

Summary
Recent studies have shown that galectin-3 (Gal-3; also known as LGALS3), a β-galactoside-binding lectin, promotes cell migration during re-epithelialization of corneal wounds. The goal of this study was to characterize the molecular mechanism by which Gal-3 stimulates cell migration. We demonstrate here that exogenous Gal-3, but not Gal-1 or Gal-8, promotes cell scattering and formation of lamellipodia in human corneal epithelial cells in a β-lactose-inhibitable manner. α3β1 integrin was identified as the major Gal-3-binding protein in corneal epithelial cells by affinity chromatography of cell lysates on a Gal-3-Sepharose column. Preincubation of cells with anti-α3 integrin function-blocking antibody significantly inhibited the induction of lamellipodia by Gal-3. Furthermore, exogenous Gal-3 activated both focal adhesion kinase, a key regulator of integrin-dependent intracellular signaling, and Rac1 GTPase, a member of the family of Rho GTPases, well known for its role in the reorganization of the actin cytoskeleton and formation of lamellipodial extensions. Experiments involving knockdown of β-1,6-N-acetylglucosaminytransferase V, an enzyme that synthesizes high-affinity glycan ligands for Gal-3, revealed that carbohydrate-mediated interaction between Gal-3 and complex N-glycans on α3β1 integrin plays a key role in Gal-3-induced lamellipodia formation. We propose that Gal-3 promotes epithelial cell migration by cross-linking MGAT5-modified complex N-glycans on α3β1 integrin and subsequently activating α3β1-integrin–Rac1 signaling to promote lamellipodia formation.
Keywords: Galectin-3, α3β1 integrin, Lamellipodia, Cell migration, Epithelium
Introduction
Re-epithelialization is an essential event during the wound healing process. Impaired or delayed re-epithelialization and associated persistent epithelial defects and ulceration, represent a serious medical problem in many organs such as cornea, skin and mucous membrane (Dignass, 2001; Ma and Dohlman, 2002; Raja et al., 2007). In the majority of the cases, failure of re-epithelialization is due to a reduced potential of the epithelium to migrate across the wound bed rather than inadequate cell proliferation (Hanna, 1966; Seiler et al., 1989). Cell migration is a complex biological process involving: (1) extension of cellular protrusions such as broad fan-shaped lamellipodia or finger-like filopodia; (2) interactions of the surface molecules of the cell protusions with the permissive sites in the underlying matrix to form transient cell-matrix adhesions; and (3) actomyosin-mediated cell contraction and forward movement accompanied by concomitant detachment of adhesions at the rear end (Ridley et al., 2003). Among the factors that regulate the process of cell migration are transmembrane integrin receptors that mediate cell-matrix adhesions and also initiate and coordinate intracellular signaling pathways, leading to cytoskeletal reorganization and generation of cell motility (Ridley et al., 2003).
Traditionally, the functions of most integrins have been investigated in the context of protein-protein interactions (Guo and Giancotti, 2004; Hynes, 2002; Lock et al., 2008). However, almost all integrins are glycosylated proteins, and, in recent years, a number of studies have revealed that integrin glycans can also modulate transmembrane signaling (reviewed by Bellis, 2004; Gu and Taniguchi, 2004). For example, α3β1-integrin-induced focal adhesion kinase (FAK) activation and migration on laminin-5/laminin-332 (LN-332) is reduced in a fucosyltransferase-null (Fut8–/–) embryonic fibroblasts, suggesting that Fut8-mediated α1,6-fucosylation of innermost N-acetyl-D-glucosamine (GlcNAc) residue on N-glycans modulates the functions of α3β1 integrin (Zhao et al., 2006). Also, β–1,6-N-acetyl-D-glucosamine (β1,6GlcNAc)-branched complex N-glycans synthesized by a glycosyltransferase, MGAT5, have been shown to modulate α5β1-integrin-dependent `inside-out' and `outside-in' signaling during cancer cell migration (Guo et al., 2005; Lagana et al., 2006). In addition, Przybylo et al. have shown that treatment of melanoma cells with Phaseolus vulgaris agglutinin (PHA-L, specific for core β1,6GlcNAc-branched N-glycans) reduced cell migration, suggesting that β1,6GlcNAc-branched glycans are involved in the process of cell motility (Przybylo et al., 2008).
In recent years, studies aimed at characterization of the mechanisms by which integrin glycans regulate cell migration have revealed that interactions between integrin glycans and carbohydrate-binding proteins, galectins, play an essential role in integrin-dependent cell adhesion and migration (Carcamo et al., 2006; Fischer et al., 2005; Friedrichs et al., 2008; Goetz et al., 2008; Lagana et al., 2006; Levy et al., 2003; Nishi et al., 2003; Zhuo et al., 2008). For example, Lagana et al. have shown that galectin-3 (Gal-3; also known as LGALS3) interactions with MGAT5-modified N-glycans at the cell surface of mammary carcinoma cells promote α5β1 integrin activation and cell motility (Lagana et al., 2006); α4β1, α5β1 and α4β7 integrins have been identified as major Gal-1 glycosylated binding partners involved in immune synapse formation, pre-B-cell-receptor clustering and activation (Rossi et al., 2006); and Gal-8 has been shown to form high-affinity interactions with β1 integrins, modulate cell-matrix interactions and promote cell spreading by activating Rho GTPases and PI3K (Diskin et al., 2009; Levy et al., 2001; Levy et al., 2003).
Studies in our laboratory have focused on the role of a structurally unique member of the galectin family, Gal-3, in the process of cell migration (Cao et al., 2002). We have demonstrated that: (1) migrating epithelia of healing mouse corneas express elevated levels of Gal-3 compared with nonmigrating epithelia of normal corneas; (2) the rate of re-epithelialization of corneal wounds is significantly slower in Gal-3-deficient mice compared with wild-type mice; and (3) exogenous Gal-3 stimulates re-epithelialization of corneal wounds in a carbohydrate-dependent manner (Cao et al., 2002). However, the molecular mechanism by which Gal-3 influences re-epithelialization of corneal wounds remains unknown. In the present study, we demonstrate for the first time that Gal-3 promotes formation of lamellipodia by activating α3β1-integrin–Rac1 signaling in epithelial cells and that carbohydrate-mediated interaction between Gal-3 and complex N-glycans on α3β1 integrin is involved in Gal-3-induced lamellipodia formation.
Results
Exogenous Gal-3 promotes cell scattering, lamellipodia formation, and cell motility
In an effort to characterize the mechanism by which Gal-3 enhances re-epithelialization of corneal wounds in vivo (Cao et al., 2002), experiments were performed to determine whether Gal-3 promotes initiation of migratory phenotype in corneal epithelium. For this, the HCLE cells were incubated in the absence or the presence of Gal-3 and the morphology of the cells, in particular lamellipodia formation, was analyzed after staining with TRITC-phalloidin. Lamellipodia are actin-rich, fan-shaped, membrane protrusions at the leading edge of motile cells (Small and Resch, 2005). As early as 30 minutes after exposure to Gal-3, 80% of the cells displayed lamellipodial membrane protrusions as opposed to 5% in control cells (Fig. 1A). The stimulatory effect of Gal-3 on lamellipodia formation was dose dependent (Fig. 1B, inset) and specifically inhibited by a competing sugar, β-lactose, but not by an irrelevant disaccharide, sucrose (Fig. 1A), suggesting that the carbohydrate recognition domain of Gal-3 is involved in the formation of lamellipodia in HCLE cells. In time-lapse video microscopy, Gal-3-treated cells showed colony dispersion and a cell scattering effect. As early as 2 minutes after stimulation with Gal-3, cell-cell dissociation was detected (supplementary material Movie 1), the scattering of colonies gradually increased, and by 10 minutes after exposure to Gal-3, formation of lamellipodia and filopodia was evident in the majority of cells. Furthermore, the cells that had dissociated from the colonies were migratory. By contrast, the cells incubated in medium alone (supplementary material Movie 2) or in the presence of Gal-3 with β-lactose (data not shown) did not show any of the morphological changes induced by Gal-3.
Fig. 1.

Exogenous Gal-3 and Gal-7, but not Gal-1, Gal-8 or plant lectins, promote formation of lamellipodia in corneal and skin epithelial cells in a carbohydrate-dependent manner. (A) HCLE cells were growth-factor-starved for 2 hours and were then incubated in serum-free medium in the presence or absence of exogenous Gal-3 (25 μg/ml) and saccharides (0.1 M) for 30 minutes. At the end of the incubation period, cells were stained with TRITC-phalloidin and were evaluated using a Leica Optigrid confocal microscope for the presence of lamellipodia (Ai). At least 250 cells were counted from several nonoverlapping microscopic fields and the percentage of cells with lamellipodia was estimated (Aii). No significant lamellipodial protrusions were detected in cells incubated in medium alone. By contrast, the majority of the cells incubated in the presence of Gal-3 had lamellipodial protrusions. The stimulatory effect of exogenous Gal-3 was inhibited by a competing saccharide, β-lactose, but not by a noncompeting disaccharide, sucrose. Data are expressed as mean ± s.e.m. (n=250 cells/group). This experiment was repeated at least five times with reproducible results (*P<0.01 compared with the control). (B) Quantification of fluorescence microscopy data showing that Gal-3 and Gal-7, but not Gal-1, Gal-8 or plant lectins, promote lamellipodia formation in HCLE cells. Inset: Cells were incubated with varying concentrations of Gal-3. (C,D) Gal-3 also promotes lamellipodia formation in primary cultures of human corneal (C) and skin epithelial (D) cells. White arrows indicate lamellipodia. Scale bar: 16 μm.
We have previously shown that Gal-3 and Gal-7, but not Gal-1, stimulate re-epithelialization of corneal wounds (Cao et al., 2002). It was, therefore, of interest to determine whether other members of galectin family are able to induce lamellipodia formation. As shown in Fig. 1B, Gal-3 and Gal-7, but not Gal-1 or Gal-8, stimulated the formation of lamellipodia. In addition, none of the plant lectins, including RCA, PNA, Con A and WGA, induced lamellipodial protrusions. Gal-3 also stimulated lamellipodia formation in primary cultures of human corneal epithelial cells (Fig. 1C) and HaCaT skin epithelial cells (Fig. 1D).
α3β1 integrin is a major Gal-3-binding protein
α3β1 integrin is known to play a key role in the formation of lamellipodia (Choma et al., 2004). To determine whether α3β1 integrin is a Gal-3-binding protein, HCLE cell lysates were chromatographed on a Gal-3 affinity column, and electrophoresis blots of bound proteins eluted with β-lactose were immunostained. In western blot analysis, a 130-kDa anti-α3-integrin-reactive component was detected in the fraction eluted from the Gal-3 column by β-lactose (Fig. 2A, β-lactose eluate) but not in the fraction eluted with sucrose (Fig. 2A, sucrose eluate). Also, matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) analysis revealed that the fraction eluted from the Gal-3 affinity column contained a number of proteins including α3 integrin (not shown). By contrast, β-lactose eluate from the Gal-7 column did not show any anti-α3-integrin-reactive component (data not shown). Double-labeled immunofluorescence experiments revealed that Gal-3 and α3 integrin are colocalized at the sites of corneal epithelial cell-cell and cell-matrix adhesions in the frozen sections of corneas (Pearson's correlation coefficient, 0.71±0.08, n=5; Fig. 2B). In cell culture, the colocalization of Gal-3 and α3 integrin was found to be concentrated at the lamella (basal region of lamellipodia) of the leading edge and at the rear of the Gal-3-induced migrating epithelial cells (Pearson's correlation coefficient, 0.8±0.05, n=5; Fig. 2C).
Fig. 2.

(A) α3 Integrin is a major Gal-3-binding protein. HCLE cell lysate was applied to a Gal-3 affinity column, the column was first eluted with a noncompeting disaccharide, sucrose, and then with a competing sugar, β-lactose. Unfractionated extract (2.5 μg protein) and the glycoproteins eluted from the Gal-3-affinity column (derived from 200 μg unfractionated extract) were resolved in reducing 10% SDS-PAGE gels and the protein blots of the gels were processed for immunostaining using a rabbit anti-human α3 integrin polyclonal antibody. Note that a major antibody-reactive 130-kDa glycoprotein (arrow) was detected in the β-lactose but not in the sucrose eluate. (B) α3 integrin and Gal-3 are colocalized in mouse corneal epithelium. Frozen sections (10 μm thick) of mouse corneas were fixed with ice-cold methanol and co-labeled with anti-human Gal-3 rat monoclonal and anti-human α3 integrin rabbit polyclonal antibodies. Gal-3 is green and α3 integrin is red. The merged image shows complete colocalization of Gal-3 and α3 integrin at both cell-cell and cell-matrix junctions. (Bottom right) The intensity plot along the line drawn in the merged image showing a high Pearson's correlation coefficients (r=0.71±0.08; n=5) indicative of substantial Gal-3 (green) colocalization with α3 integrin (red). Scale bar: 20 μm. (C) Colocalization of Gal-3 and α3 integrin in epithelial cells in culture. The HCLE cells were exposed to Gal-3 and were processed for immunostaining using Gal-3 and α3-integrin-specific monoclonal antibodies. A single section of the z-stack representing the bottom of the cell shows that Gal-3 (red) and α3 integrin (green) colocalize at cell-matrix interaction sites specifically in lamellae (base of lamellipodia) and at the rear of the migrating cell (merged image). The inset is an enlarged image of the boxed region showing discrete yellow spots indicating the colocalization of the lectin and integrin. (Bottom right) The intensity plot along the line drawn in the merged image showing a high Pearson's correlation coefficient (r=0.8±0.05; n=5) suggestive of significant colocalization of Gal-3 (red) with α3 integrin (green). Note that no staining was detected in the control samples treated with rat or mouse IgG.
An anti-α3 integrin monoclonal antibody inhibits Gal-3-induced lamellipodia formation
To determine whether α3β1 integrin is essential for Gal-3-mediated lamellipodia formation, time-lapse and immunofluorescence microscopy were performed using cells pretreated with an anti-α3 integrin mAb (P1B5). Strikingly, Gal-3-induced lamellipodia formation was inhibited in the vast majority of the cells preincubated with the P1B5 antibody (supplementary material Movie 3). Moreover, in the presence of the antibody, the cells were found to extend tufts of finger-like projections (filopodia) in the direction of motility without fan-shaped lamellipodial protrusions. By contrast, the control IgG did not block the function of Gal-3 in promoting lamellipodia formation (supplementary material Movie 4). Interestingly, the function of Gal-3 in mediating colony scattering was not prevented by P1B5 (supplementary material Movie 3). When the morphology of cells was visualized by staining with phalloidin, it was clear that Gal-3-treated cells that were preincubated with P1B5 (Fig. 3, Anti-α3 integrin + Gal-3, white *) but not control IgG (Fig. 3, IgG + Gal-3) or mAb against an irrelevant integrin (α2 integrin, clone P1E6; not shown) had a rounded morphology without lamellipodia. On average, pretreatment with the PIB5 antibody resulted in 45% fewer cells with lamellipodia compared with the cells pretreated with control IgG (Fig. 3, graph). These data indicate that α3β1 integrin is essential for Gal-3-mediated lamellipodia formation in epithelial cells.
Fig. 3.

An anti-α3 integrin but not anti-LN-332 function-blocking mAb inhibits Gal-3-induced lamellipodia formation. HCLE cells were plated on chamber slides and were incubated with 10 μg/ml of the anti-α3 integrin mAb (P1B5), anti-LN-332 mAb (P3H9-2) or a control mAb (anti-α2 integrin, Clone P1E6) for 30 minutes, prior to exposure to Gal-3. At the end of the incubation period, the cells were evaluated for the presence of lamellipodial protrusions as described in Fig. 1 legend (bottom panel). Note that preincubation with anti-α3 integrin mAb blocked Gal-3-induced lamellipodia formation in the majority of the cells (* in the anti-α3 integrin + Gal-3 panel). By contrast, preincubation of cells with anti-LN-332 mAb, or anti-α2 integrin mAb or control IgG, did not prevent Gal-3-induced lamellipodial protrusions (IgG + Gal-3 panel). The cells incubated in medium alone and medium containing Gal-3 served as negative and positive controls, respectively. Data are expressed as mean ± s.e.m. (n=250 cells/group). This experiment was repeated at least three times with reproducible results (**P<0.01 compared with Gal-3-treated cells; *P<0.01 compared with control). White arrows indicate lamellipodia. Scale bar: 16 μm.
Immunostaining experiments revealed that HCLE cells grown on FNC-coated wells for 16 hours robustly expressed LN-332, a known ligand of α3β1 integrin, which plays a role in integrin-mediated Rac activation (Choma et al., 2007; Choma et al., 2004). However, preincubation of cells with a function-blocking mAb against LN-332 (P3H9-2; 30 μg/ml) did not block the Gal-3-induced lamellipodia formation (Fig. 3, anti-LN-332 mAb + Gal-3), although it blocked HCLE cell adhesion to the matrix made by HaCaT cells by ∼50% (data not shown). These observations suggest that Gal-3-mediated interaction with α3β1 integrin to promote lamellipodia formation is independent of LN-332 functions.
Gal-3 activates α3β1-integrin–Rac1 signaling
Having established that α3β1 integrin is the major binding partner for Gal-3 in corneal epithelial cells, it was of interest to determine whether Gal-3 activates the α3β1-integrin–Rac1 signaling pathway that is known to modulate lamellipodia formation (Choma et al., 2007; Choma et al., 2004). To determine whether Gal-3 activates Rac1 in HCLE cells, a GST-PAK pulldown assay was performed. The levels of total Rac1 (GTP-bound + GDP-bound forms) protein were similar regardless of whether the cells were incubated in the presence or the absence of Gal-3. By contrast, within 30 seconds exposure to Gal-3, there was a 2.3-fold increase in the amount of activated Rac1 (Fig. 4A) and enhanced Rac1 activity was maintained up to 10 minutes (data not shown). This suggests that endogenous Rac1-GTPase activation is involved in an α3β1-integrin-mediated signal transduction cascade induced by Gal-3, leading to actin reorganization and formation of lamellipodial protrusions. Published studies have shown that Rac1-GTPase stimulates the formation of lamellipodia by promoting actin-remodeling downstream of activation of FAK in epithelial cells (Choma et al., 2007; Choma et al., 2004; Mitra and Schlaepfer, 2006). Thus, we also performed western blot analyses using anti-Y397 FAK phosphospecific antibody to determine whether Gal-3 activates FAK. These experiments revealed that FAK was activated after addition of Gal-3 demonstrating that integrin signaling pathways are activated in HCLE cells upon exposure to Gal-3 (Fig. 4B). These findings are in line with the report of Lagana et al. showing that in mammary carcinoma cells, Gal-3 activates FAK (Lagana et al., 2006).
Fig. 4.

Gal-3 activates Rac1 GTPase and FAK in corneal epithelial cells. (A) HCLE cells were growth-factor-starved and were incubated in the presence or absence of Gal-3 in serum-free medium. Cell extracts were subjected to pulldown assays using GST-PAK to examine GTP-bound Rac1 (active Rac1) levels. Precipitated GTP-Rac1 as well as total cell lysates were examined by immunoblot analysis using a mouse anti-Rac1 mAb. The intensity of each band was quantified by densitometry and the ratios of GTP-loaded Rac1 to total Rac1 were determined and normalized to cells exposed to serum-free medium alone (media control). Note that the levels of total Rac1 (GTP-bound + GDP-bound forms) were similar regardless of whether the cells were exposed to Gal-3 or not. By contrast, within 30 seconds of exposure to Gal-3, there was a 2.3-fold increase in the amount of activated Rac1 (bar graph), and enhanced Rac1 activity was maintained up to 10 minutes (data not shown). Values are mean ± s.e.m. of three separate experiments. *P<0.01 compared to media control. (B) Electrophoresis blots were probed with anti-FAK or anti-Y397 phosphorylated FAK monoclonal antibodies. Note that FAK is activated (p-FAK lanes) after exposure to Gal-3, whereas total FAK levels (FAK lanes) are similar in the control and Gal-3-exposed cells. The intensity of each band was quantified and ratios of p-FAK to total FAK was calculated and normalized to control values (bar graph). *P<0.05 when compared with control; data are mean ± s.e.m. from three independent experiments.
A reduction in β1,6GlcNAc-branched N-glycans on α3 integrin abolishes the ability of Gal-3 to promote lamellipodia formation
The MGAT5 enzyme synthesizes N-glycan intermediates, the β1,6GlcNAc-branched glycans, that are elongated with N-acetyllactosamines to create high affinity ligands for galectin-3 (Dennis et al., 2002). To evaluate the importance of Gal3–α3-integrin–glycan interactions in lectin-mediated lamellipodia formation, we analyzed the effect of MGAT5 knockdown on α3 integrin glycans and on Gal3-induced lamellipodia formation.
The expression of MGAT5 was knocked down by transfection of epithelial cells with shRNA constructs directed against MGAT5. As controls, cells were transfected with constructs expressing a nontargeting sequence. The results were analyzed by qRT-PCR. Transfection of cells with shRNA constructs directed against MGAT5 resulted in a substantial reduction (∼66%) of MGAT5 mRNA (Fig. 5A). The loss of N-glycan intermediates synthesized by MGAT5 was confirmed by the loss of the ability of cells to bind the rhodamine-labeled L-PHA lectin, which reacts specifically with core β1,6GlcNAc-branched products synthesized by MGAT5 (Cummings and Kornfeld, 1982). Consistent with the decreased mRNA levels, MGAT5 shRNA-expressing cells showed a significant reduction (∼62%) in L-PHA binding as detected by fluorescence staining (Fig. 5B, L-PHA staining). However, no change in Con A binding (Fig. 5B, Con A staining) was observed in MGAT5 knockdown cells, suggesting that the expression of high mannose or biantennary N-linked glycans was not altered after knockdown of MGAT5. The morphology and growth pattern were essentially similar between nontransfected control and MGAT5 shRNA-expressing cells.
Fig. 5.

Targeted knockdown of MGAT5 by shRNA reduces β1,6GlcNAc-branched N-glycans of α3 integrin in epithelial cells. (A) RT-qPCR. Total RNA was isolated from noninfected control, MGAT5 shRNA- and nontarget shRNA-expressing cells. (Left) RT-qPCR was performed using primers specific for human MGAT5. Left panel: original amplification plots of MGAT5 mRNAs showing significantly lower signals in cells expressing MGAT5 shRNA compared with cells expressing nontarget shRNA, and noninfected control cells. (Right) The data were normalized against GAPDH and the fold change was calculated using noninfected control cells as a calibrator (n=3). *P<0.05 compared with noninfected or nontarget shRNA-expressing cells. (B) Lectin staining. Noninfected, nontarget shRNA-, and MGAT5 shRNA-expressing cells grown on chamber slides were fixed with 4% paraformaldehyde and then stained with either rhodamine-conjugated L-PHA (specificity: core β1,6GlcNAc-branched N-glycans) or Con A (specificity: high mannose and bisected hybrid N-glycans). The staining intensity was quantified from 10 images in each experimental condition and normalized to that of noninfected cells (bar graph). Top panel shows representative micrographs of stained cells. Note that the binding of L-PHA but not Con A was reduced significantly in cells expressing MGAT5 shRNA. Data are expressed as mean ± s.e.m. The experiment was repeated three times with reproducible results. *P<0.05 compared with noninfected and nontarget shRNA-expressing cells. Scale bar: 32 μm. (C) Western blotting. To determine whether knockdown of MGAT5 expression reduces β1,6GlcNAc-branched complex N-glycans on α3β1 integrin, RIPA buffer extracts from confluent cultures of noninfected, MGAT5 shRNA-, and nontarget shRNA-expressing cells (600 μg total protein each) were incubated with agarose-bound Con A or L-PHA (1 hour, 4°C). Following incubation, the beads were washed, boiled in SDS-PAGE sample buffer (without mercaptoethanol) to release bound proteins, centrifuged, and supernatants were electrophoresed in SDS polyacrylamide gels. Samples derived from 2.5 μg original cell protein were electrophoresed in each lane. Protein blots of the gels were then processed for immunostaining with rabbit anti-α3 integrin. Note that an equivalent level of anti-α3-integrin-reactive component is present in the Con-A-bound fraction and whole cell extract (WCL). By contrast, significantly reduced level of anti-α3-integrin-reactive component was detected in the L-PHA-bound fraction in MGAT5 knockdown cells. Also, no anti-α3-integrin-reactive components were detected in the supernatants of cell extracts incubated with agarose beads alone or in the blots of cell extracts not exposed to primary antibody (not shown). Relative binding of lectins to α3 integrin was calculated by densitometric analysis (bar graph). Values are mean ± s.e.m. of three independent experiments. *P<0.05 compared with noninfected and nontarget shRNA-expressing cells.
To determine whether β1,6GlcNAc-branched glycans on α3 integrin were reduced in MGAT5 shRNA-expressing cells, pulldown experiments were conducted using agarose-conjugated plant lectins (L-PHA and Con A), and the proteins bound to the lectins were analyzed for reactivity with anti-α3 integrin by western blot analysis. The α3 integrin from MGAT5 shRNA-expressing cells showed a significant reduction in L-PHA binding compared with noninfected and nontarget shRNA-expressing cells (Fig. 5C). By contrast, the binding of α3 integrin to Con A was not affected. These data indicate that knockdown of MGAT5 gene expression leads to significant reduction in β1,6GlcNAc-branched complex N-glycans on α3 integrin. Neither the expression level of α3 integrin (Fig. 5C), nor its cell surface localization (data not shown) was affected in MGAT5 cells.
Next, we determined whether a reduction of MGAT5 enzyme activity on N-glycans of α3 integrin hinders Gal-3-induced lamellipodia formation in epithelial cells. After exposure to Gal-3, on average 79% of noninfected (Fig. 6, noninfected + Gal-3) and 71% of nontarget shRNA-expressing (Fig. 6, nontarget shRNA+ Gal-3) epithelial cells produced lamellipodia, whereas only 41% of MGAT5 shRNA-expressing cells showed lamellipodia (Fig. 6, MGAT5 shRNA + Gal-3). These data suggest that MGAT5-modified complex N-glycans on α3 integrin play a key role in the molecular mechanism leading to Gal-3-induced lamellipodial extensions in epithelial cells.
Fig. 6.
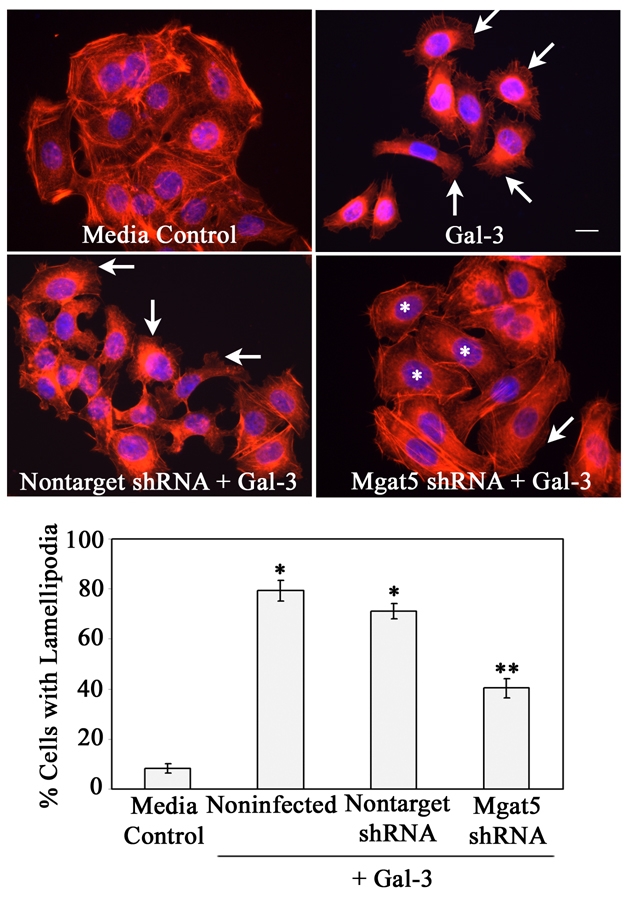
MGAT5 knockdown inhibits Gal-3-induced lamellipodia formation in epithelial cells. Epithelial cells expressing either nontarget shRNA or MGAT5 shRNA or noninfected cells were exposed to serum-free medium in the presence and absence of Gal-3 for 30 minutes. At the end of the incubation period, cells were stained with TRITC-phalloidin (red) and DAPI (blue) and were then evaluated for the presence of lamellipodial protrusions, under a fluorescent microscope. Representative images are shown in the top panel. The majority of cells incubated with Gal-3, but not with media alone, show lamellipodial protrusions (arrows). Note that the stimulatory effect of Gal-3 on lamellipodia formation was inhibited in MGAT5 shRNA-expressing cells (* in MGAT5 shRNA+Gal-3 panel), but not in the cells expressing nontarget shRNA. The percentage of cells with lamellipodial protrusions were calculated after examination of at least 250 cells from each experimental sample (bar graph). Data are expressed as mean ± s.e.m. (n=250 cells/group). The experiment was repeated three times with reproducible results. *P<0.01 compared with control; **P<0.01 compared with noninfected cells exposed to Gal-3. Scale bar: 16 μm.
Discussion
The goal of the present study was to determine the molecular mechanism by which Gal-3 promotes epithelial cell migration during corneal wound closure. The study revealed a novel role of Gal-3 in promoting lamellipodial protrusions in epithelial cells by interacting with complex N-glycans on α3β1 integrin.
Our findings that Gal-3 and Gal-7, but not Gal-1, initiate the formation of lamellipodia are in line with our published study showing that Gal-3 and Gal-7, but not Gal-1, promote epithelial cell migration during re-epithelialization of corneal wounds (Cao et al., 2002). It is of interest to note that in a recent study, based on the findings that Gal-7-null mice show a defect in re-epithelialization of skin wounds and in the distribution of cortactin (a protein localized at the lamellipodia), it has been suggested that Gal-7 modulates the formation and/or stabilization of lamellipodia (Gendronneau et al., 2008). Although all mammalian galectins specifically recognize galactose-containing glycans, each galectin has fine specificity for more complex glycans, and saccharides in the vicinity of galactose considerably influence the affinity for a given galectin (reviewed by Brewer, 2004; Hirabayashi et al., 2002; Rapoport et al., 2008). For example, the affinity of Gal-1 for the blood group A tetrasaccharide is about 100-fold lower than that for Gal-3 (Sparrow et al., 1987). Owing to fine differences in carbohydrate-binding specificities, each galectin might interact with a discrete spectrum of glycoprotein receptors, with consequent specific downstream effects. For example, Gal-8, but not Gal-1 or Gal-3, promotes apoptosis of human non-small-cell lung carcinoma cells (Hadari et al., 2000). Also, in a recent study (Diskin et al., 2009), we have demonstrated that: (1) trabecular meshwork (TM) cells from human eye adhere to and spread on Gal-8-but not on Gal-1- or Gal-3-coated wells; (ii) Gal-8 modulates the adhesion and cytoskeletal arrangement of TM cells by binding to β1 integrins and inducing Rho signaling; and (3) TM cell β1 integrins carry predominantly α2,3-sialylated glycans, which are high affinity ligands for Gal-8 but not for Gal-1 or Gal-3. Other studies have also demonstrated that glycosylation pattern distinctively modulates the recognition of cell surface glycans and biological signaling by different galectins (Stowell et al., 2008a; Stowell et al., 2008b; Toscano et al., 2007; Zhuo et al., 2008). These studies support the notion that many galectin-mediated functions are unique to a specific galectin in a particular cell type (Bao and Hughes, 1999; Diskin et al., 2009; Hadari et al., 2000). By contrast, different members of the galectin family might also have overlapping binding specificity and, thus, in some cases, multiple galectins mediate the same function in a cell type (Friedrichs et al., 2007; Maeda et al., 2003; Paclik et al., 2008; Stillman et al., 2006; Stowell et al., 2007). However, we note that when multiple galectins mediate the same function in a cell type, it is not always because of their overlapping binding specificity (Fukumori et al., 2003; Hernandez et al., 2006; Lu et al., 2007; Stillman et al., 2006; Sturm et al., 2004). For example, exogenous Gal-1, Gal-3 and Gal-9 all promote apoptosis in T-cells, but they do so by binding to distinct cell surface binding proteins to initiate distinct signal transduction events (Lu et al., 2007; Stillman et al., 2006). Our findings that α3β1 integrin is a binding partner of Gal-3 but not of Gal-7 suggests that both Gal-3 and Gal-7, which promoted lamellipodia formation in the current study, did so, by distinct mechanisms.
That the carbohydrate recognition domain and the carbohydrate-dependent function of Gal-3 are directly involved in the stimulatory effect of Gal-3 on the initiation of lamellipodia is suggested by our findings that: (1) binding of Gal-3 to α3β1 integrin, as well as the stimulatory effect of Gal-3 on lamellipodia formation, was inhibited by a competing sugar, β-lactose, but not by a noncompeting sugar, sucrose; and (2) knockdown of gene expression of a rate limiting enzyme, MGAT5, resulted in the loss of β1,6GlcNAc-branched complex N-glycans, the high-affinity ligands of Gal-3, on α3β1 integrin with a concomitant inhibition of Gal-3-induced lamellipodia formation. These findings are in contrast to a recent report (Hsu et al., 2009) showing that in a different cell type, dendritic cells, intracellular Gal-3 regulates formation of membrane ruffles and cell migration via a mechanism that is independent of carbohydrate-mediated signaling. However, there are a number of differences between the two studies. First, the lamellipodia referred to in the current study and membrane ruffles mentioned by Hsu et al. (Hsu et al., 2009), are distinct structures (Adams, 2001; Chhabra and Higgs, 2007). If the adhesions at the lamellipodia break, the leading lamella retracts and forms characteristic structures on the cell surface that are generally described as membrane ruffles. Second, in the study by Hsu et al., it was largely the function of intracellular Gal-3 that was investigated, whereas in the current study, the function of extracellular Gal-3 was investigated.
It is likely that Gal-3 influences events related to cell migration by distinct mechanisms in different cell types, and that the intra- and extracellular Gal-3 modulates cell migration by distinct mechanisms. Also, it is well established that intra- and extracellular Gal-3 modulate distinct functions (reviewed by Liu and Rabinovich, 2005). For instance, in some cell types (e.g. Burkitt lymphoma and macrophages), intracellular Gal-3 inhibits apoptosis (Hoyer et al., 2004; Hsu et al., 2000), whereas in other cell types (e.g. thymocytes, peripheral blood mononuclear cells and activated T cells) the extracellular Gal-3 promotes apoptosis (Fukumori et al., 2003; Stillman et al., 2006). It is known that secretion of galectins is tightly controlled and that a number of factors including the nature of the culture media used, the state of activation, the presence of cytokines, as well as adhesive and membrane fusion events can modulate galectin secretion (Hughes, 1999). In this respect, it is relevant to note that in dendritic cells, Gal-3 is localized intracellularly and is not released by the cells (Hsu et al., 2009), whereas corneal epithelial cells secrete Gal-3 as shown by its presence in large amounts at the site of cell-matrix and cell-cell adhesion. More importantly, our previous studies have shown that the expression of Gal-3 is enhanced at the sites of cell-matrix adhesion at the leading edge during re-epithelialization of corneal wounds (Cao et al., 2002). Thus, in the context of re-epithelialization of corneal wounds, the focus of the current study was on extracellular Gal-3.
One of the major pathways leading to lamellipodia formation in epithelial cells is triggered by LN-332 binding to α3β1 integrin that lead to the activation of FAK and Rac (Choma et al., 2007; Choma et al., 2004). Our findings that preincubation of cells with a function-blocking antibody against LN-332 did not reduce Gal-3-induced lamellipodia formation suggests that the lectin is capable of promoting lamellipodia formation independently of LN-332. In future, it will be worthwhile performing more extensive and dedicated studies, including the use of LN-332 knockout cells and LN-332-deficient cells derived from patients with junctional epidermolysis bullosa (JEB) to rigorously test the concept that in the complete absence of LN-332, other molecules such as Gal-3 can induce lamellipodia formation.
The current study has focused on characterization of the role of α3β1 integrin in Gal-3-mediated lamellipodia formation. Affinity chromatography of the HCLE cell lysates on a Gal-3-Sepharose column revealed that a number of proteins in addition to α3β1 integrin bind to Gal-3. These include α6, αv and β4 integrins, CD98, and Endo180 (C.S. and N.P., unpublished data). It is our hope that future studies will investigate to what extent, if any, additional Gal-3-binding proteins are involved in the lectin-induced cell migration and wound healing.
In summary, we have demonstrated here that Gal-3, by interacting with MGAT5-modified complex N-glycans, activates α3β1-integrin–Rac1 signaling to promote formation of lamellipodia in epithelial cells. Based on the findings presented in the current study and previously published studies showing that: (1) α3β1-integrin-mediated Rac1 activation is essential to promote lamellipodia formation (Choma et al., 2004); (2) Gal-3 cross-links cell surface receptors (e.g. EGF and TGF-β receptors) by interacting with MGAT5-modified complex N-glycans to promote their signal transduction (Partridge et al., 2004); and (3) clustering of integrins leads to activation of its intracellular signaling (Guo and Giancotti, 2004; Hynes, 2002; Lock et al., 2008), we propose the following model of Gal-3-mediated epithelial cell migration and re-epithelialization of wounds (Fig. 7). According to this model, Gal-3, by virtue of its multivalency (Fred Brewer, 2002), cross-links and clusters α3β1 integrin on the cell surface at the leading edge of the migrating epithelium. The clustering of α3β1 integrin activates FAK and Rac1 and this, in turn, promotes lamellipodia formation, cell migration, and re-epithelialization of wounds.
Fig. 7.

Proposed model of Gal-3-mediated signaling in epithelial cells leading to the formation of lamellipodial protrusions and cell migration. According to this model, Gal-3, by virtue of its multivalency (Fred Brewer, 2002), cross-links and clusters α3β1 integrin on the cell surface at the leading edge of the migrating epithelium. The clustering of α3β1 integrin activates FAK and Rac1 and this, in turn, promotes lamellipodia formation, cell migration and re-epithelialization of wounds. This model is suggested based on our findings that: (1) α3β1 integrin is a major Gal-3-binding partner and a function-blocking anti-α3 integrin mAb blocks the Gal-3-mediated lamellipodia formation, (2) Gal-3 interacts with MGAT5-modified complex N-glycans on α3β1 integrin, (3) Gal-3 activates Rac1, a member of Rho GTPases, and the published findings showing that: (i) Gal-3 cross-links cell surface receptors (e.g. EGF and TGFβ receptors) by interacting with MGAT5-modified complex N-glycans to promote their signal transduction (Partridge et al., 2004), and (ii) α3β1-integrin–Rac1 signaling is essential to promote lamellipodia formation (Choma et al., 2004).
Materials and Methods
Cell culture
Studies were carried out using telomerase reverse transcriptase-immortalized human corneal epithelial (HCLE) cells (Gipson et al., 2003). The HCLE cells were maintained in keratinocyte serum-free medium supplemented with 0.2 ng/ml epidermal growth factor (EGF), 25 μg/ml bovine pituitary extract (K-SFM; Invitrogen, Carlsbad, CA) and 0.4 mM CaCl2 at 37°C in a 5% CO2 incubator. For comparative purposes, primary cultures of human corneal epithelium and a skin keratinocyte cell line, HaCaT, were also used. Primary human corneal epithelial (HCE) cells were cultured from donor corneal rims of transplant corneas using endothelium-free explants (Ebato et al., 1988). HaCaT cells (a kind gift from Jonathan Garlick, Tufts University, Boston, MA) are spontaneously immortalized, nontransformed epithelial cells derived from adult human skin and are similar to normal skin keratinocytes (Boukamp et al., 1988). The HaCaT cells were grown in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) containing 5% fetal calf serum and 10 mM HEPES buffer.
Analysis of the effect of Gal-3 on corneal epithelial cell morphology
Recombinant full-length galectins used in this study were produced as described previously (Cooper et al., 1991; Diskin et al., 2009; Hsu et al., 1992; Kuwabara et al., 2002). Both live and conventional fluorescent microscopy techniques were used to assess Gal-3-induced migratory phenotype of corneal epithelial cells. For live microscopy, the HCLE cells (4×104 cells per dish) were grown overnight on a 35-mm glass-bottomed dish (MatTek, Ashland, MA) coated with FNC (fibronectin-collagen) Coating Mix (AthenaES, Baltimore, MD). The cells were starved of growth factor for 2 hours with keratinocyte basal medium (Lonza, Walkersville, MD) supplemented with 0.4% BSA and were then incubated in the absence or the presence of 25 μg/ml recombinant human Gal-3. The temperature of the microscope stage was maintained at 37°C using a custom-made heating chamber, and the cells were observed under a Leitz Wetzlar 32× 0.4 NA phase-contrast objective using a Leitz Diavert inverted microscope (Leitz, Germany). Images were captured every minute for up to 30 minutes using a Fujifilm Digital Camera (Fujifilm, Tokyo, Japan) attached to the eyepiece of the microscope. A series of phase-contrast images were converted to video (avi files) using the ImageJ public domain software. In some experiments, to characterize the role of the carbohydrate recognition domain, the cells were exposed to Gal-3 in the presence of a competing disaccharide, β-lactose (0.1 M) or an irrelevant disaccharide, sucrose (0.1 M). To assess the role of integrins in Gal-3-induced migratory phenotype, cells were incubated with 10 μg/ml α3 integrin function-blocking monoclonal antibody (mAb; P1B5) or control IgG (Chemicon, Temecula, CA) for 30 minutes, washed twice with PBS, and then treated with Gal-3. The cells were then observed by phase-contrast microscopy and time-lapse images were made as described above.
For conventional fluorescence microscopy, corneal epithelial cells (2×104 cells per chamber) were plated on eight-chamber culture slides (BD Biosciences, San Jose, CA) coated with FNC Coating Mix. After overnight incubation, the cells were starved of growth factor and then incubated in the absence or presence of Gal-3 (25 μg/ml) for 30 minutes. At the end of incubation period, the cells were fixed with 4% paraformaldehyde in PBS for 20 minutes, permeabilized with 0.5% Triton X-100 for 10 minutes, and stained with tetramethylrhodamine (TRITC)-conjugated phalloidin (0.1 μg/ml in PBS for 1 hour; Sigma, St Louis, MO). The chamber slides were mounted using Vectashield mounting medium (Vector Labs, Burlingame, CA) and viewed with a 40× Zeiss objective lens on a Leica Optigrid confocal microscope. Images were generated using the Volocity software (Improvision, Waltham, MA). At least 250 cells were counted from several nonoverlapping microscopic fields, and the percentage of cells with lamellipodia were estimated. To characterize the specificity of the action of Gal-3, in some experiments, the cells were exposed to other members of galectin family (Gal-1, Gal-7 or Gal-8) and various plant lectins including Ricinus communis agglutinin (RCA), wheat germ agglutinin (WGA), concanavalin A (Con A) and peanut agglutinin (PNA; Vector Labs). The α3 integrin or LN-332 function-blocking experiments were done essentially as described above for time-lapse video microscopy except that the cells were plated on eight-chamber slides and the changes in cell morphology were studied after staining with phalloidin. After completing studies with HCLE cells, the ability of Gal-3 to induce lamellipodia formation was examined in the primary cultures of human corneal epithelium and the HaCaT cells, and the results obtained using the distinct cell types were compared.
Affinity chromatography and western blot analysis
The Gal-3 affinity matrix was prepared by coupling 5 mg recombinant human Gal-3 to cyanogen-bromide-activated Sepharose 4B (Sigma) according to the manufacturer's instructions. To determine whether α3β1 integrin is a Gal-3-binding protein, the radioimmunoprecipitation (RIPA) buffer (150 μM NaCl, 50 μM Tris pH 8.0, 1% NP 40, 0.5% deoxycholate, 0.1% SDS) extract derived from ten 100-mm dishes of HCLE cells was chromatographed on a Gal-3 affinity matrix. The unbound components were removed by washing the column with PBS containing 0.1% Triton X-100 (PBST) and the bound proteins were eluted using PBST containing 0.1 M β-lactose. In some experiments, to confirm the sugar-binding specificity of the Gal-3-binding proteins, the column was eluted with PBST containing 0.1 M sucrose prior to elution with β-lactose. Fractions eluted from the column with saccharides were dialyzed against water, lyophilized, and resolved by electrophoresis in reducing 10% SDS-PAGE gel. Protein blots of the gels were subjected to western blot analysis using polyclonal rabbit anti-human α3 integrin (1:500; Chemicon) as a primary antibody and goat anti-rabbit horseradish peroxidase (HRP)-labeled IgG (1:20,000; Vector Labs) as a secondary antibody. That α3β1 integrin is a Gal-3-binding protein was further confirmed by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS; PerkinElmer Life Sciences, Waltham, MA) of the bound proteins eluted from the Gal-3 affinity column by β-lactose. The above-mentioned methods were also used to determine whether α3 integrin binds to Gal-7.
Immunolocalization of Gal-3 and α3β1 integrin
To study the colocalization of Gal-3 and α3β1 integrin in corneal tissues, frozen sections (10 μm thick) of mouse corneas were fixed in ice-cold methanol for 2 minutes, blocked with PBS containing 2% BSA and 3% goat serum and incubated with rat monoclonal anti-mouse Gal-3 (undiluted hybridoma fluid, mAb M3/38; American Type Culture Collection, Manassas, VA) and rabbit polyclonal anti-human α3 integrin (Chemicon; 1:100). In control experiments, rat IgG (Chemicon) and rabbit IgG (Vector Labs) were used (20 μg/ml, each) instead of primary antibodies. The secondary antibodies used were Alexa-Fluor-488-conjugated anti-rat IgG (1:500; Molecular Probes, Carlsbad, CA) to visualize Gal-3 (green fluorescence) and Cy3-conjugated anti-rabbit IgG (1:1500; Molecular Probes) to visualize α3 integrin (red fluorescence).
To study the Gal-3–α3 integrin colocalization in cell culture, the growth-factor-starved HCLE cells exposed to 25 μg/ml of Gal-3 were fixed with 4% paraformaldehyde, blocked with PBS containing 1% BSA, and incubated with a cocktail of rat monoclonal anti-mouse Gal-3 (1:250, mAb M3/38; Santa Cruz Biotechnology) and mouse monoclonal anti-human α3 integrin (1:50, mAb ASC-1; Chemicon). Purified rat (Jackson Immuresearch, PA) and mouse IgG (Chemicon) were used as controls. The secondary antibodies used were rhodamine-conjugated anti-rat IgG (1:75; Jackson ImmunorResearch) to visualize Gal-3 and fluorescein (FITC)-conjugated anti-mouse IgG (1:50; Jackson Immunoresearch) to visualize α3 integrin. The stained tissue sections and cell cultures were viewed by confocal microscopy using a Leica TCS SP2 laser confocal scanning microscope (Leica, Wetzler, Germany). Optical sections with double fluorescence for green and red channels were acquired from the top to the bottom of each cell at ∼0.6-μm step intervals. The NIH ImageJ program was used to create fluorescence intensity line profiles of green and red merged images of corneal tissue sections or HCLE cells. To quantify the extent of overlap between two immunolabels, Pearson's correlation coefficient (r) values were determined from three different labeled areas per overlapped image of at least five images, and mean ± s.e.m. values were calculated. The Pearson's coefficient value close to +1 is considered indicative of the overlap of the two components.
FAK activation assay
Cell lysates were prepared using RIPA buffer from HCLE cells exposed to Gal-3 for various time periods (0, 2, 5, and 10 minutes). Aliquots of cell lysates containing 35 μg of total protein were electrophoresed in a 4-12% SDS-polyacrylamide gel. Protein blots of the gel were probed with rabbit anti-FAK pY397 (clone 141-9; Invitrogen, 1:1000) or mouse anti-FAK (clone 77/FAK; 1:1000; BD Transduction Laboratories, San Jose, CA) mAb.
Rac1 activity assay
The Gal-3-induced activation of Rac1 GTPase was examined using the EZ-detect Rac1 GTPase kit (Pierce, Rockford, IL) according to the manufacturer's recommendations. Briefly, the HCLE cells were plated onto FNC Coating Mix at a density of 5×106 cells per 100 mm dish and were starved of growth factor. After exposure to Gal-3 for various time periods (30 seconds, 5 minutes and 10 minutes), the cells were washed with ice-cold PBS and then lysed with 500 μl cell lysis buffer (25 mM Tris-HCl pH 7.5, 150 mM NaCl, 5 mM MgCl2, 1% NP-40, 1 mM DTT and 5% glycerol) supplemented with a protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN). The cell lysates containing 500 μg protein were added to a column containing 20 μg GST–human-PAK1–PBD and a SwellGel Immobilized Glutathione disc. After incubation for 1 hour at 4°C with shaking, the bound proteins were eluted with 50 μl of 2× SDS sample buffer containing β-mercaptoethanol and were then electrophoresed in a 4-20% gradient of polyacrylamide gels. Protein blots of the gels were sequentially incubated with a mouse anti-human Rac1 antibody for 1 hour (1:1000; Pierce) and horse anti-mouse HRP-labeled IgG (1:50,000; Vector Labs) for 1 hour. Immunostained components were then visualized using a chemiluminescence detection system (PerkinElmer Life Sciences, Waltham, MA) and quantified using ImageJ. Unfractionated total cell lysate (20 μg total protein) was used as a loading control for the pulldown assay. Normalized Rac1 activity values were determined by dividing the amount of activated Rac1 in the pulldown fraction by the amount of total Rac1 in the loading control lane.
Silencing MGAT5 gene expression by lentiviral shRNAs
A set of five different shRNA lentiviral transduction particles targeting human MGAT5 (Sigma; catalog no. SHVRS-NM_002410) was tested for knockdown of MGAT5 expression. For this study, we used HaCaT cells, which are suitable for puromycin-based selection, as opposed to HCLE cells, which are not suitable (Gipson et al., 2003). Briefly, the cells were plated in a 96-well plate (7.5×103 cells per well), and, after overnight incubation, medium was replaced with growth medium containing 8 μg/ml hexadimethrine bromide. The cells were then infected with lentiviral particles targeting MGAT5 or a nontarget shRNA (moi: 1.0). After overnight incubation with the virus, the cells were washed and incubated in growth medium for 2 days. Pooled stable transfectants were established using puromycin selection (0.4 μg/ml puromycin in growth medium).
The extent of MGAT5 knockdown at the mRNA level was assessed by quantitative reverse transcription polymerase chain reaction (qRT-PCR). The clone selected was TRCN0000036061. qRT-PCR was performed using the Mx4000 real-time PCR machine (Stratagene, La Jolla, CA) as described by Diskin et al. (Diskin et al., 2006). Briefly, cDNA was synthesized from 2 μg total RNA using the High Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA). PCR was carried out in triplicate using inventory gene-specific primers (MGAT5: Hs00159136_m1; GAPDH: Hs99999905_m1, Applied Biosystems) and the TaqMan Universal PCR Master Mix (Applied Biosystems). Reactions performed in the absence of cDNA served as a nontemplate negative control. After an initial denaturation step (95°C for 10 minutes), the reactions were subjected to 50 cycles involving denaturation (95°C for 15 seconds) and annealing plus extension (60°C for 1 minute). The threshold cycle values (Ct) were calculated for all samples. Quantification data of the MGAT5 gene were normalized with respect to the expression of a house-keeping gene, GAPDH.
The knockdown of MGAT5 products was analyzed by immunofluorescence staining with the rhodamine-conjugated Phaseolus vulgaris leukoagglutinin (L-PHA) lectin, which reacts specifically with core β1,6GlcNAc-branched products synthesized by MGAT5 (Cummings and Kornfeld, 1982). For immunofluorescence microscopy, the noninfected, nontarget shRNA- or MGAT5 shRNA-expressing epithelial cells were cultured on chamber slides in growth medium as described above. The cells were fixed and incubated with 10 μg/ml of either rhodamine-conjugated L-PHA or an irrelevant control lectin, concanavalin A (Con A; Vector Labs) in PBS for 1 hour at room temperature. After washing with PBS, the chamber slides were mounted and subjected to fluorescence microscopy with constant exposure time. The intensity of red fluorescence was measured in at least 10 nonoverlapping microscopic fields containing an approximately equal number of cells, using ImageJ. The mean fluorescence intensity from each microscopic field was used to compare the lectin-binding capacity of different cell types.
The loss of β1,6GlcNAc-branched glycans specifically on α3 integrin was assessed by pulldown experiments. Briefly, RIPA buffer lysates of epithelial (noninfected and shRNA expressing) cells (600 μg protein) were incubated with 15 μl of Sepharose 4B (Sigma) for 1 hour at 4°C with shaking to prevent nonspecific binding. Unbound proteins were then incubated with 15 μl agarose-conjugated L-PHA (3 mg lectin/ml gel) or Con A (6 mg lectin/ml gel; Vector Labs) for 1 hour at 4°C. After washing the beads with PBS, proteins bound to the beads were released by boiling in SDS sample buffer (without β-mercaptoethanol) and electrophoresed in 12% SDS-polyacrylamide gels. Protein blots of the gels were processed for immunostaining using a rabbit anti-α3 integrin as described above. Control blots were processed the same way except that incubation with primary antibody was omitted.
Supplementary Material
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/122/20/3684/DC1
This work was supported by a National Eye Institute (NIH) Grant EY007088 (N.P.), R01AI20958 (F.-T.L.), R01EY03306 (I.K.G.), a core grant for vision research P30EY13078, New England Corneal Transplant Fund, Mass Lions Eye Research fund and a challenge grant from Research to Prevent Blindness. Deposited in PMC for release after 12 months.
References
- Adams, J. C. (2001). Cell-matrix contact structures. Cell Mol. Life Sci. 58, 371-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao, Q. and Hughes, R. C. (1999). Galectin-3 and polarized growth within collagen gels of wild-type and ricin-resistant MDCK renal epithelial cells. Glycobiology 9, 489-495. [DOI] [PubMed] [Google Scholar]
- Bellis, S. L. (2004). Variant glycosylation: an underappreciated regulatory mechanism for beta1 integrins. Biochim. Biophys. Acta 1663, 52-60. [DOI] [PubMed] [Google Scholar]
- Boukamp, P., Petrussevska, R. T., Breitkreutz, D., Hornung, J., Markham, A. and Fusenig, N. E. (1988). Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 106, 761-771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer, C. F. (2004). Thermodynamic binding studies of galectin-1, Gal-3 and -7. Glycoconj. J. 19, 459-465. [DOI] [PubMed] [Google Scholar]
- Cao, Z., Said, N., Amin, S., Wu, H. K., Bruce, A., Garate, M., Hsu, D. K., Kuwabara, I., Liu, F. T. and Panjwani, N. (2002). Galectins-3 and -7, but not galectin-1, play a role in re-epithelialization of wounds. J. Biol. Chem. 277, 42299-42305. [DOI] [PubMed] [Google Scholar]
- Carcamo, C., Pardo, E., Oyanadel, C., Bravo-Zehnder, M., Bull, P., Caceres, M., Martinez, J., Massardo, L., Jacobelli, S., Gonzalez, A. et al. (2006). Galectin-8 binds specific beta1 integrins and induces polarized spreading highlighted by asymmetric lamellipodia in Jurkat T cells. Exp. Cell Res. 312, 374-386. [DOI] [PubMed] [Google Scholar]
- Chhabra, E. S. and Higgs, H. N. (2007). The many faces of actin: matching assembly factors with cellular structures. Nat. Cell Biol. 9, 1110-1121. [DOI] [PubMed] [Google Scholar]
- Choma, D. P., Pumiglia, K. and DiPersio, C. M. (2004). Integrin alpha3beta1 directs the stabilization of a polarized lamellipodium in epithelial cells through activation of Rac1. J. Cell Sci. 117, 3947-3959. [DOI] [PubMed] [Google Scholar]
- Choma, D. P., Milano, V., Pumiglia, K. M. and DiPersio, C. M. (2007). Integrin alpha3beta1-dependent activation of FAK/Src regulates Rac1-mediated keratinocyte polarization on laminin-5. J. Invest. Dermatol. 127, 31-40. [DOI] [PubMed] [Google Scholar]
- Cooper, D. N., Massa, S. M. and Barondes, S. H. (1991). Endogenous muscle lectin inhibits myoblast adhesion to laminin. J. Cell Biol. 115, 1437-1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings, R. D. and Kornfeld, S. (1982). Characterization of the structural determinants required for the high affinity interaction of asparagine-linked oligosaccharides with immobilized Phaseolus vulgaris leukoagglutinating and erythroagglutinating lectins. J. Biol. Chem. 257, 11230-11234. [PubMed] [Google Scholar]
- Dennis, J. W., Pawling, J., Cheung, P., Partridge, E. and Demetriou, M. (2002). UDP-N-acetylglucosamine:alpha-6-D-mannoside beta1,6 N-acetylglucosaminyltransferase V (Mgat5) deficient mice. Biochim. Biophys. Acta 1573, 414-422. [DOI] [PubMed] [Google Scholar]
- Dignass, A. U. (2001). Mechanisms and modulation of intestinal epithelial repair. Inflamm. Bowel Dis. 7, 68-77. [DOI] [PubMed] [Google Scholar]
- Diskin, S., Kumar, J., Cao, Z., Schuman, J. S., Gilmartin, T., Head, S. R. and Panjwani, N. (2006). Detection of differentially expressed glycogenes in trabecular meshwork of eyes with primary open-angle glaucoma. Invest. Ophthalmol. Vis. Sci. 47, 1491-1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diskin, S., Cao, Z., Leffler, H. and Panjwani, N. (2009). The role of integrin glycosylation in galectin-8-mediated trabecular meshwork cell adhesion and spreading. Glycobiology 19, 29-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebato, B., Friend, J. and Thoft, R. A. (1988). Comparison of limbal and peripheral human corneal epithelium in tissue culture. Invest. Ophthalmol. Vis. Sci. 29, 1533-1537. [PubMed] [Google Scholar]
- Fischer, C., Sanchez-Ruderisch, H., Welzel, M., Wiedenmann, B., Sakai, T., Andre, S., Gabius, H. J., Khachigian, L., Detjen, K. M. and Rosewicz, S. (2005). Galectin-1 interacts with the {alpha}5{beta}1 fibronectin receptor to restrict carcinoma cell growth via induction of p21 and p27. J. Biol. Chem. 280, 37266-37277. [DOI] [PubMed] [Google Scholar]
- Fred Brewer, C. (2002). Binding and cross-linking properties of galectins. Biochim. Biophys. Acta 1572, 255-262. [DOI] [PubMed] [Google Scholar]
- Friedrichs, J., Torkko, J. M., Helenius, J., Teravainen, T. P., Fullekrug, J., Muller, D. J., Simons, K. and Manninen, A. (2007). Contributions of galectin-3 and -9 to epithelial cell adhesion analyzed by single cell force spectroscopy. J. Biol. Chem. 282, 29375-29383. [DOI] [PubMed] [Google Scholar]
- Friedrichs, J., Manninen, A., Muller, D. J. and Helenius, J. (2008). Galectin-3 regulates integrin alpha2beta1-mediated adhesion to collagen-I and -IV. J. Biol. Chem. 283, 32264-32272. [DOI] [PubMed] [Google Scholar]
- Fukumori, T., Takenaka, Y., Yoshii, T., Kim, H. R., Hogan, V., Inohara, H., Kagawa, S. and Raz, A. (2003). CD29 and CD7 mediate galectin-3-induced type II T-cell apoptosis. Cancer Res. 63, 8302-8311. [PubMed] [Google Scholar]
- Gendronneau, G., Sidhu, S. S., Delacour, D., Dang, T., Calonne, C., Houzelstein, D., Magnaldo, T. and Poirier, F. (2008). Galectin-7 in the control of epidermal homeostasis after injury. Mol. Biol. Cell 19, 5541-5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gipson, I. K., Spurr-Michaud, S., Argueso, P., Tisdale, A., Ng, T. F. and Russo, C. L. (2003). Mucin gene expression in immortalized human corneal-limbal and conjunctival epithelial cell lines. Invest. Ophthalmol. Vis. Sci. 44, 2496-2506. [DOI] [PubMed] [Google Scholar]
- Goetz, J. G., Joshi, B., Lajoie, P., Strugnell, S. S., Scudamore, T., Kojic, L. D. and Nabi, I. R. (2008). Concerted regulation of focal adhesion dynamics by galectin-3 and tyrosine-phosphorylated caveolin-1. J. Cell Biol. 180, 1261-1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, J. and Taniguchi, N. (2004). Regulation of integrin functions by N-glycans. Glycoconj. J. 21, 9-15. [DOI] [PubMed] [Google Scholar]
- Guo, H. B., Lee, I., Bryan, B. T. and Pierce, M. (2005). Deletion of mouse embryo fibroblast N-acetylglucosaminyltransferase V stimulates alpha5beta1 integrin expression mediated by the protein kinase C signaling pathway. J. Biol. Chem. 280, 8332-8342. [DOI] [PubMed] [Google Scholar]
- Guo, W. and Giancotti, F. G. (2004). Integrin signalling during tumour progression. Nat. Rev. Mol. Cell. Biol. 5, 816-826. [DOI] [PubMed] [Google Scholar]
- Hadari, Y. R., Arbel-Goren, R., Levy, Y., Amsterdam, A., Alon, R., Zakut, R. and Zick, Y. (2000). Galectin-8 binding to integrins inhibits cell adhesion and induces apoptosis. J. Cell Sci. 113, 2385-2397. [DOI] [PubMed] [Google Scholar]
- Hanna, C. (1966). Proliferation and migration of epithelial cells during corneal wound repair in the rabbit and the rat. Am. J. Ophthalmol. 61, 55-63. [PubMed] [Google Scholar]
- Hernandez, J. D., Nguyen, J. T., He, J., Wang, W., Ardman, B., Green, J. M., Fukuda, M. and Baum, L. G. (2006). Galectin-1 binds different CD43 glycoforms to cluster CD43 and regulate T cell death. J. Immunol. 177, 5328-5336. [DOI] [PubMed] [Google Scholar]
- Hirabayashi, J., Hashidate, T., Arata, Y., Nishi, N., Nakamura, T., Hirashima, M., Urashima, T., Oka, T., Futai, M., Muller, W. E. et al. (2002). Oligosaccharide specificity of galectins: a search by frontal affinity chromatography. Biochim. Biophys. Acta 1572, 232-254. [DOI] [PubMed] [Google Scholar]
- Hoyer, K. K., Pang, M., Gui, D., Shintaku, I. P., Kuwabara, I., Liu, F. T., Said, J. W., Baum, L. G. and Teitell, M. A. (2004). An anti-apoptotic role for galectin-3 in diffuse large B-cell lymphomas. Am. J. Pathol. 164, 893-902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, D. K., Zuberi, R. I. and Liu, F. T. (1992). Biochemical and biophysical characterization of human recombinant IgE-binding protein, an S-type animal lectin. J. Biol. Chem. 267, 14167-14174. [PubMed] [Google Scholar]
- Hsu, D. K., Yang, R. Y., Pan, Z., Yu, L., Salomon, D. R., Fung-Leung, W. P. and Liu, F. T. (2000). Targeted disruption of the galectin-3 gene results in attenuated peritoneal inflammatory responses. Am. J. Pathol. 156, 1073-1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, D. K., Chernyavsky, A. I., Chen, H. Y., Yu, L., Grando, S. A. and Liu, F. T. (2009). Endogenous galectin-3 is localized in membrane lipid rafts and regulates migration of dendritic cells. J. Invest. Dermatol. 129, 573-583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, R. C. (1999). Secretion of the galectin family of mammalian carbohydrate-binding proteins. Biochim. Biophys. Acta 1473, 172-185. [DOI] [PubMed] [Google Scholar]
- Hynes, R. O. (2002). Integrins: bidirectional, allosteric signaling machines. Cell 110, 673-687. [DOI] [PubMed] [Google Scholar]
- Kuwabara, I., Kuwabara, Y., Yang, R. Y., Schuler, M., Green, D. R., Zuraw, B. L., Hsu, D. K. and Liu, F. T. (2002). Galectin-7 (PIG1) exhibits pro-apoptotic function through JNK activation and mitochondrial cytochrome c release. J. Biol. Chem. 277, 3487-3497. [DOI] [PubMed] [Google Scholar]
- Lagana, A., Goetz, J. G., Cheung, P., Raz, A., Dennis, J. W. and Nabi, I. R. (2006). Galectin binding to Mgat5-modified N-glycans regulates fibronectin matrix remodeling in tumor cells. Mol. Cell. Biol. 26, 3181-3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy, Y., Arbel-Goren, R., Hadari, Y. R., Eshhar, S., Ronen, D., Elhanany, E., Geiger, B. and Zick, Y. (2001). Galectin-8 functions as a matricellular modulator of cell adhesion. J. Biol. Chem. 276, 31285-31295. [DOI] [PubMed] [Google Scholar]
- Levy, Y., Ronen, D., Bershadsky, A. D. and Zick, Y. (2003). Sustained induction of ERK, protein kinase B, and p70 S6 kinase regulates cell spreading and formation of F-actin microspikes upon ligation of integrins by galectin-8, a mammalian lectin. J. Biol. Chem. 278, 14533-14542. [DOI] [PubMed] [Google Scholar]
- Liu, F. T. and Rabinovich, G. A. (2005). Galectins as modulators of tumour progression. Nat. Rev. Cancer 5, 29-41. [DOI] [PubMed] [Google Scholar]
- Lock, J. G., Wehrle-Haller, B. and Stromblad, S. (2008). Cell-matrix adhesion complexes: master control machinery of cell migration. Semin. Cancer Biol. 18, 65-76. [DOI] [PubMed] [Google Scholar]
- Lu, L. H., Nakagawa, R., Kashio, Y., Ito, A., Shoji, H., Nishi, N., Hirashima, M., Yamauchi, A. and Nakamura, T. (2007). Characterization of galectin-9-induced death of Jurkat T cells. J. Biochem. 141, 157-172. [DOI] [PubMed] [Google Scholar]
- Ma, J. J. and Dohlman, C. H. (2002). Mechanisms of corneal ulceration. Ophthalmol. Clin. North Am. 15, 27-33. [DOI] [PubMed] [Google Scholar]
- Maeda, N., Kawada, N., Seki, S., Arakawa, T., Ikeda, K., Iwao, H., Okuyama, H., Hirabayashi, J., Kasai, K. and Yoshizato, K. (2003). Stimulation of proliferation of rat hepatic stellate cells by galectin-1 and galectin-3 through different intracellular signaling pathways. J. Biol. Chem. 278, 18938-18944. [DOI] [PubMed] [Google Scholar]
- Mitra, S. K. and Schlaepfer, D. D. (2006). Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 18, 516-523. [DOI] [PubMed] [Google Scholar]
- Nishi, N., Shoji, H., Seki, M., Itoh, A., Miyanaka, H., Yuube, K., Hirashima, M. and Nakamura, T. (2003). Galectin-8 modulates neutrophil function via interaction with integrin alphaM. Glycobiology 13, 755-763. [DOI] [PubMed] [Google Scholar]
- Paclik, D., Lohse, K., Wiedenmann, B., Dignass, A. U. and Sturm, A. (2008). Galectin-2 and -4, but not Galectin-1, promote intestinal epithelial wound healing in vitro through a TGF-beta-independent mechanism. Inflamm. Bowel Dis. 14, 1366-1372. [DOI] [PubMed] [Google Scholar]
- Partridge, E. A., Le Roy, C., Di Guglielmo, G. M., Pawling, J., Cheung, P., Granovsky, M., Nabi, I. R., Wrana, J. L. and Dennis, J. W. (2004). Regulation of cytokine receptors by Golgi N-glycan processing and endocytosis. Science 306, 120-124. [DOI] [PubMed] [Google Scholar]
- Przybylo, M., Pochec, E., Link-Lenczowski, P. and Litynska, A. (2008). Beta1-6 branching of cell surface glycoproteins may contribute to uveal melanoma progression by up-regulating cell motility. Mol. Vis. 14, 625-636. [PMC free article] [PubMed] [Google Scholar]
- Raja Sivamani, K., Garcia, M. S. and Isseroff, R. R. (2007). Wound re-epithelialization: modulating keratinocyte migration in wound healing. Front. Biosci. 12, 2849-2868. [DOI] [PubMed] [Google Scholar]
- Rapoport, E. M., Kurmyshkina, O. V. and Bovin, N. V. (2008). Mammalian galectins: structure, carbohydrate specificity, and functions. Biochemistry (Mosc.) 73, 393-405. [DOI] [PubMed] [Google Scholar]
- Ridley, A. J., Schwartz, M. A., Burridge, K., Firtel, R. A., Ginsberg, M. H., Borisy, G., Parsons, J. T. and Horwitz, A. R. (2003). Cell migration: integrating signals from front to back. Science 302, 1704-1709. [DOI] [PubMed] [Google Scholar]
- Rossi, B., Espeli, M., Schiff, C. and Gauthier, L. (2006). Clustering of pre-B cell integrins induces galectin-1-dependent pre-B cell receptor relocalization and activation. J. Immunol. 177, 796-803. [DOI] [PubMed] [Google Scholar]
- Seiler, W. O., Stahelin, H. B., Zolliker, R., Kallenberger, A. and Luscher, N. J. (1989). Impaired migration of epidermal cells from decubitus ulcers in cell cultures. A cause of protracted wound healing? Am. J. Clin. Pathol. 92, 430-434. [DOI] [PubMed] [Google Scholar]
- Small, J. V. and Resch, G. P. (2005). The comings and goings of actin: coupling protrusion and retraction in cell motility. Curr. Opin. Cell Biol. 17, 517-523. [DOI] [PubMed] [Google Scholar]
- Sparrow, C. P., Leffler, H. and Barondes, S. H. (1987). Multiple soluble beta-galactoside-binding lectins from human lung. J. Biol. Chem. 262, 7383-7390. [PubMed] [Google Scholar]
- Stillman, B. N., Hsu, D. K., Pang, M., Brewer, C. F., Johnson, P., Liu, F. T. and Baum, L. G. (2006). Galectin-3 and galectin-1 bind distinct cell surface glycoprotein receptors to induce T cell death. J. Immunol. 176, 778-789. [DOI] [PubMed] [Google Scholar]
- Stowell, S. R., Karmakar, S., Stowell, C. J., Dias-Baruffi, M., McEver, R. P. and Cummings, R. D. (2007). Human galectin-1, -2, and -4 induce surface exposure of phosphatidylserine in activated human neutrophils but not in activated T cells. Blood 109, 219-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowell, S. R., Arthur, C. M., Mehta, P., Slanina, K. A., Blixt, O., Leffler, H., Smith, D. F. and Cummings, R. D. (2008a). Galectin-1, -2, and -3 exhibit differential recognition of sialylated glycans and blood group antigens. J. Biol. Chem. 283, 10109-10123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowell, S. R., Arthur, C. M., Slanina, K. A., Horton, J. R., Smith, D. F. and Cummings, R. D. (2008b). Dimeric Galectin-8 induces phosphatidylserine exposure in leukocytes through polylactosamine recognition by the C-terminal domain. J. Biol. Chem. 283, 20547-20559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm, A., Lensch, M., Andre, S., Kaltner, H., Wiedenmann, B., Rosewicz, S., Dignass, A. U. and Gabius, H. J. (2004). Human galectin-2: novel inducer of T cell apoptosis with distinct profile of caspase activation. J. Immunol. 173, 3825-3837. [DOI] [PubMed] [Google Scholar]
- Toscano, M. A., Bianco, G. A., Ilarregui, J. M., Croci, D. O., Correale, J., Hernandez, J. D., Zwirner, N. W., Poirier, F., Riley, E. M., Baum, L. G. et al. (2007). Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nat. Immunol. 8, 825-834. [DOI] [PubMed] [Google Scholar]
- Zhao, Y., Itoh, S., Wang, X., Isaji, T., Miyoshi, E., Kariya, Y., Miyazaki, K., Kawasaki, N., Taniguchi, N. and Gu, J. (2006). Deletion of core fucosylation on alpha3beta1 integrin down-regulates its functions. J. Biol. Chem. 281, 38343-38350. [DOI] [PubMed] [Google Scholar]
- Zhuo, Y., Chammas, R. and Bellis, S. L. (2008). Sialylation of beta1 integrins blocks cell adhesion to galectin-3 and protects cells against galectin-3-induced apoptosis. J. Biol. Chem. 283, 22177-22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.