

Abstract
Virus infection may contribute to asthma pathogenesis. In turn, a Th2-polarized pulmonary environment may increase host susceptibility to infection. We used a cockroach antigen (CRA) model of allergic airways disease to test the hypothesis that Th2 cytokine overproduction increases susceptibility to mouse adenovirus type 1 (MAV-1). CRA sensitization led to upregulated lung expression of IL-4 and IL-13, lung cellular inflammation, and exaggerated airways mucus production. Following intranasal MAV-1 infection, lung cellular inflammation was more pronounced in CRA-sensitized mice than in unsensitized mice at 7 days post infection but not at a later time point. CRA sensitization did not significantly suppress lung IFN-γ expression, and lung IFN-γ expression was upregulated in both CRA-sensitized mice and unsensitized mice over the course of MAV-1 infection. Despite CRA-induced differences in pulmonary inflammation, MAV-1 viral loads in lung and spleen and MAV-1 gene expression in the lung did not differ between CRA-sensitized and unsensitized mice. Our data therefore suggest that MAV-1 pathogenesis is not affected directly or indirectly by the Th2 polarization associated with allergic airways disease.
Keywords: adenovirus, viral pathogenesis, asthma, Th2 cytokines
Introduction
Asthma is a chronic disorder of the airways characterized by inflammation, increased airway hyperresponsiveness, and obstruction of air flow (reviewed in Eisenbarth, Cassel, and Bottomly, 2004). Asthma prevalence continues to rise among 0 to 17 year-olds, increasing from an estimated 3.6% to 6.2% between 1980 to 1996, with the greatest increase observed in 0 to 4 year-olds (Akinbami and Schoendorf, 2002). Acute viral respiratory infections act as triggers for asthma exacerbation, with specific viruses isolated in up to 80–85% of asthma exacerbations in children (Johnston, 2007; Traves and Proud, 2007). Viral infections may also be risk factors for subsequent development of asthma. Children infected with respiratory syncytial virus (RSV) as infants are more likely to develop asthma compared to children without RSV infection (Sigurs et al., 2000), although it is unclear if asthma develops as a direct result of RSV infection or if the virus targets children who are already predisposed to develop obstructive airway disease (Peebles, 2004; Perez-Yarza et al., 2007). Some reports suggest that persistent adenovirus infection serves as a risk factor for childhood asthma (Macek et al., 1994; Marin et al., 2000).
Chronic inflammation characteristic of asthma is dependent on prototypic T-helper type (Th)2 cytokines such as interleukin (IL)-4 and IL-13 (Chatila, 2004). IL-4 and IL-13 activate the intracellular signal transducer and activator of transcription (STAT)6, one key mediator in pathways linked to allergic inflammation and mucus production (Chatila, 2004). Data from mouse models suggest that the Th2-polarized microenvironment in asthma may act as a risk factor for infection. For instance, clearance of Pseudomonas aeruginosa from the lungs is impaired in mice with allergic airways inflammation induced by ovalbumin sensitization (Beisswenger et al., 2006). There is also evidence that susceptibility to viral infection is increased in the presence of elevated levels of Th2 cytokines. Mice treated with IL-4 following influenza virus infection show increased viral titers in the lungs compared to controls, and clearance of virus from the lungs is impaired (Moran et al., 1996). Mice constitutively overexpressing IL-4 in the lungs also exhibit a delay in RSV clearance (Fischer et al., 1997).
Studies of human adenovirus pathogenesis are limited by the strict species-specificities of the adenoviruses. Little is known regarding the effects of Th2 cytokines on adenovirus pathogenesis. Mouse adenovirus type 1 (MAV-1) provides an excellent tool to study the pathogenesis of an adenovirus in its natural host. We have previously established MAV-1 as a model of adenovirus respiratory infection (Weinberg et al., 2007; Weinberg et al., 2005). Here we used MAV-1 to test the hypothesis that Th2 polarization increases susceptibility to respiratory infection with an adenovirus.We report the effects of in vivo Th2 polarization on MAV-1 pathogenesis using allergic airways sensitization with cockroach antigen (CRA). Our results suggest that MAV-1 is relatively resistant to the effects of exaggerated Th2 cytokine production.
RESULTS
Effects of Th2 Polarization on MAV-1-Induced Pulmonary Inflammation
Cockroach antigen (CRA) sensitization has been established as a model of allergic airways disease, with increased airway hyperreactivity, mucus production, peribronchial inflammation, and upregulation of lung IL-4 and IL-13 in sensitized mice compared to control animals (Berlin, Hogaboam, and Lukacs, 2006). We used CRA to modify the pulmonary environment in mice prior to infection with MAV-1. After an initial systemic sensitization, mice were repeatedly challenged intranasally (i.n.) with CRA over an 18-day period prior to infection and during the course of infection (summarized in Figure 1a) in order to localize the response to the airways. We used RT-qPCR to assess the effect of CRA sensitization on expression in the lung of IL-4 and IL-13 (Figure 1b). Following sensitization but prior to infection, expression of each Th2 cytokine was significantly increased in the lungs of CRA-sensitized mice compared to unsensitized control animals that received phosphate-buffered saline (PBS; 68 ± 19 fold change from PBS-treated control animals for IL-4; 1595 ± 591 fold change for IL-13; mean ± S.E.M.; P<0.01 in each case). These data are similar to studies using CRA sensitization-induced allergic airways disease in which IL-4 and IL-13 protein levels were increased in both lung homogenate and bronchoalveolar lavage fluid of CRA-sensitized mice compared to unsensitized controls (Lindell et al., 2008). Lung expression of both IL-4 and IL-13 continued to be increased in CRA-sensitized mice compared to unsensitized control mice at all time points following MAV-1 infection (data not shown).
Figure 1.
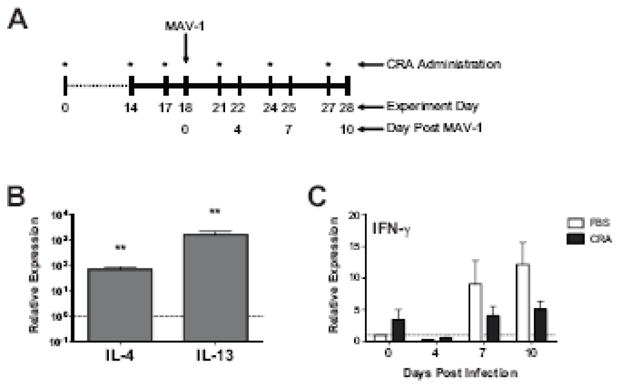
Cytokine expression in the lungs of CRA-sensitized mice. (A) Experimental protocol for in vivo CRA sensitization and infection. Animals were sensitized intraperitoneally and subcutaneously with CRA at day 0 and subsequently received intranasal CRA at the time points indicated with an asterisk. Unsensitized control mice were instead treated with PBS at each time point. All animals were infected intranasally with MAV-1 on day 18. Animals were euthanized and organs were harvested prior to infection on day 18 and then at 4, 7 and 10 d.p.i. (corresponding to experiment days 22, 25 and 28). (B) RT-qPCR was used to assess IL-4 and IL-13 expression in the lungs of CRA-sensitized and unsensitized control mice on day 18, immediately prior to MAV-1 infection. Data for five mice per group are presented as means ± S.E.M. **P<0.01 compared to unsensitized mice. (C) RT-qPCR was used to assess IFN-γ expression in the lungs of CRA-sensitized and unsensitized control mice prior to infection and then at the indicated time points post infection. Data for CRA-sensitized mice are presented as fold change from expression levels measured in unsensitized control mice, which are set at 1 for each time point (indicated with horizontal dashed line). Data for five to ten mice per group are combined from three independent experiments and presented as means ± S.E.M.
We also assessed lung expression of interferon (IFN)-γ following the initial 18-day CRA sensitization and throughout the course of MAV-1 infection (Figure 1c). Somewhat surprisingly, CRA sensitization did not initially suppress IFN-γ expression in the lungs. Following sensitization but prior to infection, lung IFN-γ expression was instead slightly upregulated in CRA-sensitized mice compared to unsensitized control mice (Figure 1c, day 0; 3.4 ± 1.6 fold change from PBS-treated control animals), although this difference was not statistically significant. Lung IFN-γ expression was upregulated in both CRA-sensitized and unsensitized control mice following MAV-1 infection. IFN-γ expression was slightly lower in CRA-sensitized mice compared to unsensitized control mice following MAV-1 infection, but there were no statistically significant differences between groups at any time point.
Similar to our earlier findings (Weinberg et al., 2005), MAV-1 infection of unsensitized mice induced a mild pneumonitis (Figure 2a, left panels). Mononuclear cells began to accumulate around medium and large airways of infected mice as early as 4 d.p.i., and increasing peribronchial infiltrates were noted at 7 and 10 d.p.i. By itself, CRA sensitization also induced a cellular inflammatory response in the lungs, with inflammatory cells surrounding airways of sensitized mice (Figure 2a, uppermost right panel). Cellular inflammation became more extensive in CRA-sensitized mice over the course of MAV-1 infection (Figure 2a, right panels). MAV-1-induced cellular inflammation was more pronounced in the lungs of CRA-sensitized mice compared to control mice at 4 and 7 d.p.i. By 10 d.p.i., lungs of MAV-1-infected control mice were histologically similar to lungs of MAV-1-infected, CRA-sensitized mice.
Figure 2.

Cellular inflammation in lungs of CRA-sensitized, MAV-1 infected mice. Mice were sensitized with CRA or treated with PBS according to the schedule outlined in Figure 1a. All mice were then infected intranasally with MAV-1. Lungs were harvested from CRA-sensitized and unsensitized control mice at the indicated time points. (A) Paraffin-embedded lung sections were stained with hematoxylin and eosin to evaluate cellular infiltrates. Images are characteristic of a minimum of two mice per group per time point. Scale bars, 100 μm. (B) Pathology index scores were generated to quantify cellular inflammation in the lungs of CRA-sensitized and unsensitized control mice. Data are presented as means ± S.E.M. at each time point (n=2 at 0 and 10 d.p.i., n=4 at 4 and 7 d.p.i.). *P<0.05 compared to unsensitized mice at the same time point.
Pathology index scores (Table 1) quantifying lung inflammation in available lung sections reflected the qualitative assessments of lung inflammation described above. As shown in Figure 2b, lung pathology increased over time in both CRA-sensitized and unsensitized control mice over the course of MAV-1 infection. Pathology was greater in CRA-sensitized mice compared to unsensitized control mice at every time point before and after MAV-1 infection, but only at 7 d.p.i. was this difference statistically significant.
Table 1.
Quantification of cellular inflammation in histologic specimens.
| Scorea | Cellular Infiltrates Around Airway Lumens | Pneumonitis |
|---|---|---|
| 0 | No infiltrates | No infiltrates |
| 1 | 1 to 3 cell diameters thick | Increased cells visible only at high power |
| 2 | 4 to 10 cell diameters thick | Easily seen cellular Infiltrates |
| 3 | >10 cell diameters thick | Extensive consolidation by inflammatory cells |
A score from 0 to 3 was given for each of the two categories. The score for each category was multiplied by a number reflecting the extent of involvement in the specimen (5% to 25% = 1, >25% to 50% = 2, >50% = 3). The final pathology index score was obtained by adding together values for each category, resulting in a total score that could range from 0 to 18.
Mucus Production in Airways of CRA-Sensitized and MAV-1-Infected Mice
Mucus overproduction is a common feature of asthma (Evans and Koo, 2008), and respiratory viral infection is often associated with upregulated mucus production in the airways (see, for instance, Inoue et al., 2006). There are no reports that specifically detail mucus production induced by infection with either a human adenovirus or MAV-1. We first assessed airway mucus production using Periodic acid-Schiff (PAS) staining. No mucus production was evident in lungs of unsensitized, uninfected mice (Figure 3a, upper left panel). At 10 d.p.i., scattered sparse staining in respiratory epithelial cells suggested that mucus production was induced to a limited extent by MAV-1 infection (Figure 3a, bottom left panel). Prior to infection, mucus production evaluated by PAS staining was dramatically enhanced in the airways of CRA-sensitized mice compared to unsensitized control mice (Figure 3a, compare upper left and upper right panels). Mucus production became even more pronounced in respiratory epithelial cells over the course of MAV-1 infection in the setting of ongoing CRA exposure (Figure 3a, right panels). These qualitative assessments correlated with quantitative measurements of PAS staining in the lungs. The percentage of airways with PAS staining in respiratory epithelial cells was consistently greater in CRA-sensitized mice than in unsensitized control mice, both before and following MAV-1 infection (Figure 3b). The percentage of PAS-positive airway lumens increased over the first 7 d.p.i. in CRA-sensitized mice but remained very low in unsensitized control mice over the course of infection.
Figure 3.

Mucus production in lungs of CRA-sensitized, MAV-1 infected mice. Mice were sensitized with CRA or treated with PBS according to the schedule outlined in Figure 1a. All mice were then infected intranasally with MAV-1. Lungs were harvested from CRA-sensitized and unsensitized control mice at the indicated time points. (A) Lung sections were stained with PAS to evaluate mucus production in the lungs. Pronounced staining of mucus is present in respiratory epithelial cells lining the airways of CRA-sensitized mice (right panels). Scattered staining is present in the respiratory epithelial cells lining the airways of PBS-treated, MAV-1-infected mice at 7 and 10 d.p.i. (left panels, arrows indicate examples of PAS staining). Images are characteristic of a minimum of two mice per group per time point. Scale bars, 100 μm. (B) Lung sections were examined to quantify the percentage of airway lumens with positive PAS staining. Data are presented as means ± S.E.M. at each time point (n=2 at 0 and 10 d.p.i., n=4 at 4 and 7 d.p.i.). *P<0.05 and ***P<0.001 compared to unsensitized mice at the same time point. (C) RT-qPCR was used to assess muc5ac and gob5 expression in the lungs of CRA-sensitized and unsensitized control mice on day 18, immediately prior to MAV-1 infection. Data for CRA-sensitized mice are presented as fold change from expression levels measured in unsensitized control mice, which are set at 1 for each time point (indicated with horizontal dashed line). Data for five mice per group are presented as means ± S.E.M. **P<0.01 compared to unsensitized mice at the same time point.
We confirmed the exaggerated mucus production in CRA-sensitized mice using RT-qPCR to measure the expression of muc5ac and gob5, genes encoding major components of mucus in the airways (Figure 3c). Following the initial 18-day CRA sensitization protocol, the expression of each gene was upregulated in CRA-sensitized mice compared to PBS-treated mice (18.9 ± 9.1 fold change from PBS-treated control animals for muc5ac; 8.1 ± 2.7 for gob5; P<0.01 in each case).
Effects of CRA Sensitization on MAV-1 Viral Loads and Viral Gene Expression
To determine if the Th2-skewed immunologic environment in CRA-sensitized mice increased susceptibility to MAV-1 respiratory infection, we used qPCR to measure MAV-1 viral loads at various time points following infection (Figure 4a). BALB/c mice are typically much more resistant to MAV-1 than are other inbred mouse strains (Guida et al., 1995; Spindler et al., 2001), and we were not able to consistently isolate infectious virus from the lungs of BALB/c mice using plaque assay (data not shown). We therefore used qPCR measurements of MAV-1 DNA as a more sensitive way to quantify viral loads in the lungs of infected mice. CRA sensitization did not affect the amount of viral DNA detected in the lungs at an early time point (4 d.p.i.), a time point corresponding to the typical peak of lung viral loads (7 d.p.i.), or a later time point when viral DNA is being cleared from the lungs (10 d.p.i.). To provide an additional correlate of viral activity in the lungs, we used RT-qPCR to quantify the expression of MAV-1 hexon (Figure 4b). Similar to the pattern observed with DNA viral loads, there was no statistically significant difference between viral gene expression in the lungs of CRA-sensitized mice compared to control mice at any time point.
Figure 4.
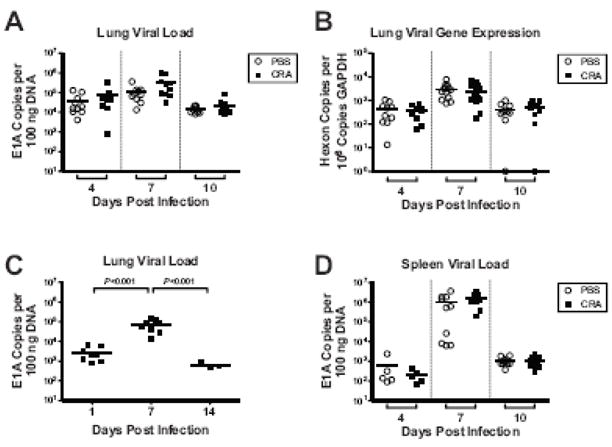
Effects of CRA sensitization on MAV-1 viral loads and viral gene expression. (A, B and D) Mice were sensitized with CRA or treated with PBS according to the schedule outlined in Figure 1a. All mice were then infected intranasally with MAV-1. DNA and RNA were extracted from lungs and DNA was extracted from spleens harvested at the indicated time points. (C) In a separate experiment, naïve mice were infected intranasally with MAV-1 and DNA was extracted from lungs at the indicated time points. qPCR was used to quantify MAV-1 genome copies in DNA from lung (A,C) and spleen (D). RT-qPCR was used to quantify copy numbers of MAV-1 hexon and GAPDH transcripts in the lungs (B). Individual circles represent values for individual mice and horizontal bars represent means for each group. DNA viral load data are expressed as copies of MAV-1 genome per 100 ng of input DNA. Viral gene expression data are expressed as copies of MAV-1 hexon per 106 copies of GAPDH. Indicated P values were obtained by two-way ANOVA with Bonferroni’s multiple comparison tests. Where not indicated, P values were > 0.05.
Of note, the viral loads that were measured at 7 d.p.i. in CRA-sensitized and unsensitized control mice in these experiments (Figure 4a) were similar in magnitude to viral loads measured at 7 d.p.i. in separate experiments demonstrating a time course of MAV-1 respiratory infection (Figure 4c). Lung viral loads at 7 d.p.i. in the CRA experiments (Figure 4a) were approximately two logs greater than those measured in separate experiments at 1 d.p.i. (Figure 4c). This accumulation of viral DNA, in conjunction with the detection of increasing amounts of viral gene expression in the lung (Fig. 4b) and the dissemination of virus to the spleen at a later time point (Fig. 4d and text below), all strongly suggest that MAV-1 actively replicates in the lungs of BALB/c mice.
Lastly, to determine whether CRA-sensitization affected the dissemination of MAV-1 out of the lung, we measured viral loads in the spleen, which is an additional target organ for MAV-1 following i.n. inoculation (Kajon, Brown, and Spindler, 1998). We detected no statistically significant differences in spleen viral loads in CRA-sensitized mice compared to PBS-treated control mice at any time point (Figure 4d). Together, these data suggest that CRA sensitization and the resulting Th2-skewed immunologic environment in the lung had no effect on viral replication in the lung, clearance of virus from the lung, or viral dissemination out of the lung.
Discussion
Aberrant inflammatory responses in the lung such as the Th2-polarized environment seen in asthma may alter an individual’s susceptibility to a variety of pathogens. Previous work in animal and cell culture models suggests that Th2-skewed inflammatory responses alter susceptibility to some bacterial and viral pathogens.For instance, preincubation in vitro of primary human bronchial epithelial cells with recombinant IL-4 or IL-13 increased the amount of Pseudomonas aeruginosa recovered at 24 h.p.i. (Beisswenger et al., 2006). In the same study, allergic sensitization of mice with ovalbumin increased the amount of viable bacteria recovered from the lungs compared to amounts recovered from unsensitized animals. Treatment of mice with IL-4 delays the clearance of influenza virus from the lungs (Moran et al., 1996). Similarly, IL-4-overexpressing transgenic mice show delayed clearance of RSV following i.n. infection (Fischer et al., 1997). This effect is associated with diminished RSV-specific cytolytic activity of lung lymphocytes.
Similar studies examining the effect of Th2 polarization on human adenovirus pathogenesis have not yet been done, in part because of the strict species-specificity of the adenoviruses. In this study, we investigated the effects of Th2 polarization on MAV-1 pathogenesis. In a mouse model of allergic airways disease, CRA sensitization substantially upregulated lung expression of IL-4 and IL-13, induced a cellular inflammatory response in the lungs, and increased airway mucus production. MAV-1 viral gene expression in the lungs and MAV-1 viral loads in lungs and spleens were not affected by CRA sensitization, despite dramatic differences in pulmonary inflammation at baseline and the slight suppression of lung IFN-γ responses at later time points in CRA-sensitized animals. Taken together, our data suggest that MAV-1 is relatively resistant to the effects of classical Th2 cytokines in the lungs.
In theory, there are many ways in which exaggerated Th2 cytokine signaling could directly influence viral pathogenesis. STAT6 phosphorylation is the major intracellular event triggered by binding of IL-4 or IL-13 to the IL4Rα receptor (Heller et al., 2004; Kuperman et al., 1998). Phosphorylated STAT6 homodimerizes and translocates to the nucleus, where it initiates gene transcription by binding to specific DNA motifs (Heller et al., 2004; Kuperman et al., 1998). Transcriptional activation by STAT6 involves interactions with the cofactors cAMP response element-binding binding protein (CBP) and p300 (Gingras et al., 1999; McDonald and Reich, 1999). Human adenovirus E1A, which also binds to CBP and p300, inhibits CBP/p300-mediated STAT6 coactivation and represses IL-4-induced target gene activity (Gingras et al., 1999; McDonald and Reich, 1999). While adenovirus infection may thus inhibit subsequent cytokine-stimulated STAT6 activity, it is plausible that viral gene transcription could be enhanced by STAT6 activated by IL-4 and/or IL-13. Multiple transcription factors in addition to STAT6 are activated in the presence of “proasthmatic cytokines” such as IL-4 and IL-13 (Panebra et al., 2007), so other pathways may contribute to Th2 cytokine enhancement of adenovirus replication. It is also possible that IL-4 and IL-13 suppress the production of cytokines or other factors that inhibit viral replication in an autocrine or paracrine manner. Th2 cytokine-mediated increases in the expression of a virus receptor, as has been demonstrated for ICAM-1, a rhinovirus receptor (Bianco et al., 1998; Bianco et al., 2000), could also contribute to changes in viral pathogenesis.
There are also many ways in which Th2 polarization could indirectly influence viral pathogenesis, modulating host immune responses in ways that could be conducive to viral replication. For instance, alveolar macrophages (AMs) play a critical role in mediating the clearance of adenovirus from the lungs via phagocytosis (Carey et al., 2007; Zsengeller et al., 2000) in addition to recruiting and activating other cells to the site of infection (Zhou et al., 1994; Zsengeller et al., 2000). IL-4 can interfere with AM function (Zhou et al., 1994), potentially contributing to increased viral replication in the lungs. IL-4 and IL-13 may also affect viral pathogenesis by altering lymphocyte function. Th2 cytokines inhibit the function of Th1 lymphocytes through interference with memory CTL expansion or recruitment of specific memory CD8+ T cells (Bot et al., 2000). Specific roles for T lymphocytes have not yet been defined in MAV-1 respiratory infection, and in this study we did not phenotype the inflammatory cells infiltrating the lungs of infected mice in order to determine if T lymphocytes contribute to MAV-1-induced responses in the lungs. However, T lymphocytes are involved in MAV-1 pathogenesis following i.p. infection, playing a role in the acute immunopathology observed in MAV-1 encephalomyelitis and contributing to long-term host survival following intraperitoneal (i.p.) infection (Moore, Brown, and Spindler, 2003). In our model of allergic airways disease, increased IL-4 and IL-13 in the lungs could foster an atmosphere in which lymphocytes critical for the control of viral infection are not effectively recruited to the airways, are impaired in their function, or both. Based on our in vivo data, however, any such change induced by CRA sensitization was not sufficient to substantially alter MAV-1 pathogenesis.
It is interesting that lung IFN-γ expression was modestly upregulated, although not to a statistically significant degree, following CRA sensitization. This subtle finding is not unique to our report, as CRA-associated IFN-γ upregulation in the lung was reported in a study using a similar mouse model of chronic CRA exposure (Lindell et al., 2008). The particular cell types producing increased levels of IFN-γ in this model have not yet been identified (Nicholas Lukacs, personal communication). MAV-1 is relatively resistant to the effects of type I and type II IFNs in mouse L929 cells (Kajon and Spindler, 2000). However, mice deficient for IFN-γ production are more susceptible to systemic MAV-1 infection (Kathy Spindler, personal communication), suggesting that IFN-γ may contribute to the control of MAV-1 infection in some in vivo settings. IFN-γ overproduction caused by latent gammaherpesvirus infection is associated with decreased MAV-1 lung viral loads (Nguyen et al., 2008). IFN-γ expression was readily detected in both CRA-sensitized mice and unsensitized control mice following MAV-1 infection. It is possible that this IFN-γ expression was sufficient to contribute to control of MAV-1 such that no differences in viral loads or viral gene expression were observed.
Mucus overproduction in the lung is a common feature of asthma (Evans and Koo, 2008). The results of our study suggest that acute MAV-1 infection did not induce mucus production in the lungs of unsensitized mice. Mucus production increased in the lungs of CRA-sensitized mice following MAV-1 infection, but not in the lungs of unsensitized, MAV-1-infected mice. It is possible that allergic airways disease induced by CRA sensitization primes the lungs for exaggerated MAV-1-induced mucus production. Likewise, this type of synergy between CRA sensitization and MAV-1 infection, though not specifically evaluated in these studies, may account for some of the observed differences in pulmonary inflammation between unsensitized and CRA-sensitized animals (as with lung IFN-γ expression in Figure 1c and lung cellular inflammation in Figure 2). However, it seems more likely that increased mucus production following MAV-1 infection was due to the ongoing exposure to CRA, a potent stimulus for mucus production by itself.
While increased mucus production could interfere with efficient clearance of a pathogen from the lungs, it is also possible that mucus overproduction could inhibit respiratory infection by interfering with the access of a virus to its target receptor on the luminal surface of a respiratory epithelial cell. MAV-1 does not use the mouse equivalent of the coxsackie-adenovirus receptor used by many human adenovirus serotypes (Lenaerts et al., 2006). Similar to human adenovirus type 5, integrin and heparan sulfate glycosaminoglycans play a role in interactions between MAV-1 and target cells (Raman et al., 2009). Consistent with the ability of mucus to inhibit virus access to target receptors, MAV-1 infection of mice deficient in Muc1, a tethered mucin present on the luminal surface of respiratory epithelial cells (Gendler, 2001), results in higher lung viral loads than in wild type mice (Ngyuen and Weinberg, manuscript in preparation). Inhibitory effects of mucus overproduction could counterbalance any pro-viral effects of Th2 cytokine overproduction induced by CRA sensitization, leading to an apparent overall lack of an effect on MAV-1 pathogenesis as we found in this study.
In summary, we demonstrate that CRA sensitization of mice leads to a Th2-polarized pulmonary environment, with exaggerated expression of IL-4 and IL-13 and increased mucus production in the lung. In addition, CRA sensitization was associated with the suppression of IFN-γ responses induced by MAV-1 respiratory infection. Unlike previous reports describing increased susceptibility to respiratory infection with bacterial and unrelated viral pathogens in settings of increased Th2 cytokine levels, Th2 polarization induced by CRA sensitization did not increase susceptibility to MAV-1 infection. Thus, while CRA sensitization induced robust changes in pulmonary inflammation characteristic of allergic airways disease, these changes in pulmonary immune function did not substantially affect the pathogenesis of MAV-1 respiratory infection.
Materials and Methods
Virus and Cell Lines
MAV-1 was grown in NIH 3T6 fibroblasts and titers were determined by plaque assay as previously described (Cauthen, Welton, and Spindler, 2007).
Cockroach Antigen Sensitization and In Vivo Infection
All animal work was approved by the University of Michigan Committee on the Use and Care of Animals. An allergic airways model using CRA sensitization (summarized in Figure 1a) was modified from that previously described (Berlin, Hogaboam, and Lukacs, 2006). Four to six week-old female BALB/c mice (Jackson Laboratories) were housed in specific pathogen-free conditions. Under ketamine/xylazine anesthesia, mice were sensitized i.p. and subcutaneously (s.q.) with a 1:1 emulsion of CRA (~2 μg/mouse, Hollister-Steir) and incomplete Freund’s Adjuvant (Sigma). On days 14 and 17 following the initial sensitization, the mice were anesthetized and challenged i.n. with CRA (~0.8 μg/mouse) diluted to 40 μl with phosphate buffered saline (PBS). On day 18, mice were anesthetized and inoculated i.n. with 105 PFU of MAV-1 diluted to a total volume of 40 μl with sterile phosphate-buffered saline (PBS). Mice received additional i.n. challenges of CRA on days 21, 24, and 27, corresponding to 3, 6, and 9 d.p.i. Unsensitized control mice were inoculated with equivalent amounts of PBS instead of CRA at each time point. At baseline following sensitization but before infection, and then again at 4, 7 and 10 d.p.i., organs were harvested and snap-frozen on dry ice and stored at −80°C. In separate experiments, unsensitized mice were infected with 105 PFU of MAV-1 as detailed above. Organs were harvested and snap-frozen at baseline prior to infection and then at multiple time points post-infection,
DNA and RNA Isolation from Mouse Organs
DNA was extracted from the middle lobe of the right lung and approximately 30 mg of the spleen using the Dneasy® Tissue Kit (Qiagen Inc.). Total RNA was extracted from lungs as previously described (Nguyen et al., 2008). In brief, approximately one-third of each lung was homogenized using sterile glass beads in a mini Beadbeater (Biospec Products) for 30 seconds in 1 ml of TRIzol. RNA was then isolated from the homogenates according to the manufacturer’s protocol.
Analysis of Viral Load and Viral Gene Expression
MAV-1 viral loads were measured in organs using quantitative real-time PCR (qPCR) as previously described (Nguyen et al., 2008). Primers and probe were used to detect a 59-bp region of the MAV-1 EIA gene as follows: forward 5′-GCACTCCATGGCAGGATTCT-3′, reverse 5′-GGTCGAAGCAGACGGTTCTTC-3′, and probe 5′-FAM-TACTGCCACTTCTGC-MGB-3′. 5 μl of extracted DNA were added to reactions containing TaqMan Universal PCR Mix (Applied Biosystems), forward and reverse primers (each at 200 nM final concentration) and probe (200 nM final concentration) in a 25 μl reaction volume. Analysis on an ABI Prism 7300 machine (Applied Biosystems) consisted of 40 cycles of 15 seconds at 90°C and 60 seconds at 60°C. Standard curves generated using known amounts of plasmid containing the MAV-1 EIA gene were used to convert cycle threshold values for experimental samples to copy numbers of EIA DNA. Results were standardized to the ng amount of input DNA. Each sample was assayed in triplicate.
Expression of the MAV-1 hexon gene was measured using RT-qPCR. 2.5 μg of RNA were reverse transcribed using MMLV reverse transcriptase (Invitrogen) in 20 μl reactions according to the manufacturer’s instructions. Primers and probe adapted from Lenaerts et al. (2005) were used to detect a 167-bp region of the MAV-1 hexon gene as follows: forward 5′-GGCCAACACTACCGACACTT-3′, reverse 5′-TTTTGTCCTGTGGCATTTGA-3′, and probe 5′-FAM-CATTCCAGCCAACTTATGGCTCGGC-TAMRA-3′. 2 μl of cDNA were added to a reaction containing TaqMan Universal PCR Mix (Applied Biosystems), forward and reverse primers (each at 200 nM final concentration) and probe (200 nM final concentration) in a 25 μl reaction volume. Separate reactions were prepared with 2 μl of cDNA, 1.25 μl of 20x mouse GAPDH gene expression assay (Applied Biosystems) and TaqMan Universal PCR Mix in a 25 μl reaction volume. In each case, RT-qPCR analysis consisted of 40 cycles of 15 seconds at 90°C and 60 seconds at 60°C. Standard curves generated using known amounts of plasmid containing the MAV-1 hexon gene or the mouse GAPDH gene were used to convert cycle threshold values for experimental samples to copy numbers of hexon and GAPDH transcripts, respectively. Copy numbers of viral transcripts were normalized to copy numbers of GAPDH transcripts for each sample. In a subset of samples prepared without using reverse transcriptase, we verified that trace amounts of contaminating DNA in the RNA samples contributed only a negligible amount to hexon copies numbers detected using RT-qPCR.
Analysis of Cytokine and Mucus Gene Expression
Cytokine gene expression was quantified using RT-qPCR. cDNA prepared as described above was amplified using gene expression assays for mouse IL-4, IL-13 and GAPDH (Applied Biosystems). 2 μl of cDNA were added to reactions containing TaqMan Universal PCR Mix and 1.25 μl each of 20x gene expression assays for the target cytokine and GAPDH. IFN-γ was detected using primers as follows: forward 5′-AAAGAGATAATCTGGCTCTGC-3′ and reverse 5′-GCTCTGAGACAATGAACGCT-3′. Expression of gob5 was detected using the primers: forward 5′-GAGTGGGCTCACTTCCGATG-3′ and reverse 5′-GCTGAACACCTCACTGCTTGG-3′; expression of muc5ac was detected using the primers: forward 5′-CCAGCACCATCTCTACAACCC-3′ and reverse 5′-GCAAAGCTCCTGTTTGCACTC-3′. For IFN-γ, gob5, and muc5ac, 2 μl of cDNA were added to reactions containing TaqMan Power SYBR Green PCR Mix (Applied Biosystems) and forward and reverse primers (each at 200 nM final concentration) in a 25 μl reaction volume. Separate reactions were prepared with TaqMan Power SYBR Green PCR Mix (Applied Biosystems) and primers for mouse GAPDH (forward 5′-TGCACCACCAACTGCTTAG-3′ and reverse 5′-GGATGCAGGGATGATGTTC-3′ at 167 nM each). RT-qPCR analysis consisted of 40 cycles of 15 seconds at 90°C and 60 seconds at 60°C. Quantification of target gene expression was normalized to GAPDH and expressed as fold change from control groups using the comparative CT method (Fink et al., 1998). We used ΔT values to calculate significance between groups and used 2−ΔΔT values for data presentation (Patrizio et al., 2007; Yuan et al., 2006).
Histology
For two or more mice per experimental group at each time point, the left lung was inflated with PBS to preserve lung architecture and fixed in 10% formalin. Organs were embedded in paraffin and 5 μm sections were cut for histopathology. Sections were stained with hematoxylin and eosin to evaluate cellular infiltrates. PAS staining was performed to evaluate mucus production. Slides were viewed through a Laborlux 12 microscope (Leitz). Digital images were obtained with an EC3 digital imaging system (Leica Microsystems) using Leica Acquisition Suite software (Leica Microsystems). Final images were assembled using Adobe Illustrator (Adobe Systems).
To quantify mucus production, slides were examined in a blinded fashion in order to count total airway lumens in a lung section (minimum of 25 per section) and airway lumens with PAS staining in the epithelium. These values were used to calculate the percentage of PAS-staining airways in each lung section. To quantify cellular inflammation in the lungs, slides were examined in a blinded fashion to determine a pathology index as previously described (Cooke et al., 1996). Separate scores were generated for the severity of cellular infiltrates around airway lumens and pneumonitis (alveolar and interstitial infiltrates) according to the parameters outlined in Table 1. Each score was multiplied by a number reflecting the extent of involvement in the lung (5% to 25% = 1, >25% to 50% = 2, >50% = 3). The final pathology index was obtained by adding together the values for cellular infiltrates around airway lumens and pneumonitis, resulting in a value that could range from 0 to 18.
Statistics
Analysis of data for statistical significance was conducted using Prism 3 for Macintosh (GraphPad Software, Incorporated). For viral load and viral gene expression data, differences between groups at a given time point were analyzed using the Mann-Whitney rank sum test. Differences in lung viral loads between time points (Fig. 4C) were analyzed using two-way analysis of variance (ANOVA) followed by Bonferroni’s multiple comparison tests. For cytokine and mucus gene expression data, differences between ΔT values were analyzed using the Mann-Whitney rank sum test for comparisons of two groups or two-way ANOVA for comparison of more than two groups followed by Bonferroni’s multiple comparison tests to analyze differences between two groups at each time point. For quantified PAS staining data and histological scoring, differences between multiple groups were analyzed using two-way ANOVA with Bonferroni’s multiple comparison tests to analyze differences between two groups at each time point. P values less than 0.05 were considered statistically significant.
Acknowledgments
We thank Kathy Spindler for helpful review of the manuscript, Nick Lukacs and Aaron Berlin for technical advice regarding the CRA experiments, and Shanna Ashley for technical advice regarding plaque assays. This research was supported by a University of Michigan Child Health Research Center Junior Investigator Award (K12 HD28820) and by K08 HL083103 to Jason Weinberg. The University of Michigan Research Histology and IHC Laboratory is supported in part by the National Institutes of Health through the University of Michigan’s Cancer Center Support Grant (P30 CA46592).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Victoria E. Anderson, Email: victoria.anderson@rmpdc.org.
Y N. Nguyen, Email: nhuy@umich.edu.
Jason B. Weinberg, Email: jbwein@umich.edu.
References
- Akinbami LJ, Schoendorf KC. Trends in childhood asthma: prevalence, health care utilization, and mortality. Pediatrics. 2002;110(2 Pt 1):315–22. doi: 10.1542/peds.110.2.315. [DOI] [PubMed] [Google Scholar]
- Beisswenger C, Kandler K, Hess C, Garn H, Felgentreff K, Wegmann M, Renz H, Vogelmeier C, Bals R. Allergic airway inflammation inhibits pulmonary antibacterial host defense. J Immunol. 2006;177(3):1833–1837. doi: 10.4049/jimmunol.177.3.1833. [DOI] [PubMed] [Google Scholar]
- Berlin AA, Hogaboam CM, Lukacs NW. Inhibition of SCF attenuates peribronchial remodeling in chronic cockroach allergen-induced asthma. Lab Invest. 2006;86(6):557–65. doi: 10.1038/labinvest.3700419. [DOI] [PubMed] [Google Scholar]
- Bianco A, Sethi SK, Allen JT, Knight RA, Spiteri MA. Th2 cytokines exert a dominant influence on epithelial cell expression of the major group human rhinovirus receptor, ICAM-1. Eur Respir J. 1998;12(3):619–26. doi: 10.1183/09031936.98.12030619. [DOI] [PubMed] [Google Scholar]
- Bianco A, Whiteman SC, Sethi SK, Allen JT, Knight RA, Spiteri MA. Expression of intercellular adhesion molecule-1 (ICAM-1) in nasal epithelial cells of atopic subjects: a mechanism for increased rhinovirus infection? Clin Exp Immunol. 2000;121(2):339–45. doi: 10.1046/j.1365-2249.2000.01301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bot A, Holz A, Christen U, Wolfe T, Temann A, Flavell R, von Herrath M. Local IL-4 expression in the lung reduces pulmonary influenza-virus-specific secondary cytotoxic T cell responses. Virology. 2000;269(1):66–77. doi: 10.1006/viro.2000.0187. [DOI] [PubMed] [Google Scholar]
- Carey B, Staudt MK, Bonaminio D, van der Loo JCM, Trapnell BC. PU.1 redirects adenovirus to lysosomes in alveolar macrophages, uncoupling internalization from infection. J Immunol. 2007;178(4):2440–2447. doi: 10.4049/jimmunol.178.4.2440. [DOI] [PubMed] [Google Scholar]
- Cauthen AN, Welton AR, Spindler KR. Construction of mouse adenovirus type 1 mutants. Methods Mol Med. 2007;130:41–59. doi: 10.1385/1-59745-166-5:41. [DOI] [PubMed] [Google Scholar]
- Chatila TA. Interleukin-4 receptor signaling pathways in asthma pathogenesis. Trends Mol Med. 2004;10(10):493–499. doi: 10.1016/j.molmed.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Cooke KR, Kobzik L, Martin TR, Brewer J, Delmonte J, Jr, Crawford JM, Ferrara JL. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood. 1996;88(8):3230–3239. [PubMed] [Google Scholar]
- Eisenbarth SC, Cassel S, Bottomly K. Understanding asthma pathogenesis: linking innate and adaptive immunity. Curr Opin Pediatr. 2004;16(6):659–66. doi: 10.1097/01.mop.0000145920.00101.e4. [DOI] [PubMed] [Google Scholar]
- Evans CM, Koo JS. Airway mucus: The good, the bad, the sticky. Pharmacol Ther. 2008 doi: 10.1016/j.pharmthera.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink L, Seeger W, Ermert L, Hanze J, Stahl U, Grimminger F, Kummer W, Bohle RM. Real-time quantitative RT-PCR after laser-assisted cell picking. Nat Med. 1998;4(11):1329–33. doi: 10.1038/3327. [DOI] [PubMed] [Google Scholar]
- Fischer JE, Johnson JE, Kuli-Zade RK, Johnson TR, Aung S, Parker RA, Graham BS. Overexpression of interleukin-4 delays virus clearance in mice infected with respiratory syncytial virus. J Virol. 1997;71(11):8672–8677. doi: 10.1128/jvi.71.11.8672-8677.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendler SJ. MUC1, the renaissance molecule. J Mammary Gland Biol Neoplasia. 2001;6(3):339–53. doi: 10.1023/a:1011379725811. [DOI] [PubMed] [Google Scholar]
- Gingras S, Simard J, Groner B, Pfitzner E. p300/CBP is required for transcriptional induction by interleukin-4 and interacts with Stat6. Nucleic Acids Res. 1999;27(13):2722–9. doi: 10.1093/nar/27.13.2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guida JD, Fejer G, Pirofski LA, Brosnan CF, Horwitz MS. Mouse adenovirus type 1 causes a fatal hemorrhagic encephalomyelitis in adult C57BL/6 but not BALB/c mice. J Virol. 1995;69(12):7674–7681. doi: 10.1128/jvi.69.12.7674-7681.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller NM, Matsukura S, Georas SN, Boothby MR, Rothman PB, Stellato C, Schleimer RP. Interferon-gamma inhibits STAT6 signal transduction and gene expression in human airway epithelial cells. Am J Respir Cell Mol Biol. 2004;31(5):573–82. doi: 10.1165/rcmb.2004-0195OC. [DOI] [PubMed] [Google Scholar]
- Inoue D, Yamaya M, Kubo H, Sasaki T, Hosoda M, Numasaki M, Tomioka Y, Yasuda H, Sekizawa K, Nishimura H, Sasaki H. Mechanisms of mucin production by rhinovirus infection in cultured human airway epithelial cells. Respir Physiol Neurobiol. 2006;154(3):484–99. doi: 10.1016/j.resp.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Johnston SL. Innate immunity in the pathogenesis of virus-induced asthma exacerbations. Proc Am Thorac Soc. 2007;4(3):267–70. doi: 10.1513/pats.200701-030AW. [DOI] [PubMed] [Google Scholar]
- Kajon AE, Brown CC, Spindler KR. Distribution of mouse adenovirus type 1 in intraperitoneally and intranasally infected adult outbred mice. J Virol. 1998;72(2):1219–1223. doi: 10.1128/jvi.72.2.1219-1223.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajon AE, Spindler KR. Mouse adenovirus type 1 replication in vitro is resistant to interferon. Virology. 2000;274(1):213–219. doi: 10.1006/viro.2000.0459. [DOI] [PubMed] [Google Scholar]
- Kuperman D, Schofield B, Wills-Karp M, Grusby MJ. Signal transducer and activator of transcription factor 6 (Stat6)-deficient mice are protected from antigen-induced airway hyperresponsiveness and mucus production. J Exp Med. 1998;187(6):939–48. doi: 10.1084/jem.187.6.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenaerts L, Daelemans D, Geukens N, De Clercq E, Naesens L. Mouse adenovirus type 1 attachment is not mediated by the coxsackie-adenovirus receptor. FEBS Lett. 2006;580(16):3937–3942. doi: 10.1016/j.febslet.2006.06.027. [DOI] [PubMed] [Google Scholar]
- Lenaerts L, Verbeken E, De Clercq E, Naesens L. Mouse adenovirus type 1 infection in SCID mice: an experimental model for antiviral therapy of systemic adenovirus infections. Antimicrob Agents Chemother. 2005;49(11):4689–4699. doi: 10.1128/AAC.49.11.4689-4699.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindell DM, Berlin AA, Schaller MA, Lukacs NW. B cell antigen presentation promotes Th2 responses and immunopathology during chronic allergic lung disease. PLoS ONE. 2008;3(9):e3129. doi: 10.1371/journal.pone.0003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macek V, Sorli J, Kopriva S, Marin J. Persistent adenoviral infection and chronic airway obstruction in children. Am J Respir Crit Care Med. 1994;150(1):7–10. doi: 10.1164/ajrccm.150.1.8025775. [DOI] [PubMed] [Google Scholar]
- Marin J, Jeler-Kacar D, Levstek V, Macek V. Persistence of viruses in upper respiratory tract of children with asthma. J Infect. 2000;41(1):69–72. doi: 10.1053/jinf.2000.0688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald C, Reich NC. Cooperation of the transcriptional coactivators CBP and p300 with Stat6. J Interferon Cytokine Res. 1999;19(7):711–22. doi: 10.1089/107999099313550. [DOI] [PubMed] [Google Scholar]
- Moore ML, Brown CC, Spindler KR. T cells cause acute immunopathology and are required for long-term survival in mouse adenovirus type 1-induced encephalomyelitis. J Virol. 2003;77(18):10060–10070. doi: 10.1128/JVI.77.18.10060-10070.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran TM, Isobe H, Fernandez-Sesma A, Schulman JL. Interleukin-4 causes delayed virus clearance in influenza virus-infected mice. J Virol. 1996;70(8):5230–5235. doi: 10.1128/jvi.70.8.5230-5235.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen Y, McGuffie BA, Anderson VE, Weinberg JB. Gammaherpesvirus modulation of mouse adenovirus type 1 pathogenesis. Virology. 2008;380(2):182–90. doi: 10.1016/j.virol.2008.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panebra A, Schwarb MR, Glinka CB, Liggett SB. Heterogeneity of transcription factor expression and regulation in human airway epithelial and smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2007;293(2):L453–62. doi: 10.1152/ajplung.00084.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrizio M, Musumeci M, Stati T, Fasanaro P, Palazzesi S, Catalano L, Marano G. Propranolol causes a paradoxical enhancement of cardiomyocyte foetal gene response to hypertrophic stimuli. Br J Pharmacol. 2007;152(2):216–22. doi: 10.1038/sj.bjp.0707350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peebles RS., Jr Viral infections, atopy, and asthma: is there a causal relationship? J Allergy Clin Immunol. 2004;113(1 Suppl):S15–8. doi: 10.1016/j.jaci.2003.10.033. [DOI] [PubMed] [Google Scholar]
- Perez-Yarza EG, Moreno A, Lazaro P, Mejias A, Ramilo O. The association between respiratory syncytial virus infection and the development of childhood asthma: a systematic review of the literature. Pediatr Infect Dis J. 2007;26(8):733–9. doi: 10.1097/INF.0b013e3180618c42. [DOI] [PubMed] [Google Scholar]
- Raman S, Hsu TH, Ashley SL, Spindler KR. Usage of integrin and heparan sulfate as receptors for mouse adenovirus type 1. J Virol. 2009;83(7):2831–8. doi: 10.1128/JVI.02368-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurs N, Bjarnason R, Sigurbergsson F, Kjellman B. Respiratory syncytial virus bronchiolitis in infancy is an important risk factor for asthma and allergy at age 7. Am J Respir Crit Care Med. 2000;161(5):1501–1507. doi: 10.1164/ajrccm.161.5.9906076. [DOI] [PubMed] [Google Scholar]
- Spindler KR, Fang L, Moore ML, Hirsch GN, Brown CC, Kajon A. SJL/J mice are highly susceptible to infection by mouse adenovirus type 1. J Virol. 2001;75(24):12039–12046. doi: 10.1128/JVI.75.24.12039-12046.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traves SL, Proud D. Viral-associated exacerbations of asthma and COPD. Curr Opin Pharmacol. 2007;7(3):252–8. doi: 10.1016/j.coph.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg JB, Jensen DR, Gralinski LE, Lake AR, Stempfle GS, Spindler KR. Contributions of E1A to mouse adenovirus type 1 pathogenesis following intranasal inoculation. Virology. 2007;357(1):54–67. doi: 10.1016/j.virol.2006.08.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg JB, Stempfle GS, Wilkinson JE, Younger JG, Spindler KR. Acute respiratory infection with mouse adenovirus type 1. Virology. 2005;340(2):245–254. doi: 10.1016/j.virol.2005.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan JS, Reed A, Chen F, Stewart CN., Jr Statistical analysis of real-time PCR data. BMC Bioinformatics. 2006;7:85. doi: 10.1186/1471-2105-7-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Lin G, Baarsch MJ, Scamurra RW, Murtaugh MP. Interleukin-4 suppresses inflammatory cytokine gene transcription in porcine macrophages. J Leukoc Biol. 1994;56(4):507–513. doi: 10.1002/jlb.56.4.507. [DOI] [PubMed] [Google Scholar]
- Zsengeller Z, Otake K, Hossain S-A, Berclaz P-Y, Trapnell BC. Internalization of adenovirus by alveolar macrophages initiates early proinflammatory signaling during acute respiratory tract infection. J Virol. 2000;74(20):9655–9667. doi: 10.1128/jvi.74.20.9655-9667.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]