

Abstract
The generalized master equations (GMEs) that contain multiple time scales have been derived quantum mechanically. The GME method has then been applied to a model of charge migration in proteins that invokes the hole hopping between local amino acid sites driven by the torsional motions of the floppy backbones. This model is then applied to analyze the experimental results for sequence-dependent long-range hole transport in DNA reported by Meggers et al. [Meggers, E., Michel-Beyerle, M. E., & Giese, B. (1998) J. Am. Chem. Soc. 120, 12950–12955]. The model has also been applied to analyze the experimental results of femtosecond dynamics of DNA-mediated electron transfer reported by Zewail and co-workers [Wan, C., Fiebig, T., Kelley, S. O., Treadway, C. R., Barton, J. K. & Zewail, A. H. (1999) Proc. Natl. Acad. Sci. USA 96, 6014–6019]. The initial events in the dynamics of protein folding have begun to attract attention. The GME obtained in this paper will be applicable to this problem.
1. Introduction
Charge migration is a very important process in biomolecules and other large polymeric systems. It has been found that charge migration in proteins or similar systems is highly efficient, but the mechanistic origin is still not well understood even though various models have been advanced (1, 2).
Recently we have demonstrated that the photoionization of positive charge at a specific chromophore in a series of polypeptides can lead to facile migration of this charge over long peptide chains (3–5); we have studied neutral peptides of natural amino acids of the type (X)n-Y (n = 1, 2, 3, where Y denotes the aromatic amino acid) in the gas phase. They have been prepared by laser desorption and supersonic cooling. Local ionization is performed by resonant laser excitation in aromatic acid Y located at the C terminus. Subsequent UV photofragmentation of the cation is shown to directly reflect the prior charge migration in these large molecules. The distance and direction of the charge migration is determined to a first order by the ionization energies of the individual amino acids, rather than the ionization potential of the entire supramolecule (6). It has been found that the charge migration can be blocked by as small a local barrier as 0.2–0.3 eV (differences in ionization potentials between different neighboring amino acids). In this work, we shall propose a model that is compatible with experiments and supported by ab initio (6) and molecular dynamics (7) calculations. The charge is initially localized at the chromophore in the form of an electronic hole in its ground electronic state of the cation. After photoexcitation of the cation, the electron is promoted into a charge transfer (CT) state (or by a photoexcitation to a localized state followed by internal conversion to a CT state) and the new hole thus created can hop between local sites in the chain. We propose that the coupling strength between local charge states, which is responsible for charge transfer sites, varies with torsional motion, which is the result of the floppy peptide backbone. When the wiggling molecule attains a favorable conformation, the coupling strength is strong enough to ensure high probability of charge transfer between two adjacent amino acids.
Because of the importance of DNA damage and its repair, diverse biophysical and biochemical studies have sought to understand the electron transfer (ET) in DNA (8–11). The strong resemblance of the base-pair stack of DNA to conductive one-dimensional aromatic crystals has prompted the proposal that long-range charge transport might proceed through DNA. Recently Zewail and co-workers (8) have reported with femtosecond resolution the direct observation in DNA of ultrafast ET, initiated by excitation of tethered ethidium, the intercalated electron acceptor. The electron donor is 7-deazaguanine, a modified base, placed at various, fixed, distances from the acceptor. The ultrafast ET between these reactants in DNA has been observed with time constants of 5 ps and 75 ps and was found to be essentially independent of the donor–acceptor separation (10–17 Å). Zewail and co-workers assigned the 5-ps decay component to direct ET from 7-deazaguanine and the slower, 75-ps, component to the process that requires the reorientation of tethered ethidium before ET. In other words, in DNA the motion of the base pairs controls the time scale and the rate of charge transport.
From the above discussion, one can see that charge (or electron) transfer in biological systems often takes place in at least two modes—e.g., rotation and electron (or hole) transfer. It is the purpose of this paper to present a formalism to treat this type of charge transport.
2. General Theory
We shall propose the model described by the following generalized master equations (GMEs)
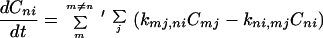 |
2-1 |
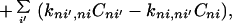 |
where (n, m) denote the sites of proteins and (i, j) represent the rotational states of the protein chain. Here Cni represents the probability to find the charge at the nth site of proteins with the rotational bond state i of the protein chain. The derivation of the above GME by the quantum mechanical approach is given in Section 3. For simplicity, the loss channels (if they exist) have been ignored. An important case will be
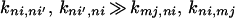 |
2-2 |
where m ≠ n. In this case, we have
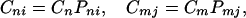 |
2-3 |
where (Pni, Pmj) represent the equilibrium distribution, and Eq. 2-1 becomes
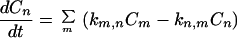 |
2-4 |
where
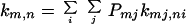 |
2-5 |
and
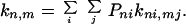 |
2-6 |
It should be noted that in Eq. 2-1 at least two different time scales described by (kmj,ni, kni,mj) and (kni′,ni, kni,ni′) are involved. For the case in which one time scale is much shorter than the other, the equilibrium approximation like that given by Eq. 2-3 can be introduced and the new set of master equations will involve only the slow time scales. The rate constants involved in the new set of master equations represent the rate constants averaged over the short time-scale states.
From Eqs. 2-1 and 2-4, we can see that in this case the charge migration (or ET) is through hopping. If the hopping takes place only through nearest neighbors and all the protein sites are equivalent, then
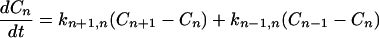 |
2-7 |
where
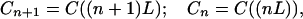 |
2-8 |
and L denotes the length between equivalent sites along the chain. Applying the Taylor expansion to Cn+1 and Cn−1 in terms of L yields
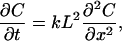 |
2-9 |
where k = kn−1,n = kn+1,n and Cn = C. That is, under this condition the charge migration can be described by a diffusion process. This type of charge migration has a special feature in which at equilibrium the charge distribution will be uniform among the protein sites. This is true provided that there is no absorbing boundary at one end and no reflecting boundary at the other.
We now discuss another type of charge migration. That is, the charge migration can take place only at a certain rotation state, say, ℓ. In this case, from Eq. 2-1 we obtain
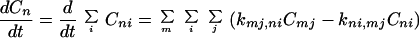 |
2-10 |
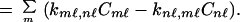 |
Furthermore, if the charge migration can take place only between the nearest neighbor and only in one direction, then
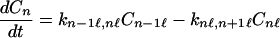 |
2-11 |
and
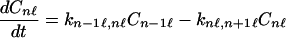 |
2-12 |
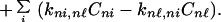 |
That is, here we have assumed that the charge migrates from the n − 1 site to the n site, and from the n site to the n + 1 site. This set of equations is very difficult to solve in general. However, for the case in which the rotational motion is much faster than charge migration, then again Eq. 2-3 can be used to obtain
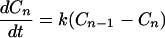 |
2-13 |
where k = kn−1ℓ,nℓPn−1ℓ = knℓ,n+1ℓPnℓ.
It should be noted that in Eq. 2-7 charge can migrate both forward and backward, whereas in Eq. 2-11 or Eq. 2-12 charge can only migrate forward, that is, from the n site to the n + 1 site.
Eq. 2-13 can easily be solved by using the Laplace transformation method to obtain
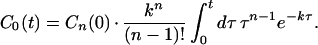 |
2-14 |
Notice that C0(0) = 0 and C0(∞) = Cn(0). That is, in this case, the yield is very high even though the charge migration rate is slowed down because of the presence of Pnℓ or Pn−1ℓ in k. For example, if n = 3, then Eq. 2-14 becomes
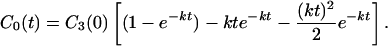 |
2-15 |
Eq. 2-15 shows that the yield of charge migration is very high (unity) and exhibits only a very weak distance dependence through k rather than kn. We realize that the above model (i.e., Eqs. 2-13 and 2-14) is too ideal, assuming that the charge migration is unidirectional and allows for no decay (i.e., ignoring the loss channels). However, it serves the purpose of demonstrating how a long-distance charge transfer can be accomplished effectively in multiple steps. It should be noted that, using this type of model, we can also describe the real time-dependent behaviors of the charge migration in the protein systems. Because of the existence of reversible processes in the charge migration, the yield of charge migration cannot be as high as that given by Eq. 2-13.
Recently long-range hole transport in DNA has attracted considerable attention (8–11). Guanine (G) bases are a target for oxidative damage in DNA (8). This damage is often the consequence of an oxidation of G to a guanine radical cation (G⋅+) (9, 10) that reacts further with H2O or O2 (11). Barton and co-workers (12) and Gasper and Schuster (13) have observed that oxidation damage can occur at G bases that are far away from the oxidant. Meggers et al. (14) have studied a hole transport process in DNA in which a guanine radical cation (G⋅+) was site-selectively generated in double-stranded DNA and the charge transfer in different oligonucleotides was investigated. Their results are reproduced in Fig. 1. They analyzed the hole transfer from G⋅+ to a GGG unit through one, two, three, and four A⋅T base pairs and found that the transfer efficiency decreases by about one order of magnitude with each intervening A⋅T base pair.
Figure 1.

Histograms demonstrating that with increasing numbers of intervening A⋅T base pairs the ratio of damage GGG/G23 or GGG/G23A24 decreases.
We shall analyze the results reported by Meggers et al. as follows, with G denoting the relative concentration of G, etc.:
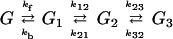 |
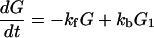 |
2-16 |
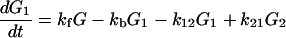 |
2-17 |
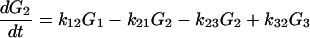 |
2-18 |
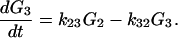 |
2-19 |
At steady or equilibrium state we find that
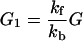 |
2-20 |
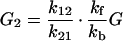 |
2-21 |
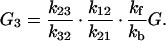 |
2-22 |
From Fig. 1 we can see that for the case of G23TGGG, kf/kb > 1, k12/k21 > 1, and k23/k32 ≪ 1 so that (k23/k32)⋅(k12/k21)⋅(kf/kb) < 1. It should be noted that this last inequality is only conditionally valid. Its validity is compatible with the experimental results shown in Fig. 1. In other words, from the ratios of individual peaks in Fig. 1 one can determine kf/kb, k12/k21, and k23/k32, that is, the free energy differences of different charge transport processes.
The fact that the relative intensities of the three peaks in various systems are approximately constant could happen, if the interconversion among the Gs is fast compared to their input rate.
From Eq. 2-13 we can see that if the reversible processes do not exist, then the charge transport yield will be unity. On the other hand, from Eqs. 2-16–2-19we can see that if the reversible processes do exist, then as long as all of the ratios of forward rate to backward rate are much larger than unity, the charge transport will still be very efficient. In other words, the charge transport can be made ineffective if the intermediate group is modified chemically (e.g., ionization potential) so that one of the ratios is much smaller than unity.
Next we shall compare the charge transfer by the so-called superexchange mechanism in one step versus the charge transfer by hopping in multiple steps. For the case in which there exists no reversible process, in real-time measurements an exponential decay would be observed in the super-exchange mechanism case, while multi-exponential decays would be involved in the multistep hopping mechanism. In this case, the charge transfer yield will be nearly unity in both mechanisms, but the distance dependence of the charge transfer rate constant would be different in the two mechanisms.
3. Quantum Mechanical Foundations
In this section, we shall provide the quantum mechanical formulation of the model presented in this paper. In refs. 15–17, the general formulations of the density matrix method have been presented. In this section, we shall show the application of the formalisms to a model of charge migration in biological systems. A main feature of this model is that processes with different time scales are involved. We start with the stochastic Liouville equation (15)
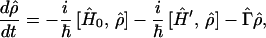 |
3-1 |
where Ĥ0 and Ĥ′ denote the zeroth-order Hamiltonian and interaction Hamiltonian, respectively, and Γ̂ represents the so-called damping operator due to the interaction between the system and heat bath, which is responsible for describing the relaxation and dephasing processes of the system. Using M and N as basis sets, we obtain
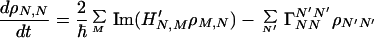 |
3-2 |
and
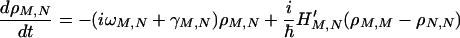 |
3-3 |
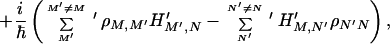 |
where −ΓNNN′N′ (N′ ≠ N) denotes the relaxation rate constant for N′ → N, while γM,N represents the dephasing rate constant. Notice that ΓNNNN describes the total relaxation of the N state, i.e., ΓNNNN = − ΣN′N′≠N ΓN′N′NN. These types of relations make sure that the principle of detailed balance is satisfied, and for the system embedded in a heat bath it can reach thermal equilibrium properly.
Using the Markoff approximation and applying the perturbation method to Eq. 3-1 by regarding Ĥ′ as a perturbation, we find, to the second-order approximation
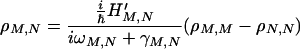 |
3-4 |
and
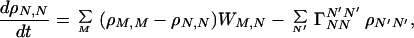 |
3-5 |
where
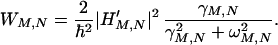 |
3-6 |
So far the electronic motion and nuclear motion of molecules have not been explicitly taken into consideration. For this purpose, the adiabatic approximation can be used.
According to the adiabatic approximation, we can make the changes M → mvj and N → nui, where (v, u) denote the vibrational states, while (n, m) represent the electronic (or site) states and (i, j) denote the rotational (or other) states. For the case in which vibrational relaxation is much faster than charge migration (or electron transfer), the thermal distribution (Pnu, Pmv) for vibrational states can be used. In this case, Eq. 3-5 reduces to Eq. 2-1 with the following correspondences,
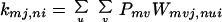 |
3-7 |
and
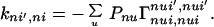 |
3-8 |
Notice that the rate constant kmj,ni can be written as
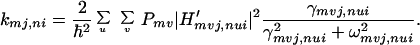 |
3-9 |
Using the Condon type approximation, which is used to separate the vibration from the rotation and electronic motions, we obtain
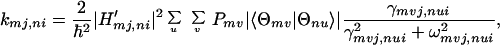 |
3-10 |
where H′mj,ni denotes the electronic interaction matrix element. It should be noted that kmj,ni denotes the charge transfer rate constant for m → n for the particular rotational configuration (i, j). At this particular configuration, the interaction energy Ĥ′ and the energy gap ωmj,ni may depend on (i, j). In this case (i.e., in the second-order approximation with respect to Ĥ′), kmj,ni describes the charge transfer through space (or direct charge transfer) and |〈Θmv|Θnu〉|2 represents the Franck–Condon factors. If the dephasing constant γmvj,nui is small or negligible, then the Lorentzian reduces to a delta function, i.e.,
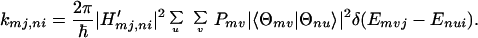 |
3-11 |
A main feature of our model here is the energetics (Emvj, Enui) depends on the (i, j) states (i.e., rotational or configurational states). That is, the energy gap in the transfer rate constant depends on (i, j) states.
As in Eq. 2-1, the notations n, m denote the sites and the notations i, j represent the rotational configurations (or states) associated with particular sites.
From Eq. 3-8 we can see that the rate constant kni′,ni for rotational motion is assumed to be induced by the heat bath through Γ̂; kni′,ni can also be induced intramolecularly through Ĥ′ like H′nu′i′,nui.
In previous papers (16, 17), we have shown that carrying out the perturbation calculation to the fourth order of approximations, for the case in which the electronic states of the bridge groups are higher than the donor electronic state one would obtain the conventional superexchange mechanism (i.e., the Raman type). On the other hand, if the electronic states of the bridge groups are lower than the donor state, one would obtain the resonance Raman type of electron transfer. This type of electron transfer can be as effective as (or even more effective than) the process involving the hopping through the bridge group.
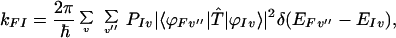 |
3-12 |
where I and F denote the initial and final electronic states, respectively, and
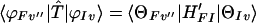 |
3-13 |
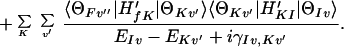 |
For the case in which the manifolds {Kv′} are much higher than the manifolds {Iv}, we can use the Plazcek approximation EIv − EKv′ ≅ EI − EK to obtain
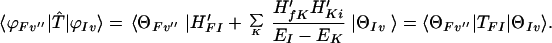 |
3-14 |
That is, in this case (similar to the Raman scattering) the second term gives us the conventional superexchange electron transfer.
On the other hand, for the case EIv > EKv′ we obtain
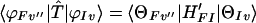 |
3-15 |
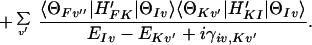 |
The second term of this expression gives us the resonance superexchange ET (i.e., the resonance Raman type ET). For example, if we are interested in D* BA → D+BA−, then the resonance superexchange ET can take place by invoking the state D+B−A as the resonance intermediate state. As in the case of resonance Raman scattering, the resonance superexchange ET can be enhanced by several orders of magnitude. The resonance superexchange ET and stepwise ET are usually in competition. Only for the case in which the former is faster than or comparable with the latter do we have to consider the resonance superexchange ET.
The exact master equations for charge migration (or ET) like Eq. 3-5 can be derived as follows. We shall rewrite Eq. 3-1 as
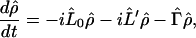 |
3-16 |
where (L̂0, L̂′) represent the Liouville operators corresponding to Ĥ0 and Ĥ′, respectively. Applying the Laplace transformation to Eq. 3-1 yields
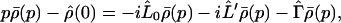 |
3-17 |
where ρ̂(0) denotes the density matrix at t = 0 and
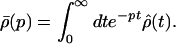 |
3-18 |
We shall introduce the projection operator D̂ for the purpose of picking up the diagonal elements of ρ̂,
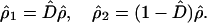 |
3-19 |
Here ρ̂1 contains diagonal elements, while ρ̂2 contains the off-diagonal elements.
Applying D̂ and (1 − D̂) separately to Eq. 3-8 yields
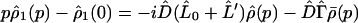 |
3-20 |
and
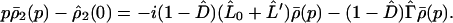 |
3-21 |
Eliminating ρ̄2(p) from Eq. 3-20, we obtain
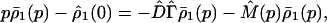 |
3-22 |
where M̄(p) represents the so-called memory kernel,
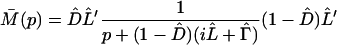 |
3-23 |
and L̂′ = L̂ − iΓ̂. Here the random phase approximation has been used, i.e. ρ̂2(0) = 0. Applying the inverse Laplace transformation to Eq. 3-22 yields
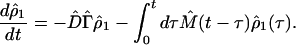 |
3-24 |
Using the perturbation method to the second-order approximation with respect to Ĥ′ and the Markoff approximation, Eq. 3-24 will reduce to Eq. 3-5. Higher-order calculations can be carried out similarly by using Eq. 3-24.
From the above discussion, we can see the charge migration (or ET) due to the strong electron correlation can be treated by using our formulation in the density matrix method (i.e., using Eq. 3-1). In this case, Ĥ0 and Ĥ′ will contain only the electronic degrees of freedom; in this case the electron transfer takes place before any nuclear motion. The ordinary ET does not concern electron correlation.
4. Discussion
In this section, to show the application of the model discussed in this paper we shall consider a simple two-site and two-state model described in the following
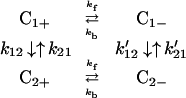 |
where (k12, k21) and (k′12, k′21) denote the charge migration (or ET) and (kf, kb) describe, for example, the rotation of protein backbones. Here for simplicity we assume that the charge migration takes place between C1+ and C2−, and that between C1− and C2+ can be ignored.
Notice that
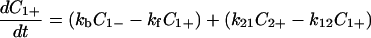 |
4-1 |
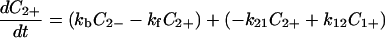 |
4-2 |
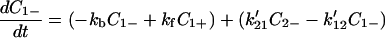 |
4-3 |
and
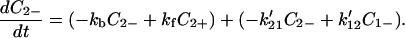 |
4-4 |
For simplicity, we shall assume that
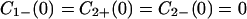 |
4-5 |
and
 |
4-6 |
Various cases can be considered. For example, for the case in which kb = 0, we find
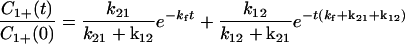 |
4-7 |
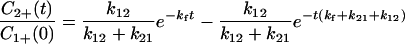 |
4-8 |
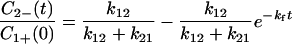 |
4-9 |
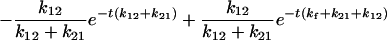 |
and
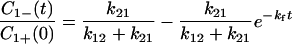 |
4-10 |
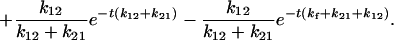 |
In this case, as t → ∞, C1+ → 0, C2+ → 0, 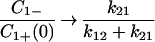 and
and 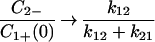 .
.
Next we shall attempt to analyze the experimental results of femtosecond dynamics of DNA-mediated electron transfer (7). For this purpose, we shall assume that C1− represents the inactive states, that is, k′12 = 0 and k′21 = 0. Furthermore, for simplicity we assume that C2+ and C2− do not communicate and that k21 is negligible. In this case, Eqs. 4-1 and 4-2 reduce to
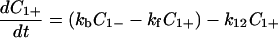 |
4-11 |
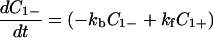 |
4-12 |
and
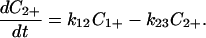 |
4-13 |
Here k23 denotes the ET rate constant for the process initiated from C2+ in case it exists.
From Eqs. 4-11 to 4-13, we can see that C1− and C1+ are involved only in Eqs. 4-11 and 4-12 and not in Eq. 4-13. k23 appears only in Eq. 4-13. That is, only C1− and C1+ are coupled through Eqs. 4-11 and 4-12. The solution of Eqs. 4-11 and 4-12 yields
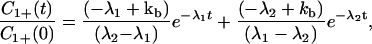 |
4-14 |
where λ1 and λ2 denote the slow time constant and fast time constant, respectively,
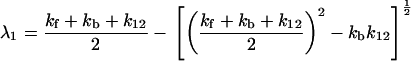 |
4-15 |
and
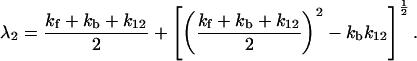 |
4-16 |
Here we have assumed that C1+ represents the photoexcited initial state; that is, C1+(0) ≠ 0, C1−(0) = 0, C2+(0) = 0. The charge transfer takes place between C1+ and C2+, and C1− represents the dark states. Zewail and co-workers (8) followed the time evolution of C1+(t) and found that 1/λ1 = 75 ps and 1/λ2 = 5 ps. Here for simplicity we have ignored the 2-ns component. For the 5Z case, they found that
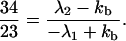 |
4-17 |
It follows that 1/kb ∼ 11 ps. Similarly for the 7Z case, we obtain 1/kb ∼ 9.1 ps and for the 9Z case, we obtain 1/kb ∼ 12 ps. As can be seen from the above analysis, the rates of kb are nearly the same and the ET rate is not too much faster than that of kb if kb ∼ kf.
Recently Bixon et al. have studied the long-range charge hopping in DNA (ref. 19 and the references therein). In our theoretical treatment of long-range charge migration in proteins and DNA, the torsional motion of floppy backbones is emphasized to play a very important role in the hole hopping between local amino acid sites in proteins. We have derived the generalized master equations which can describe the time evolution of the charge migration (and/or other dynamical processes) in complex systems. We emphasize that the long-range charge transfer can be effectively accomplished dynamically in multisteps. It is to be noted that the GME can be employed directly to analyze the time-resolved and/or steady-state experimental results.
To understand how proteins fold up into their compact three-dimensional forms is a central problem in modern structural biology and has attracted considerable experimental and theoretical attention (20, 21). Most experimental studies on the dynamics of protein folding have been confined to time scales of 1 ms and longer (20). Yet it is obvious that many phenomena that are obligatory elements of the folding processes occur on much faster time scales. For example, it is now clear that the formation of secondary and tertiary structures can occur on nanosecond and microsecond times, respectively. Thus it is obvious that theoretically to treat the dynamics of protein folding one has to deal with the processes ranging from picoseconds to seconds or longer. The GME approach presented in this paper is an ideal tool for this purpose.
In conclusion, in this paper, we have derived quantum mechanical GMEs that can be used in treating processes with various time scales. In particular, we have applied this GME method to develop a model of charge transport in proteins that invokes the hopping between local amino acid sites assisted or driven by the torsional motions of the floppy backbones.
Acknowledgments
We thank Prof. M. Ratner for helpful comments and suggestions. This work was supported in part by the National Science Council of the Republic of China and the Academia Sinica.
Abbreviations
- ET
electron transfer
- GME
generalized master equation
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.140196597.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.140196597
References
- 1.Jortner J, Ratner M, editors. Molecular Electronics: Chemistry for the 21st Century. Oxford: Blackwell Scientific; 1997. [Google Scholar]
- 2.Schlag E W, Lin S H, Weinkauf R, Rentzepis P M. Proc Natl Acad Sci USA. 1998;95:1358–1362. doi: 10.1073/pnas.95.4.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weinkauf R, Schanen P, Yang D, Soukara S, Schlag E W. J Phys Chem. 1995;99:11255–11267. [Google Scholar]
- 4.Weinkauf R, Schanen P, Metsala A, Schlag E W, Bürgle M, Kessler H. J Phys Chem. 1996;100:18567–18580. [Google Scholar]
- 5.Weinkauf R, Schlag E W, Martinez T J, Levine R D. J Phys Chem A. 1997;101:7702–7710. [Google Scholar]
- 6.Baranov L Ya, Schlag E W. Z Naturforsch. 1999;54a:387–392. [Google Scholar]
- 7.Schlag, E. W., Sheu, S.-Y., Yang, D.-Y., Selze, H. L. & Lin, S. H. (2000) J. Phys. Chem. A, in press.
- 8.Wan C, Fiebig T, Kelley S O, Treadway C R, Barton J K, Zewail A H. Proc Natl Acad Sci USA. 1999;96:6014–6019. doi: 10.1073/pnas.96.11.6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stemp E D A, Arkin M R, Barton J K. J Am Chem Soc. 1997;119:2921–2925. [Google Scholar]
- 10.Seidel C A M, Schulz A, Sauer M H M. J Phys Chem. 1996;100:5541–5553. [Google Scholar]
- 11.Sugiyama H. J Am Chem Soc. 1998;120:7373–7374. [Google Scholar]
- 12.Hall D B, Holmin E R, Barton J K. Nature (London) 1996;382:731–735. doi: 10.1038/382731a0. [DOI] [PubMed] [Google Scholar]
- 13.Gasper S M, Schuster G B. J Am Chem Soc. 1997;119:12762–12771. [Google Scholar]
- 14.Meggers E, Michel-Beyerle M E, Giese B. J Am Chem Soc. 1998;120:12950–12955. [Google Scholar]
- 15.Lin S H, Alden R G, Islampour R, Ma H, Villaeys A A. Density Matrix Method and Femtosecond Processes. Teaneck, NJ: World Scientific; 1991. [Google Scholar]
- 16.Lin S H. J Chem Phys. 1989;90:7103–7113. [Google Scholar]
- 17.Lin S H, Alden R G, Tang C K, Fujimura Y, Sugawara M. In: Mode Selective Chemistry. Jortner J, Levine R D, Pullman B, editors. Dordrecht, The Netherlands: Kluwer; 1991. pp. 467–484. [Google Scholar]
- 18.Van Brederode M E, Jones M R, Van Mourik F, Van Stokkum I H M, Van Grondelle R. Biochemistry. 1997;36:6855–6861. doi: 10.1021/bi9703756. [DOI] [PubMed] [Google Scholar]
- 19.Bixon M, Giese B, Wessely S, Langenbacher T, Michel-Beyerle M E, Jortner J. Proc Natl Acad Sci USA. 1999;96:11713–11716. doi: 10.1073/pnas.96.21.11713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pain R H, editor. Mechanisms of Protein Folding. Oxford: IRL; 1994. [Google Scholar]
- 21.Callender R H, Dyer R B, Gilmanshin R, Woodruff W H. Annu Rev Phys Chem. 1998;49:173–202. doi: 10.1146/annurev.physchem.49.1.173. [DOI] [PubMed] [Google Scholar]