM pictures of P2 + DTAB (1:1) complex at pH 8 (left) and pH 4 (right)
Conclusions
In summary, we have shown that: (i) polyelectrolytes are effective counterions for small molecule surfactants to generate non-covalent polymeric assemblies with significant decrease in CACs; (ii) By staying at a concentration in between the CAC of the surfactant and its complex with a disulfide containing polyelectrolyte, external stimuli, such as glutathione, can disassemble these aggregates. The disassembly is achieved because the polyvalent interaction between the polyelectrolyte and surfactant is changed to a monovalent interaction through a reductive disulfide bond cleavage reaction. (iii) Disassembly of these aggregates can also been achieved by high salt concentrations and lower pH conditions wherein the electrostatic interactions between the polymer and the surfactant is weakened by the stimuli. Stoichiometric polyelectrolyte-surfactant complexes have been reported to be insoluble in water.13a, 13b Most of the studies related to polyelectrolyte-surfactant complexes13 in aqueous solution deal with block/random copolymers containing both water soluble nonionic and ionic units along with the oppositely charged surfactant molecules. Those systems resemble closely to the noncovalent block copolymers, unlike the homopolymeric system, reported here. In our system also, we find precipitation in higher concentrations; however in the concentration range where we have done the assembly-disassembly studies, we found the complexes to be optically clear in water. Further it is also conceivable that in dilute solutions, even though the feed ratio of the surfactant: polyelectrolyte is 1:1, incomplete complexation may leave few carboxylate units free which may stabilize the resulting complex. Detail studies to investigate these structural details of the aggregates are underway. Although neither the biocompatibility of these structures nor the CACs are optimized for applications such as drug delivery, it is intriguing to think about the possibilities of using such noncovalent amphiphilic assemblies for such vehicles.
Acknowledgement
We thank the NIGMS of the NIH for support and the NSF-MRSEC for infrastructure.
References and Notes
- 1.Evans DF, Wennerstrom H. The Colloidal Domain. 2nd ed. New York: Wiley-VCH; 1999. [Google Scholar]
- 2.Fleischer G. J. Phys. Chem. 1993;97:517. [Google Scholar]
- 3.Rui Y, Wang S, Low PS, Thompson DH. J. Am. Chem. Soc. 1998;120:11213. [Google Scholar]; (b) Goodwin AP, Mynar JL, Ma Y, Fleming GR, Frechet JMJ. J. Am. Chem. Soc. 2005;127:9952. doi: 10.1021/ja0523035. [DOI] [PubMed] [Google Scholar]; (c) Jaeger DA, Zeng X. Langmuir. 2003;19:8721. and references therein. [Google Scholar]; (d) Menger FM, Gabrielson K. J. Am. Chem. Soc. 1994;116:1567. [Google Scholar]; (e) Heskins M, Guillet JE. J. Macromol. Sci. Chem. 1968:1441. [Google Scholar]; (f) Jiang J, Tong X, Zhao Y. J. Am. Chem. Soc. 2005;127:8290. doi: 10.1021/ja0521019. [DOI] [PubMed] [Google Scholar]; (g) Eastoe J, Vesperinas A. Soft Matter. 2005;1:338. doi: 10.1039/b510877m. [DOI] [PubMed] [Google Scholar]; (h) Heller J, Barr J, Ng SY, Abdellauoi KS, Gunny R. Avd. Drug. Deliv. Rev. 2002;54:1015. doi: 10.1016/s0169-409x(02)00055-8. [DOI] [PubMed] [Google Scholar]; (i) Jiang XG, Lavender CA, Woodcock JW, Zhao B. Macromolecules. 2008;41:2632. [Google Scholar]; (j) Rijcken CJF, Soga O, Hennink WE, van Nostrum CFJ. Controlled Release. 2007;120:131. doi: 10.1016/j.jconrel.2007.03.023. [DOI] [PubMed] [Google Scholar]; (k) Lowe AB, Torres M, Wang R. J. Polym. Sci Part A: Polym Chem. 2007;45:5864. [Google Scholar]; (l) Feng C, Shen Z, Gu L, Zhang S, Li L, Lu G, Huang X. J. Polym. Sci Part A: Polym Chem. 2008;46:5638. [Google Scholar]
- 4.Saito G, Swanson JA, Lee K-D. Avd. Drug. Deliv. Rev. 2003;55:199. doi: 10.1016/s0169-409x(02)00179-5. [DOI] [PubMed] [Google Scholar]
- 5.Lamoureux GV, Whitesides GM. J. Org. Chem. 1993;58:633. and references therein. [Google Scholar]
- 6.For disulfide containing amphiphilic structures see Nolan D, Darcy R, Ravoo BJ. Langmuir. 2003;19:4469. Kakizawa Y, Harada A, Kataoka K. J. Am. Chem. Soc. 1999;121:11247. Saily VMJ, Ryhanen SJ, Lankinen H, Luciani P, Mancini G, Parry J, Mikko, Kinnunen PKJ. Langmuir. 2006;22:956. doi: 10.1021/la052398o. Zhang J, Jing B, Tokutake N, Regen ST. Biochemistry. 2005;44:3598. doi: 10.1021/bi048258f. Jong LI, Abbott NL. Langmuir. 1998;14:2235. Ringsdorf H, Schlarb B, Venzmer J. Angew. Chem. Int. Ed. 1988;27:113. Li Y, Lokitz BS, Armes SP, McCormick CL. Macromolcecules. 2006;39:2726.
- 7.Ghosh S, Irvin K, Thayumanavan S. Langmuir. 2007;23:7916. doi: 10.1021/la700981z. [DOI] [PubMed] [Google Scholar]
- 8.(a) Basu S, Vutukuri DR, Shyamroy S, Sandanaraj BS, Thayumanavan S. J. Am. Chem.Soc. 2004;126:9890. doi: 10.1021/ja047816a. [DOI] [PubMed] [Google Scholar]; (b) Savariar EN, Aathimankandan SV, Thayumanavan S. J. Am.Chem. Soc. 2006;128:16224. doi: 10.1021/ja065213o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Savariar EN, Ghosh S, González DC, Thayumanavan S. J. Am. Chem. Soc. 2008;130:5416. doi: 10.1021/ja800164z. [DOI] [PubMed] [Google Scholar]
- 10.For synthesis of polymer P1a see Ghosh S, Basu S, Thayumanavan S. Macromolecules. 2006;39:5595.
- 11.Fowler SD, Greenspan P. J. Histochem. Cytochem. 1985;33:833. doi: 10.1177/33.8.4020099. [DOI] [PubMed] [Google Scholar]
- 12.Kim B, Peppas NA. J. Biomater. Sci. Polym. Edn. 2002;13:1271. doi: 10.1163/156856202320893000. [DOI] [PubMed] [Google Scholar]
- 13.(a) Goddard ED, Ananthapadmanabhan KP. Interactions of Surfactants with Polymers and Proteins. Boca Raton, FL: CRC Press; 1993. [Google Scholar]; (b) Macknight WJ, Ponomarenko EA, Tirrell DA. Acc. Chem. Res. 1998;31:781. [Google Scholar]; (c) Zhou S, Chu B. Adv. Mater. 2000;12:545. [Google Scholar]; (d) Bronich TK, Kabanov AV. Macromolecules. 1997;30:3519. [Google Scholar]; (e) Bronich TK, Cherry T, Vinogradov SV, Eisenberg A, Kabanov VA, Kabanov AV. Langmuir. 1998;14:6101. [Google Scholar]; (f) Kabanov AV, Bronich TK, Kabanov VA, Yu K, Eisenberg A. J. Am. Chem. Soc. 1998;120:9941. [Google Scholar]; (g) Wang C, Tam KC. Langmuir. 2002;18:6484. [Google Scholar]; (h) Mizusaki M, Morishima Y, Yoshida K, Dubin P. Langmuir. 1997;13:6941. [Google Scholar]; (i) Hashidzume A, Yoshida K, Morishima Y, Dubin P. J. Phys. Chem. B. 2002;106:2007. [Google Scholar]; (j) Hayakawa K, Tanaka R, Kurawaki J, Kusumoto Y, Satake I. Langmuir. 1999;15:4213. [Google Scholar]; (k) Zhou S, Burger C, Chu B. J. Phys. Chem. B. 2004;108:10819. [Google Scholar]; (l) Hayakawa K, Satake I, Kwak JCT. Colloid Polym Sci. 1994;272:876. [Google Scholar]