

Abstract
Heterozygous activating mutations in exon 9 of SH3BP2 have been found in most patients with cherubism, an unusual genetic syndrome characterized by excessive remodeling of the mandible and maxilla due to spontaneous and excessive osteoclastic bone resorption. Osteoclasts differentiate after binding of sRANKL to RANK induces a number of downstream signaling effects, including activation of the calcineurin/NFAT (nuclear factor of activated T cells) pathway. Here we have investigated the functional significance of SH3BP2 protein on osteoclastogenesis in the presence of sRANKL. Our results indicate that SH3BP2 both increases nuclear NFATc1 in sRANKL treated RAW 264.7 preosteoclast cells and enhances expression of tartrate resistant acid phosphatase (TRAP), a specific marker of osteoclast differentiation. Moreover, overexpression of SH3BP2 in RAW 264.7 cells potentiates sRANKL stimulated phosphorylation of PLCγ1 and 2, thus providing a mechanistic pathway for the rapid translocation of NFATc1 into the nucleus and increased osteoclastogenesis in cherubism.
Keywords: Cherubism, SH3BP2, NFAT, osteoclast, mutation, CLINICAL/PEDIATRICS, MODELING AND REMODELING, MOLECULAR PATHWAYS, TEETH & DENTAL APPLICATIONS, TRANSCRIPTIONAL FACTORS
INTRODUCTION
Cherubism (OMIM 118400) is an autosomal dominant disorder characterized by extensive bone remodeling within the mandible and/or maxilla. The onset of cherubism is early, age 2–5 years, at which time patients develop characteristic giant cell lesions in the maxilla and mandible and substantial facial swelling and cervical lymphadenopathy.[1–3]The localized nature of the bone lesions in patients with cherubism is unexpected as the disorder is associated with heterozygous germline mutations in the gene that is widely expressed throughout the osteoimmune system.[1, 2, 4–7] SH3BP2 is a complex adaptor protein, with an N-terminal pleckstrin homology, a ten amino acid Src Homology 3 (SH3) binding domain, and a C-terminal SH2 domain.[8]
RANKL initiates the development and function of osteoclasts[9, 10] and among the various transcriptional regulators that are activated by RANKL signaling in osteoclast precursor cells is NFATc1, a member of a small family of ubiquitous transcription factors.[11–13] NFAT phosphoproteins, extant in the cytoplasm, are activated by calcineurin, a Ca2+/calmodulin-dependent protein phosphatase which leads to NFAT translocation into the nucleus.[14, 15] NFATc1 plays a critical role in osteoclast development. Ishida et al. first showed that NFATc1 was highly induced after sRANKL treatment of RAW 264.7 osteoclast precursors, and that inhibiting NFATc1 activity by either antisense oligonucleotides or the calcineurin inhibitor cyclosporine A suppressed osteoclast differentiation in vitro.[16] Similarly, Takayanagi et al. demonstrated that NFATc1 is induced in osteoclast precursors by sRANKL stimulation using DNA microarray analysis and that embryonic stem cells deficient in NFATc1 were unable to differentiate into osteoclasts in vitro.[17] More recently, Hirotani et al showed that forced expression of a constitutively active form of NFATc1 is sufficient to induce RAW 264.7 osteoclast precursor cells to undergo differentiation into mature osteoclasts.[18] These results suggest that NFATc1 acts coordinately to regulate osteoclast differentiation although it is still obscure what molecules lie upstream of NFATc1 activation.[19–21] Previous studies in T cells have shown that overexpression of SH3BP2 activates transcription of a hybrid reporter gene in which luciferase expression is regulated by a promoter region containing the NFAT binding sequence from the interleukin 2 (IL-2) gene.[22–24] All SH3BP2 mutations described to date in patients with cherubism occur in exon 9 and correspond to a six amino acid region of the SH3BP2 protein.[1, 7, 25, 26] The precise function of this region remains unclear, but recent work from our laboratory suggests that these missense mutations lead to a gain-of-function rather than a loss of activity.[25] This is consistent with prior observations that deletions of 4p16.3 in patients with Wolf-Hirschhorn syndrome, which result in loss of one copy of SH3BP2, do not result in a bone resorptive phenotype.[27–29] Here we report that SH3BP2 participates in a phosphatidyl-inositol specific phospholipase C (PI-PLC), inositol phosphate-3 receptor (IP3R), and calcineurin dependent signaling cascade that activates calcineurin-induced nuclear translocation of NFATc1, leading to increased differentiation of TRAP-positive osteoclastic cells.
MATERIALS AND METHODS
Cell Culture
HEK 293 T cells, Saos2 cells and RAW 264.7 cells (American Type Culture Collection) and Jurkat T Ag cells (a generous gift from Professor Gerald Crabtree, Stanford University, Palo Alto, Ca.) were maintained at 37°C, 5% CO2 in growth media: DMEM or RPMI (Invitrogen) supplemented with 10% fetal bovine serum and 1% penicillin and streptomycin (Invitrogen). For osteoclastogenesis assays, cells were resuspended in fresh medium and cultured for 8 days in 96-well plates at a cell density of 1000 cells/well. Soluble RANKL (sRANKL) (Peprotech) was used at 100 ng/ml as indicated and cyclosporine A (Csa) (Calbiochem) at 0.4–2 μM, 2-APB (Calbiochem) at 75 μM, U-73122 and U73343 at 3 μM, as indicated. Csa, 2-APB, U-73122, or U73343 were added to the medium at the time of cell transfection and maintained in growth media thereafter.
Plasmids and Transfection of RAW 264.7 cells
We obtained the mouse SH3BP2 cDNA in pMT3 plasmid from Professor Marcel Deckert (University of Nice, France)[22, 23] and NFAT luciferase reporter from Professor Gerald Crabtree.[30] RAW 264.7 cells at 40–70% confluence were transiently transfected with SH3BP2 cDNA, a hybrid reporter gene in which luciferase expression is under the control of a 3X minimal IL-2 promoter containing the NFAT binding site (NFAT-luc)[30], and a Renilla luciferase reporter as a control for transfection efficiency (Promega). Transfection was performed by electroporation using a Nucleofector (Amaxa). 24 hours after transfection, luminescence was evaluated with a luminometer (Sirius Berthold). To create stable cell lines, RAW264.7 cells were co-transfected with plasmids encoding the NFAT-luciferase reporter gene and the neomycin resistance gene and cultured in the presence of G418 (500 μg/ml).
Immunofluorescent staining
RAW 264.7 cells were grown on cover slips in 6-well plates after Amaxa nucleofection as described above. After incubation at 37°C for 24 hours, the cells were fixed in 4% paraformaldehyde for 30 min, treated with 0.1% Triton X-100 for 5 min, and incubated in 5% BSA/PBS for 30 min. Cells were then stained with affinity-purified mouse anti-human NFATc1 (Abcam, 1:200) and affinity purified rabbit anti-human SH3BP2 (custom made, 1:1000) for 1h in PBS. Cells were then incubated first with 4 μg/ml Alexa 488 goat anti-mouse secondary antibody for 1 hour in PBS, washed and incubated with 4 μg/ml Alexa 568 goat anti-rabbit secondary antibody for 1 hour in PBS. The slides were then treated with DAPI, mounted and examined under confocal microscopy. Cells were photographed with a Leica TCS-SP spectral laser scanning confocal microscope with lasers set up to simultaneously visualize fluorescence at different wavelengths to compare subcellular localization of the mouse SH3BP2 and NFATc1 immunogens.
Determination of Osteoclast Differentiation
Osteoclast differentiation was assessed by measuring the cellular enzymatic activity of TRAP, a marker of terminal osteoclast differentiation. Specifically, cells were incubated in 0.1 ml of phosphatase substrate (3.7 mM p-nitrophenyl phosphate in 50 mM citrate buffer pH 4.8) in the presence of 10 mM sodium tartrate at 25°C for 30 minutes. After incubation, 0.1 ml of NaOH was added to each well and the absorbance was read at 410 nm on a microplate reader.
Immunoblot Analysis
For SH3BP2 immunoblots whole cell lysate was prepared in passive lysis buffer (Promega) and was assayed for total protein using BCA reagent (Bio-Rad). Protein samples (50 μg) were electrophoresed through 4–12% bis-tris polyacrylamide gels at 200 volts for 1 hour in MOPS buffer and transferred from the gel to a PVDF membrane. After an overnight incubation with 5% non fat dried milk (Carnation) at 4°C, the membrane was incubated with a specific polyclonal antiserum to SH3BP2 (Rockland Immunochemicals)(1:2000) in 5% nonfat dried milk, or incubated with antiserum to β actin (1:2000 in 5% nonfat dried milk) which was used as a control to evaluate loading per lane. Antibody binding was detected with horseradish peroxidase-conjugated secondary antibodies and enhanced chemiluminescence (ECL, Amersham).
To analyze phosphorylated proteins, cells were scraped in lysis buffer (20 mM Tris-HCL, pH7.5, 20 mM p-nitrophenyl phosphate, 1 mM EGTA, 50 mM sodium fluoride, 50 μM sodium orthovanadate, and proteinase inhibitor cocktail) and lysed by freezing and thawing 3 times. Samples were electrophoresed on a 4–12% bis-tris gel (Invitrogen), 200 volts for 40 minutes in MOPS buffer and transferred to PVDF membranes, at 100 volts for 1hour. Membranes were then blocked in 5% skim milk for 1 hour and incubated in anti-PLCγ2, phospho-specific Tyr1217 antibody or anti-Syk, phospho-specific Tyr524–525 antibody (Oncogene) overnight at 4°C. Alternatively, the membrane was stripped and, incubated with PLCγ2, Syk (Cell Signaling), SH3BP2 (custom made antibody) and β-actin antibodies (Sigma) at 4°C overnight.
Statistics
Statistical evaluation of the data used the analysis of variance (ANOVA) and Fisher’s protected least significant difference multiple comparison test to determine significance between groups.
RESULTS and DISCUSSION
In previous studies we found that RAW 264.7 cells express native SH3BP2, as determined by RNA blot and protein immunoblot, at levels similar to those in Jurkat T Ag cells, HEK 293T cells, Saos2 cells and human giant cell tumors confirming that SH3BP2 could potentially participate in signaling pathways that induce osteoclastogenesis.[31]
Since SH3BP2 is known to stimulate NFAT activity, and NFATc1 is a master regulator of osteoclastogenesis, we examined NFATc1 expression by immunoblot in response to sRANKL and then by immunofluorescence in sRANKL-stimulated RAW 264.7 cells that had been transiently transfected with SH3BP2. SH3BP2, in sRANKL stimulated cells, did not increase the total amount of NFATc1 further but rather increased NFATc1 nuclear translocation (Figure 1a) compared to a negative control stimulated with sRANKL but with empty vector transfected rather than SH3BP2 (Figure 1b).
FIG. 1.
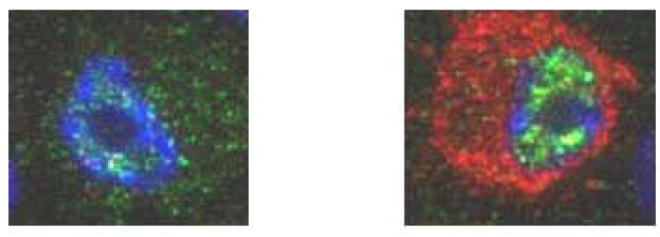


A SH3BP2 INCREASES THE NUCLEAR TRANSLOCATION OF NFATC1. RAW 264.7 cells were transiently transfected with murine SH3BP2 (red) and NFATc1 (green) translocated to the nucleus (A) compared to cells not transfected with SH3BP2 (B). SH3BP2 did not increase the total amount of NFATc1 in cells however SH3BP2 with sRANKL (100ng/ml) increased NFATc1 activation by increasing the translocation.
C. SH3BP2 INCREASES NFAT ACTIVITY. The figure demonstrates that in RAW 264.7 cells stimulated with sRANKL (100ng/ml), murine SH3BP2 cDNA significantly increases NFAT activity compared to the pMT3 vector only (p<0.0003), sRANKL (p<0.0001) or SH3BP2 alone (p<0.0001). This experiment was repeated four times with similar results.
D. TRAP ACTIVITY IS INCREASED IN THE PRESENCE OF SH3BP2. RAW 264.7 cells were transiently transfected with murine SH3BP2 and stimulated with sRANKL (100ng/ml). In RANKL stimulated (100ng/ml) and SH3BP2 transfected cells TRAP activity was significantly increased compared to the vector only (pMT3 vector) control p<0.0001, SH3BP2 alone (p<0.0001) and sRANKL stimulation alone (p=0.001). # indicates p<0.02 and ## indicates p<0.0001. This experiment was repeated three times with similar results.
There was no consistent activation of NFAT-luc in SH3BP2-transfected RAW 264.7 cells that were cultured in the absence of sRANKL. However, when sRANKL (100 ng/ml) was added to cultured cells transfected with the wild-type SH3BP2 cDNA, NFAT-luc activity increased to 2–3 fold greater than in cells that had been transiently transfected with empty vector and treated with sRANKL (Figure 1c) (p<0.0001). To determine whether over expression of SH3BP2 had an effect on osteoclastogenesis, we measured TRAP activity after RAW264.7 cells had been cultured for one week in the absence or presence of 100 ng/ml of sRANKL, after transfection with either an empty vector or mouse SH3BP2 (Figure 1d). TRAP activity was significantly increased by treatment with sRANKL, and there was an even greater increase in sRANKL-induced TRAP activity in RAW264.7 cells that had been transiently transfected with SH3BP2 compared to empty vector (p<0.001)(Figure 1d). These results correlated with the synergistic effect of SH3BP2 and sRANKL on activation of the NFAT luciferase reporter gene (Figure 1c). However the NFAT activation was not increased with transient transfection of NFAT-luc alone and sRANKL treatment. Moreover, previous data showed that NFATc1 is increased significantly in response to sRANKL and that NFATc1 is the predominant NFAT isoform and is critical to RANKL induced osteoclastogenesis in RAW 264.7 cells.[16, 18] Therefore we stably transfected RAW 264.7 cells with NFAT luciferase (to increase the number of NFAT luciferase reporter cells) and then treated the cells with sRANKL and noted a six-fold increase in NFAT luciferase in these RAW 264.7 cells stably transfected with NFAT luciferase (p<0.0001).[16–19] These results indicate that the transient transfections with the NFAT luciferase reporter, while sensitive enough to detect the effect of SH3BP2 on NFAT in RAW 264.7 cells treated with sRANKL, were not sensitive enough to detect the effect of sRANKL alone on NFAT. Stable transfection of RAW 264.7 cells with NFAT luciferase (to increase the percentage of cells with the NFAT-luc reporter) was required to see the effect of sRANKL on NFAT luciferase in the absence of SH3BP2.
In order to determine if the SH3BP2 effect was directly on NFAT translocation or upstream in the activation pathway, we used Cyclosporine A to inhibit calcineurin, a calcium-calmodulin dependent phosphatase that activates NFATc1 by dephosphorylation. Cyclosporine A treatment inhibited the sRANKL-stimulated SH3BP2-induced NFAT luciferase increase by 50% at low concentration (400 nM, p<0.0001) and 65% at high concentration (2μM, p<0.0001), indicating that SH3BP2 was acting in the signaling pathway that led to activation of calcineurin (Figure 2a). To further examine the importance of the PLC/calcineurin pathway for the SH3BP2 effect on NFAT activation PI-PLC inhibition was examined. A specific PI-PLC inhibitor (73122) at a dose of 3 μM decreased NFAT activation by 64%. The decrease in NFAT-luc activity was nearly down to the level of NFAT-luc activity without SH3BP2 transfection or sRANKL treatment (p<0.04) (Figure 2b).
FIG. 2.

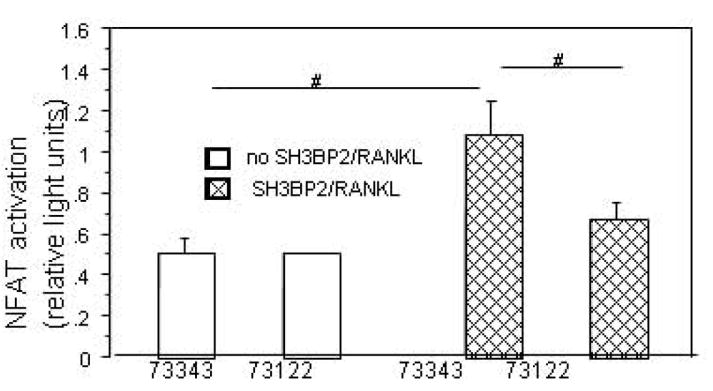
A. CYCLOSPORINE A INHIBITS SH3BP2/sRANKL INDUCED NFAT ACTIVITY. RAW 264.7, transiently transfected with murine SH3BP2 and stimulated with sRANKL (100ng/ml), were treated with Cyclosporine A. The calcineurin inhibitor (cyclosporine A) inhibits SH3BP2/sRANKL indicating that SH3BP2 is acting upstream of calcineurin. ## indicates p< 0.0001.
B. The PI-PLC inhibitor (U-73122) has a mild inhibitory effect on NFAT activation. RAW 264.7 cells were transiently transfected with murine SH3BP2, stimulated with RANKL and treated with U-73122 or U-77343 (the inactive analog of U-73122)[23] The bar graph demonstrates an inhibition of NFAT activity with U-73122 at a dose of 3μM compared to U-73343. # indicates p<0.04.
In order to further test the hypothesis that the SH3BP2 effect on NFAT involves an IP3 pathway, we examined the effect of IP3 signaling inhibition on sRANKL-stimulated RAW 264.7 cells that had been transfected with SH3BP2. The IP3R inhibitor, 2-APB reduced NFAT-luc activity by 71% (p<0.0001) (Figure 3a). Similarly, one week of treatment with 2-APB decreased TRAP activity by over 100-fold in sRANKL-stimulated SH3BP2 transfected RAW 264.7 cells (p<0.02) (Figure 3b).
FIG. 3.


A. 2-APB inhibits NFAT activation. 2-APB, the known IP3R inhibitor, inhibits NFAT activity in all groups (with or without murine SH3BP2 and/or sRANKL) in RAW 264.7 cells (# indicates p<0.02 and ## indicates p<0.0001).
B. TRAP staining and TRAP activity were nearly eliminated by 2-APB. 2-APB inhibited TRAP activity in RAW 264.7 cells after one week of culture indicating an inhibition of osteoclastogenesis. # indicates p<0.02 and ## indicates p<0.005.
To examine the mechanism for the SH3BP2-induced increase in NFAT luciferase and TRAP, candidate proteins were examined by immunoblot. Lysates from sRANKL-stimulated SH3BP2 transfected cells were analyzed by immunoblot and ImageJ. An increase (ImageJ, immunoblot) in phosphorylated PLCγ2 (65%) as well as phosphorylated Syk (75%) relative to that in sRANKL-stimulated cells transfected with the vector only (pMT3) was found in three separate experiments (Figure 4). These findings implicate SH3BP2 stimulation of NFAT-luc and osteoclastogenesis by a Syk/PLCγ pathway.
FIG. 4.
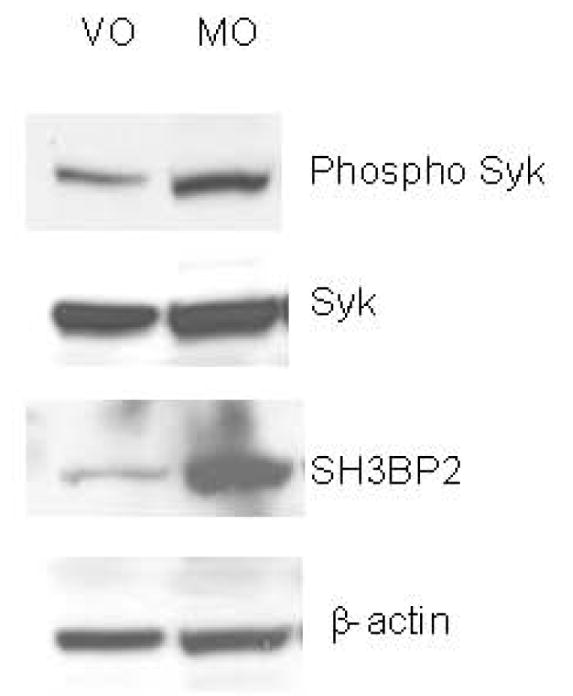
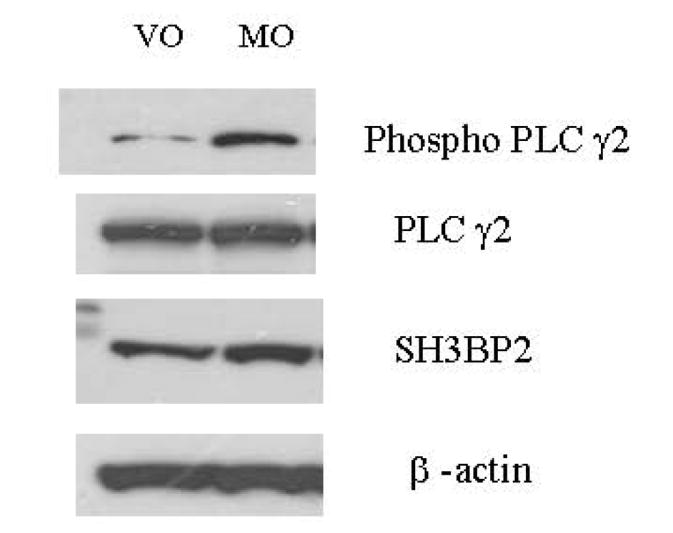
SH3BP2 INCREASES PHOSPHORYLATION OF PLCγ2 AND SYK.
A. The figure demonstrates that SH3BP2 increases the phosphorylation but not the total amount of Syk. B. The figure demonstrates that SH3BP2 increases the PLCγ2 phosphorylation but not the total amount of PLCγ2. β-actin was used as a control to ensure that equal protein was loaded in each lane. This experiment was repeated three times with similar results. VO represents cells transiently transfected with vector only and Mo represents cells transiently transfected with murine SH3BP2.
As SH3BP2 has no known protein kinase activity, it is tempting to speculate that SH3BP2 binds PLCγ2 and then acts as a scaffold to bring these molecules into close proximity with protein tyrosine kinases (e.g. Syk kinase) that can phosphorylate and thereby activate PLCγ.[17, 19, 32] Moreover PLCγ2 has recently been shown to mediate sRANKL-induced osteoclastogenesis by its effects on NFATc1 after Dap12/Fc receptor stimulation.[33] A large previous study examined the role of the P416R SH3BP2 mutation in a knockin mouse model.[34] In this model there was increased Syk phosphorylation in osteoclast precursors and a bone resorptive phenotype in the homozygous knockin mouse.[34]
This study is the first to assess the function of SH3BP2 in RAW 264.7 osteoclast precursor cells. It still is unclear which cell or combination of cells is responsible for the resorptive giant cell lesions in cherubism, but osteoclasts do respond to overexpression of SH3BP2 by increasing NFAT activity. Based on the fact that SH3BP2 overexpression leads to an increase in TRAP and NFAT activity in RAW 264.7 cells shown in this study, we propose that the resorptive lesions in cherubism result from the effect of the gain-of-function SH3BP2 mutations in osteoclast precursors. SH3BP2 activating mutations, here simulated by forced wild-type SH3BP2 expression, activate PLCγ which increases NFATc1 translocation thereby increasing osteoclastogenesis and giant cell tumor formation.
Acknowledgments
This work was supported by in part by US Public Health Service Research grants K08-AR47661 (SAL), DK34281 and DK56178 (MAL), and General Clinical Research Center Grant RR0035. Dr. Lietman is the recipient of and this work is supported by a Career Development Award from the Orthopaedic Research and Education Foundation.
Footnotes
Conflicts of interest: All authors have no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ueki Y, Tiziani V, Santanna C, Fukai N, Maulik C, Garfinkle J, Ninomiya C, doAmaral C, Peters H, Habal M, Rhee-Morris L, Doss JB, Kreiborg S, Olsen BR, Reichenberger E. Mutations in the gene encoding c-Abl-binding protein SH3BP2 cause cherubism. Nat Genet. 2001;28:125–126. doi: 10.1038/88832. [DOI] [PubMed] [Google Scholar]
- 2.Schultze-Mosgau S, Holbach LM, Wiltfang J. Cherubism: clinical evidence and therapy. J Craniofac Surg. 2003;14:201–206. doi: 10.1097/00001665-200303000-00012. discussion 207–208. [DOI] [PubMed] [Google Scholar]
- 3.Pulse CL, Moses MS, Greenman D, Rosenberg SN, Zegarelli DJ. Cherubism: case reports and literature review. Dent Today. 2001;20:100–103. [PubMed] [Google Scholar]
- 4.Petschler M, Stiller M, Hoffmeister B, Witkowski R, Opitz C, Bill JS, Peters H. [Clinical and molecular genetic observations on families with cherubism over three generations] Mund Kiefer Gesichtschir. 2003;7:83–87. doi: 10.1007/s10006-002-0444-x. [DOI] [PubMed] [Google Scholar]
- 5.Lannon DA, Earley MJ. Cherubism and its charlatans. Br J Plast Surg. 2001;54:708–711. doi: 10.1054/bjps.2001.3701. [DOI] [PubMed] [Google Scholar]
- 6.Ladhani S, Sundaram P, Joshi JM. Sleep disordered breathing in an adult with cherubism. Thorax. 2003;58:552. doi: 10.1136/thorax.58.6.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lo B, Faiyaz-Ul-Haque M, Kennedy S, Aviv R, Tsui LC, Teebi AS. Novel mutation in the gene encoding c-Abl-binding protein SH3BP2 causes cherubism. Am J Med Genet. 2003;121A:37–40. doi: 10.1002/ajmg.a.20226. [DOI] [PubMed] [Google Scholar]
- 8.Ren R, Mayer BJ, Cicchetti P, Baltimore D. Identification of a ten-amino acid proline-rich SH3 binding site. Science. 1993;259:1157–1161. doi: 10.1126/science.8438166. [DOI] [PubMed] [Google Scholar]
- 9.Wagner EF, Matsuo K. Signalling in osteoclasts and the role of Fos/AP1 proteins. Ann Rheum Dis. 2003;62(Suppl 2):ii83–85. doi: 10.1136/ard.62.suppl_2.ii83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miyamoto T, Ohneda O, Arai F, Iwamoto K, Okada S, Takagi K, Anderson DM, Suda T. Bifurcation of osteoclasts and dendritic cells from common progenitors. Blood. 2001;98:2544–2554. doi: 10.1182/blood.v98.8.2544. [DOI] [PubMed] [Google Scholar]
- 11.Winslow MM, Pan M, Starbuck M, Gallo EM, Deng L, Karsenty G, Crabtree GR. Calcineurin/NFAT signaling in osteoblasts regulates bone mass. Dev Cell. 2006;10:771–782. doi: 10.1016/j.devcel.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 12.Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17:2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- 13.Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- 14.Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell. 2002;109(Suppl):S67–79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- 15.Lee M, Park J. Regulation of NFAT activation: a potential therapeutic target for immunosuppression. Molecules and cells. 2006;22:1–7. [PubMed] [Google Scholar]
- 16.Ishida N, Hayashi K, Hoshijima M, Ogawa T, Koga S, Miyatake Y, Kumegawa M, Kimura T, Takeya T. Large scale gene expression analysis of osteoclastogenesis in vitro and elucidation of NFAT2 as a key regulator. J Biol Chem. 2002;277:41147–41156. doi: 10.1074/jbc.M205063200. [DOI] [PubMed] [Google Scholar]
- 17.Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue J, Wagner EF, Mak TW, Kodama T, Taniguchi T. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3:889–901. doi: 10.1016/s1534-5807(02)00369-6. [DOI] [PubMed] [Google Scholar]
- 18.Hirotani H, Tuohy NA, Woo JT, Stern PH, Clipstone NA. The calcineurin/nuclear factor of activated T cells signaling pathway regulates osteoclastogenesis in RAW264.7 cells. J Biol Chem. 2004;279:13984–13992. doi: 10.1074/jbc.M213067200. [DOI] [PubMed] [Google Scholar]
- 19.Takayanagi H. Mechanistic insight into osteoclast differentiation in osteoimmunology. J Mol Med. 2005;83:170–179. doi: 10.1007/s00109-004-0612-6. [DOI] [PubMed] [Google Scholar]
- 20.Paloneva J, Mandelin J, Kiialainen A, Bohling T, Prudlo J, Hakola P, Haltia M, Konttinen YT, Peltonen L. DAP12/TREM2 deficiency results in impaired osteoclast differentiation and osteoporotic features. J Exp Med. 2003;198:669–675. doi: 10.1084/jem.20030027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paloneva J, Manninen T, Christman G, Hovanes K, Mandelin J, Adolfsson R, Bianchin M, Bird T, Miranda R, Salmaggi A, Tranebjaerg L, Konttinen Y, Peltonen L. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet. 2002;71:656–662. doi: 10.1086/342259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deckert M, Tartare-Deckert S, Hernandez J, Rottapel R, Altman A. Adaptor function for the Syk kinases-interacting protein 3BP2 in IL-2 gene activation. Immunity. 1998;9:595–605. doi: 10.1016/s1074-7613(00)80657-3. [DOI] [PubMed] [Google Scholar]
- 23.Foucault I, Liu YC, Bernard A, Deckert M. The chaperone protein 14-3-3 interacts with 3BP2/SH3BP2 and regulates its adapter function. J Biol Chem. 2003;278:7146–7153. doi: 10.1074/jbc.M209509200. [DOI] [PubMed] [Google Scholar]
- 24.Foucault I, Le Bras S, Charvet C, Moon C, Altman A, Deckert M. The adaptor protein 3BP2 associates with VAV guanine nucleotide exchange factors to regulate NFAT activation by the B-cell antigen receptor. Blood. 2005;105:1106–1113. doi: 10.1182/blood-2003-08-2965. [DOI] [PubMed] [Google Scholar]
- 25.Lietman SA, Kalinchinko N, Deng X, Kohanski R, Levine MA. Identification of a novel mutation of SH3BP2 in cherubism and demonstration that SH3BP2 mutations lead to increased NFAT activation. Hum Mutat. 2006;27:717–718. doi: 10.1002/humu.9433. [DOI] [PubMed] [Google Scholar]
- 26.Imai Y, Kanno K, Moriya T, Kayano S, Seino H, Matsubara Y, Yamada A. A missense mutation in the SH3BP2 gene on chromosome 4p16.3 found in a case of nonfamilial cherubism. Cleft Palate Craniofac J. 2003;40:632–638. doi: 10.1597/1545-1569_2003_040_0632_ammits_2.0.co_2. [DOI] [PubMed] [Google Scholar]
- 27.Bergemann AD, Cole F, Hirschhorn K. The etiology of Wolf-Hirschhorn syndrome. Trends Genet. 2005;21:188–195. doi: 10.1016/j.tig.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 28.Battaglia A, Carey JC. Health supervision and anticipatory guidance of individuals with Wolf-Hirschhorn syndrome. Am J Med Genet. 1999;89:111–115. doi: 10.1002/(sici)1096-8628(19990625)89:2<111::aid-ajmg9>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 29.Battaglia A, Carey JC, Cederholm P, Viskochil DH, Brothman AR, Galasso C. Natural history of Wolf-Hirschhorn syndrome: experience with 15 cases. Pediatrics. 1999;103:830–836. doi: 10.1542/peds.103.4.830. [DOI] [PubMed] [Google Scholar]
- 30.Graef IA, Chen F, Chen L, Kuo A, Crabtree GR. Signals transduced by Ca(2+)/calcineurin and NFATc3/c4 pattern the developing vasculature. Cell. 2001;105:863–875. doi: 10.1016/s0092-8674(01)00396-8. [DOI] [PubMed] [Google Scholar]
- 31.Lietman SA, Prescott NL, Hicks DG, Westra WH, Levine MA. SH3BP2 is rarely mutated in exon 9 in giant cell lesions outside cherubism. Clin Orthop Relat Res. 2007;459:22–27. doi: 10.1097/BLO.0b013e31804b4131. [DOI] [PubMed] [Google Scholar]
- 32.Hur EM, Park YS, Lee BD, Jang IH, Kim HS, Kim TD, Suh PG, Ryu SH, Kim KT. Sensitization of epidermal growth factor-induced signaling by bradykinin is mediated by c-Src. Implications for a role of lipid microdomains. J Biol Chem. 2004;279:5852–5860. doi: 10.1074/jbc.M311687200. [DOI] [PubMed] [Google Scholar]
- 33.Mao D, Epple H, Uthgenannt B, Novack DV, Faccio R. PLCgamma2 regulates osteoclastogenesis via its interaction with ITAM proteins and GAB2. J Clin Invest. 2006;116:2869–2879. doi: 10.1172/JCI28775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ueki Y, Lin CY, Senoo M, Ebihara T, Agata N, Onji M, Saheki Y, Kawai T, Mukherjee PM, Reichenberger E, Olsen BR. Increased Myeloid Cell Responses to M-CSF and RANKL Cause Bone Loss and Inflammation in SH3BP2 “Cherubism” Mice. Cell. 2007;128:71–83. doi: 10.1016/j.cell.2006.10.047. [DOI] [PubMed] [Google Scholar]