

Abstract
Dematin is an actin-binding protein originally identified in the junctional complex of the erythrocyte plasma membrane, and is present in many nonerythroid cells. Dematin headpiece knockout mice display a spherical red cell phenotype and develop a compensated anemia. Dematin has two domains: a 315-residue, proline-rich “core” domain and a 68-residue carboxyl-terminal villin-type “headpiece” domain. Expression of full-length dematin in E. coli as a GST recombinant protein results in truncation within a proline, glutamic acid, serine, threonine rich region (PEST). Therefore, we designed a mutant construct that replaces the PEST sequence. The modified dematin has high actin binding activity as determined by actin sedimentation assays. Negative stain electron microscopy demonstrates that the modified dematin also exhibits actin bundling activity like that of native dematin. Circular dichroism (CD) and NMR spectral analysis, however, show little secondary structure in the modified dematin. The lack of secondary structure is also observed in native dematin purified from human red blood cells. 15N-HSQC NMR spectra of modified dematin indicate that the headpiece domain is fully folded whereas the core region is primarily unfolded. Our finding suggests that the core is natively unfolded and may serve as a scaffold to organize the components of the junctional complex.
Keywords: erythrocyte dematin, band 4.9, PEST sequence, NMR, headpiece domain, natively unfolded protein, actin binding protein
Introduction
The red blood cell has a highly specialized plasma membrane and underlying cytoskeleton that confers cell flexibility and deformability. Dematin, also known as band 4.9 or protein 4.9, is an actin binding protein and has been localized to the junctional complex of the erythrocyte cytoskeleton.1,2 Dematin is a member of the villin-type headpiece family, which includes actin-associated proteins that have a ∼70-residue “headpiece” domain at the extreme C-terminus of a variety of large, nonhomologous “core” domains.3 The core domain of dematin is homologous only to Limatin/abLIM, which is involved in recruiting signaling molecules to the cytoskeleton.4,5,6 Northern blot analysis shows that dematin is present in many tissues including human platelets, heart, brain, and lens tissue.7 Knockout mice that lack the headpiece domain but still produce the dematin core domain have a fragile red cell phenotype with spherical erythrocytes, and develop a compensated anemia, indicating an important role for dematin headpiece in maintaining the mechanical properties of the red cell membrane.8 The dematin gene is located on human chromosome 8p21.1,9 a region often implicated in prostate cancer. In fact, the overexpression of dematin changes the tumorigenic phenotype of PC-3 cell line back to prostate cells of normal appearance.10
Dematin has two major isoforms due to alternate RNA splicing with apparent molecular weights of 48 and 52 kDa. The 52 kDa isoform contains an additional 22 amino acid insert within the headpiece domain, as compared to the 48-kDa isoform.9 Dematin forms a trimeric complex composed of, on average, two 48 kDa units and one 52 kDa unit, in vivo.11 There is approximately one trimer of dematin per junctional complex in the human red blood cell. Dematin binds and bundles F-actin in vitro.11 Importantly, it contains the only headpiece domain that is regulated by phosphorylation, at serine 381. Although the functional role of this phosphorylation in vivo is unknown, it results in the loss of F-actin bundling activity in vitro.12 Interestingly, the isolated construct of dematin headpiece domain maintains F-actin binding activity when phosphorylated, although the actin-binding affinity is reduced threefold compared with that of unphosphorylated headpiece.3
The nuclear magnetic resonance (NMR) structures of the 68-residue dematin headpiece (DHP), corresponding to the 48 kDa form of dematin headpiece, as well as a mutant headpiece that mimics the phosphorylated form have been solved.13,14 The structure of DHP is similar to that of villin headpiece, containing an N-terminal subdomain composed of loops and turns, and a C-terminal subdomain of three α helices. Here, we investigated the structure of the large, N-terminal core domain and its effect on the structure of the headpiece domain. The 48 kDa subunit of dematin was examined by a variety of biochemical and biophysical techniques. Under experimental conditions where dematin has both F-actin binding and bundling activity, the core domain of dematin is largely devoid of secondary structure. A small amount of α-helical structure primarily arising from the fully folded headpiece domain is observed. These results suggest that the core domain of dematin exhibits properties typical of a natively unfolded protein, while the headpiece domain is folded in a conformation essentially identical to its native structure. We discuss a possible model for dematin assembly and regulation of its actin bundling activity in vivo.
Results
PEST-region induced proteolysis
To examine the structure of the unique core domain of dematin, we first decided to express the full-length 48 kDa isoform of dematin fused to a cleavable GST-tag in order to simplify its purification in E. coli. Unfortunately, this construct was prone to significant intracellular proteolysis within the sequence enriched in proline, glutamic acid, serine, and threonine (PEST).9 PEST domains are targeted for rapid destruction in eukaryotes,15 although there is no evidence that the PEST degradation pathway is active in prokaryotes. The PEST region spans residues 89–102 (Figs. 1 and 2) in dematin. To overcome the degradation of recombinant dematin, multiple mutagenesis strategies were tested. The first strategy was to mutate E98 to K98, to test if the swapped charge affects the PEST signal.15 However, expression of this mutant in E. coli still showed proteolysis. A second construct was then designed by replacing three consecutive serine and threonine residues with alanines in the PEST region,15 changing the encoded protein from 89KSTSPPPSPEVWAD102 to 89KAAAPPPSPEVWAD102. This mutant was also cleaved during bacterial expression. Deletion of the entire PEST domain (residues 89–102) prevented proteolysis, but removed significant amounts of sequence that could affect the structure of the core domain. Therefore, in the final strategy, mutating all proline, glutamic acid, serine and threonine residues in the region, was successful in preventing proteolysis. Thus, 89KSTSPPPSPEVWAD102 was replaced with 89KAAAGGGAGKVWAD102 in the construct termed PEST-replaced dematin (rD) (Fig. 2). The rD construct was used to further investigate the structure of full-length dematin. Following bacterial expression, rD was purified by a GST affinity column. A 69 kDa band that likely corresponds to the prokaryotic HSP70 chaperone protein, DnaK, coelutes with dematin from the GST column. Addition of fresh ATP to the buffers used in washing the GST column at room temperature resulted in removal of the contaminating DnaK.16 DnaK recognizes unfolded or misfolded proteins and its strong binding is consistent with the presence of unfolded regions in our expressed dematin construct.
Figure 1.

Amino acid sequence of the 48 kDa form of dematin. The first 315 amino acids belong to the N-terminal core domain. The C-terminal headpiece domain, residues 316–383, is shown in blue. The proline residues are highlighted in red. The PEST region, residues 89–102, is highlighted in green. The negatively charged region, residues 216–229, is in yellow.
Figure 2.
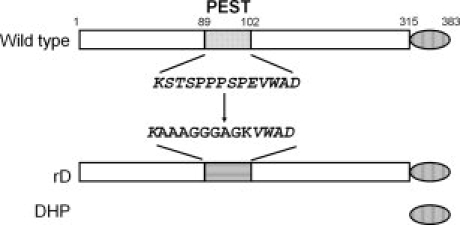
Dematin constructs. Top schematic shows wild type sequence highlighting the PEST sequence (residues 89–102) and the C-terminal headpiece domain (residues 316–383). The wild type PEST sequence is shown below the wild type dematin. The mutated PEST sequence in rD is shown below the wild type PEST sequence.
Actin binding and bundling by rD
To test whether rD expressed in E. coli retains the activity of wild type dematin isolated from red blood cells, its actin binding and bundling activities were measured. Actin binding was demonstrated using an actin sedimentation assay and actin bundling was observed by negative stain electron microscopy. Actin sedimentation assays indicate that rD retains actin binding affinity (Fig. 3). The rD cosediments with F-actin in the pellets of the binding assays. The amount of rD in the actin pellet increases with increasing rD concentration from 2 μM to 16 μM in the reaction (Fig. 3 Lanes 2–6). In the absence of actin, only trace amounts of rD are seen in the pellet fraction (Fig. 3 lane 7). Unfortunately, we are unable to quantitate our sedimentation assays as the maximum solublitity of dematin (∼50 μM) is well below concentration required for saturation. However it is clear that rD binds actin, in vitro, as previously described for dematin isolated from human red blood cells.9
Figure 3.
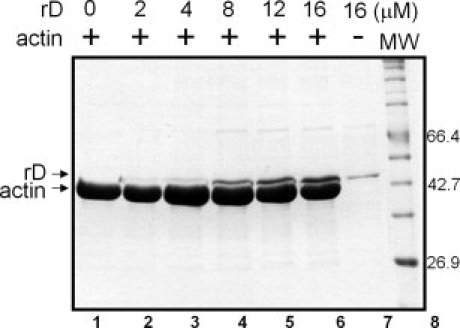
Actin sedimentation assays of rD. Increasing concentrations of rD were incubated with 22 μM F-actin for 1 hr at 4°C. After centrifugation for 1 h at 100,000g at 4°C, the pellets were harvested and run on SDS-PAGE gel. Lanes 1–6: Pellets of the reactions containing increasing concentrations of rD. Lane 7: 16 μM rD sedimented in the absence of actin. Lane 8: Protein molecular weight markers.
Actin bundling assays were performed to verify the actin crosslinking activity of the rD construct. In negative stain electron microscopy images, actin bundles up to 50 nm in diameter were observed when F-actin was incubated with rD at a 1:1 weight ratio (Fig. 4). As rD has a molecular weight close to actin, the ratio of rD and actin in actin bundling assays is almost 1:1. Bundles are also observed at lower molar ratios of rD to actin (1:2) (Supp. Info. Fig. 1). Thus, rD has actin binding and bundling activity, which is consistent with wild type dematin isolated from erythrocytes in vitro.12
Figure 4.
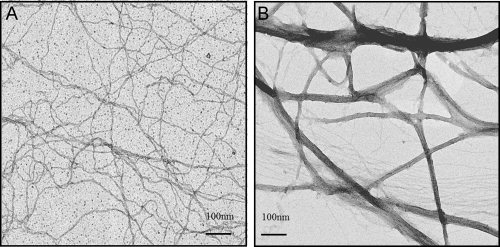
Actin bundling assay of rD. After incubation for 2 h, 0.2 mg/mL of actin (A) or 0.2 mg/mL actin and 0.2 mg/mL rD (B) in bundling buffer were loaded onto carbon coated and glow discharged grids and stained with 1% uranyl acetate. Micrographs were taken at ×45,000 magnification at room temperature.
Secondary structure analysis of dematin
To determine the secondary structure of dematin, the far-UV CD spectrum of rD was acquired in a buffer that mimics physiological conditions (Fig. 5). The spectrum has a strong negative peak at 201 nm indicating a largely random coil conformation. To insure that this random structure was not due to rD misfolding in our E. coli expression and purification system, we also collected CD spectra of dematin purified from human erythrocytes (RBC dematin).12 Both proteins have similar CD spectra with approximately 80% random coil structure. There is a slight but reproducible difference in the wavelength of maximal CD between rD and RBC dematin (201 and 203 nm, respectively). This may be explained by the fact that RBC dematin is a mixture of both the 48 kDa and 52 kDa forms. In addition, the rD construct is completely unphosphorylated, whereas RBC dematin is partially phosphorylated.1,12
Figure 5.
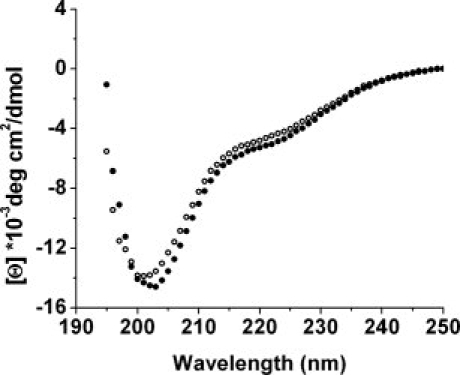
Far UV circular dichroism spectra of dematin. Open circles: dematin purified from human erythrocyte; Filled circles: rD. The concentration of both samples was 3 μM in 200 mM NaCl, 10 mM phosphate, and pH 7.0 at room temperature. Each spectrum is the average of 3 scans with a 15 s averaging time at each nm, in a 1 mm path length cell. The spectra were corrected by subtraction of a scan of buffer alone.
Thermal unfolding experiments monitored by CD showed that constructs containing the core domain are stable up to about 40°C at which point they begin to unfold and irreversibly aggregate (not shown). The thermal unfolding of the isolated dematin headpiece domain has been previously shown to be reversible.3
NMR spectroscopy indicates that the headpiece domain of rD is folded
The 15N HSQC spectrum provides all resonances of N-H groups in a protein sequence, showing crosspeaks at the 15N and 1H resonances of each N-H group. The 15N HSQC spectrum of rD displays the characteristics expected from the CD spectra: a cluster of resonances at random coil chemical shifts between 7.9 ppm to 8.6 ppm [Fig. 6(B)]. However, another set of dispersed crosspeaks in the spectrum indicates a well-folded region. The dispersed crosspeaks can be identified as arising from the C-terminal headpiece domain by overlaying the spectra of rD and the isolated dematin headpiece construct (DHP) (Fig. 6).13 The chemical shifts of headpiece residues are essentially identical with those of isolated DHP [Fig. 6(A)], indicating that the headpiece domain is independently folded in the context of the whole protein. The exact correlation of the resonances of the headpiece domain in rD and DHP indicate that their conformations are identical. Furthermore, the presence of the narrow resonances of the headpiece domain in rD indicates that the headpiece domain does not interact with the core domain.
Figure 6.
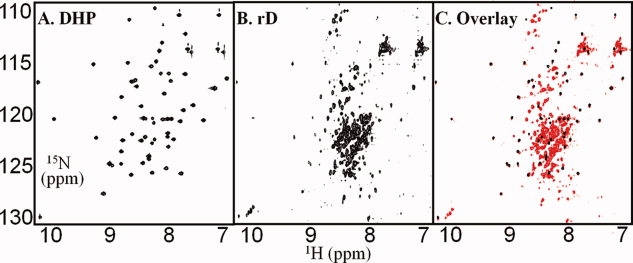
15N HSQC spectra of dematin constructs. (A) dematin headpiece (DHP) [data from Frank et al. 200413, the spectrum was taken at 20°C, and pH 6.0; (B) rD (∼ 50 μM); (C) Overlay of the spectra of DHP (black) and rD (red). Spectra of rD were acquired at 20°C, and pH 7.0, as 256 increments of 2048 complex data points and the average of 128 scans per increment.
Counting the number of resonances from the core domain of rD indicates that crosspeaks for about 160 to 180 residues are missing. These regions are likely in intermediate exchange on the NMR time scale, and therefore too broad to be detected in HSQC spectra. These missing resonances may be involved in the trimer formation by dematin. Thus, our data indicate that rD has a well-folded headpiece domain and a primarily, natively unfolded core domain.
Discussion
To examine the structure of the unique core domain of dematin and to determine if the core and headpiece domains interact with each other, we designed a PEST sequence replaced mutant dematin (rD) for biophysical and biochemical analysis. The 48 kDa form of dematin has 41 proline residues spread-out in the sequence (Fig. 1), an occurrence of 10.7%, which is twice the normal proline content (5.2%).17 The high percentage of proline is consistent with the low secondary structure content we observed by CD (Fig. 5). There is a 14-residue stretch containing 12 negative charges in the region from Glu216 to Glu229, whose significance is unknown at present. A PEST degradation sequence is found in the core domain from Lys89 to Asp102. This region is responsible for dematin degradation in bacteria and may also serve as a regulated degradation tag in eukaryotic cells. Replacement of 90STS92 in the sequence with alanines did not prevent proteolysis, nor did changing the single glutamic acid residue to lysine. However, replacement of all prolines with glycines, all serines and threonines to alanines, and glutamic acid to lysine prevents proteolysis.
The rD has the same actin binding and bundling activities in vitro as wild-type dematin. The CD spectrum of rD indicates that it contains approximately 80% random coil structure with the remaining 20% made up of α-helical and β-sheet structure. To insure that the lack of secondary structure of rD was not due our prokaryotic expression system, we also examined dematin purified from human red blood cells. The CD spectra of rD and dematin from human erythrocytes are essentially the same indicating that both proteins are largely natively unfolded. The natively unfolded structure we observed is consistent with predictions that much of dematin would be unfolded by the predictor of natural denatured regions program, PONDR.18
NMR spectroscopy was used to determine if the C-terminal headpiece domain was folded independently from the core domain as has been seen with villin.19 15N-HSQC spectrum of rD has a set of clustered resonances at random coil chemical shifts between 7.9 ppm and 8.6 ppm and another set of crosspeaks at dispersed chemical shifts. These disperse peaks can be identified as arising from the C-terminal folded headpiece domain by comparison to the spectra of the isolated DHP domain.13 There is a near perfect correlation of the 15N and 1H chemical shifts of the isolated DHP domain and those from rD. Thus, the headpiece domain conformation is unaltered by the core domain, except at its extreme N-terminus where the linkage to the core occurs. Furthermore, the sharp resonances of the headpiece domain of rD and lack of chemical shift changes indicate that it does not interact with the core domain.
After excluding the resonances arising from the headpiece domain, there are still some 160–180 expected resonances from the backbone amides of the core domain of rD that are missing in the 15N-HSQC spectrum. These regions are likely in intermediate exchange on the NMR time scale, and therefore too broad to be detected in HSQC spectra. Alternatively, these missing resonances could also be involved in intermolecular interactions that result in the trimerization that has been reported for dematin.11 In the limiting (and unlikely) case where all of the 160–180 residues formed a compact, trimeric structure composed of loops and turns without significant canonical secondary structure, the molecular weight of that region would be between 50 and 60 kDa which would also result in significant line broadening. Most likely, the dematin core contains regions of fully unfolded structure (visible as sharp resonances in the HSQC spectrum) and regions interconverting between multiple conformations on the millisecond time scale (corresponding to the missing resonances broadened by intermediate exchange). The region of the core domain required for the trimerization of dematin remains to be localized.
Why is the core domain of dematin natively unfolded? The classic notion is that proteins require a well-defined globular structure to be functional.20 But more and more evidence is accumulating that disordered proteins can also be biologically active.21,22 For example, human securin, also called PTTG1 (pituitary tumor transforming gene 1 product), under physiological conditions, is devoid of tertiary and secondary structure except for a small amount of poly-(L-proline) type II structure.23 Tat (transactivator of transcription), a small RNA-binding protein that plays a central role in the regulation of human immunodeficiency virus type 1 replication, is another example of a natively unfolded protein.24 We propose that the core domain of dematin may act as a scaffold. Regions of the natively unfolded core domain self-associate to form a trimer. The three headpiece domains are dispersed outside the trimer. Either both of the core and headpiece, or only the headpiece domain, binds with actin filaments to form actin bundles. Thus, although intrinsically unstructured, dematin retains F-actin binding affinity and bundling activity.
Methods
Dematin expression constructs
Constructs were prepared from the 48 kDa form of human dematin fused to glutathione S-transferase (GST tag) in the pGEX-2T plasmid (GE Healthcare)9 using QuikChange mutagenesis according to the manufacturer's directions (Stratagene). Eight extra residues at the C-terminus of the original vector (384GSPGIHRD391) were removed by mutating the G384 codon (GGA) to a stop codon (TGA). In addition, a PreScission protease cleavage site was inserted between the GST tag and the dematin sequence.
To generate the dematin constructs with PEST mutations,15,25 the following primers were used: PEST deleted mutation, 5′-CGCTCGCTGTCACCCAGCCGGTCGCCTGG-3′ and 5′-CCAGGCGACCGGCTGGGTGACAGCGAGCG-3′ were used to completely delete the PEST region (89KSTSPPPSPEVWAD102); E98K charge-swap mutation, 5′-CCCCCACCATCCCCAAAGGTGTGGGCGGACAGCC-3′ and 5′-GGCTGTCCGCCCACACCTTTGGGGATGGTGGGGG-3′; Mutation of 90STS92 to 90AAA92, 5′-CGCTGTCACCCAAAGCCGCAGCGCCCCCACCATCCCC-3′ and 5′-GGGGATGGTGGGGGCGCTGCGGCTTTGGGTGACAGCG-3′; PEST replaced mutation (rD), 5′-GCGCTCGCTGTCACCCAAAGCCGCAGCGGGGGGCGGAGCCGGGAAGGTGTGGGCGGACAGCCGG-3′, and 5′-CCGGCTGTCCGCCCACACCTTCCCGGCTCCGCCCCCCGCTGCGGCTTTGGGTGACAGCGAGCGC-3′ were used to replace 89KSTSPPPSPEVWAD102 with 89KAAAGGGAGKVWAD102 (Figs. 1 and 2). All constructs were verified by nucleotide sequencing.
Protein expression and purification
Escherichia coli BL21 cells (Novagen) were cultured in Luria broth (LB) containing 100 μg/mL ampicillin at 37°C to an optical density at 600 nm of ∼0.6. Protein expression was induced by addition of isopropyl-1-thio-β-d-galactoside (IPTG) to a final concentration of 0.8 mM and further incubated for 3 h. For isotopically enriched preparations, 4 L of cells cultured in LB to an OD600 of 0.6 were harvested, resuspended and induced with IPTG in 1 L of M9 minimal media supplemented with 1 g/L 15N-labeled ammonium chloride for 4 h at 37°C using the protocol described by Marley et al.26 The cells were harvested by centrifugation at ∼4000g and cell pellets from 1 L cultures were resuspended in 20 mL cell lysis buffer (200 mM sodium chloride, 10 mM phosphate, pH 7.0, 1 mg/mL lysozyme, 1 mM phenylmethylsulphonyl fluoride, 1 mM benzamidine, 1 mM ethylenediaminetetraacetic acid, and 5 mM β-mercaptoethanol (β-ME)). The bacterial suspension was sonicated for 2 min on ice (Branson Sonic Power Co.), and immediately centrifuged at 20,000g at 4°C for 25 min. The supernatants were applied to glutathione agarose column (Sigma) at 4°C (10 mL supernatant to 1 mL of column volume). The column was washed 6 times by 5 column volumes of fresh ATP wash buffer (5 mM adenosine 5′-triphosphate (ATP), 5 mM magnesium chloride, 200 mM sodium chloride, 1 mM sodium azide, and 10 mM phosphate, pH 7.0) at room temperature, followed by 3 column volumes of cleavage buffer (200 mM sodium chloride, 1 mM sodium azide, 1 mM 1,4-Dithiothreitol (DTT), and 10 mM phosphate, pH 7.0) at 4°C. Typically, 40 units of PreScission protease (GE Healthcare) were added in 1 column volume of cleavage buffer at 4°C for on-column cleavage. Cleaved dematin was eluted with cleavage buffer after 4 h, whereas the GST tag and GST-tagged PreScission protease were retained on the column.
Actin binding assay
Actin was purified from chicken pectoral muscle using standard procedures,27 and stored as F-actin at 4°C. Actin was freshly dialyzed against G buffer (5 mM tris(hydroxymethylaminino)methane (TRIS), pH 8.0, 0.2 mM calcium chloride, 0.5 mM DTT, 0.2 mM ATP, 0.1 mM sodium azide).28 The G-actin concentration was determined by absorbance at 290 nm using an extinction coefficient of 26,640 L/M/cm.28 Actin sedimentation assays were performed at 4°C.3 The rD protein was centrifuged for 1 h at 100,000g at 4°C to remove potential aggregates before preparing sedimentation reactions. G-actin (300 μM) was first incubated in F-buffer (10 mM TRIS, pH 8.0, 1 mM magnesium chloride, 100 mM sodium chloride, 0.1 mM ATP, 0.2 mM DTT, 3 mM sodium azide, and 0.1 mM calcium chloride) overnight to insure complete polymerization. F-actin (final concentration 22 μM) was separately mixed with increasing concentrations of rD. The total volumes of the reactions were adjusted to 50 μL by reaction buffer (200 mM sodium chloride, 1 mM sodium azide, 1 mM DTT, and 10 mM phosphate, pH 7.0). As a control for protein sedimentation in the absence of actin, a sample of 16 μM rD alone was used. After incubation for 1 h, the samples were centrifuged for 1 h at 100,000g at 4°C in a prechilled Beckman A-100 rotor. Supernatants were carefully isolated from the pellets and 20 μL of sample loading buffer (0.5% bromophenol blue, 50 mM TRIS, pH 7.5, 2% sodium dodecylsulfate (SDS), 5% β-ME, and 12% glycerol) was added. The pellets were soaked in 20 μL of sample loading buffer for 1 h at room temperature followed by the addition of 48 μL of reaction buffer to equalize the volume with that of the supernatant. The samples were analyzed on 12.5% SDS-PAGE gels and visualized by Coomassie blue staining.
Actin bundling assay
Actin was freshly dialyzed against bundling buffer (50 mM TRIS, pH 7.5, 2 mM magnesium chloride, 0.04 mM calcium chloride, 0.7 mM EGTA, 75 mM sodium chloride, 0.02% sodium azide, 1 mM DTT, and 1 mM ATP) at 4°C. The rD construct was added to a final concentration of 0.2 mg/mL with F-actin (final 0.2 mg/mL) and incubated for 2 h at room temperature in 50 μL aliquots. The samples were loaded onto carbon-coated and glow-discharged copper grids (SPI Supplies) for 1 min, washed with 10 drops of wash buffer (6.7 mM TRIS, pH 7.5, and 50 mM sodium chloride), and stained with 1% uranyl acetate for 20 s.29 All samples were imaged on a Philips CM12 transmission electron microscope operated at 120 kV with a LaB6 filament and recorded on SO-163 EM (Kodak) film at 45,000× magnification under minimal electron dose conditions. The film was processed with undiluted Kodak D-19 developer for 12 min, followed by Kodak rapid fixer for 5 min. Electron micrographs were digitized on a Creo IO Smart2 Scanner (Global Imaging) at 1270 dpi.
Far UV circular dichroism
Circular dichroism (CD) measurements were obtained with an AVIV model 215 spectrometer. CD spectra were recorded using a 1 mm path length cuvette and protein concentrations of 3 μM in 10 mM phosphate, pH 7.0, 200 mM sodium chloride at 20°C. Wavelength spectra are the average of 3 scans from 260 to 190 nm with 1 nm steps, and an averaging time of 15 s. All spectra were baseline corrected against the same cell with buffer alone. The signal was converted to mean residue ellipticity. To estimate the secondary structure content from the CD spectra, the ellipticity value [Θ]m at the wavelength of 222 nm was used to determine the α-helix content.30
Nuclear magnetic resonance spectroscopy
NMR samples contained ∼50 μM15N-labeled dematin constructs in the buffer (10 mM phosphate, pH 7.0, 200 mM sodium chloride, 10% D2O, 0.1 mM 3-trimethylsilyl tetradeutero sodium propionate (TMSP), and 0.01% sodium azide). The pH was adjusted to 7.0 without correction for the effect of the D2O on the measured pH value. Heteronuclear single quantum coherence (HSQC) NMR spectra were acquired at 20°C on a Bruker DMX500 spectrometer. The sweep widths were 6000 and 1500 Hz for 1H and 15N, respectively. Spectra were acquired as 256 increments of 2048 complex data points and the average of 128 scans per increment. A Watergate pulse sequence was used for water suppression.31 All 1H and 15N chemical shifts are referenced against TMSP.32 The NMR data were processed using NMRPipe and NMRDraw,33 and analyzed with NMRView.34
Glossary
Abbreviations:
- β-ME
β-mercaptoethanol: CD, Circular Dichroism
- DHP
dematin headpiece
- DTT
1,4-Dithiothreitol
- HSQC
heteronuclear single quantum coherence
- IPTG
isopropyl-1-thio-β-D-galactoside
- LB
Luria broth
- NMR
nuclear magnetic resonance
- PEST
proline, glutamic acid, serine, and threonine rich sequence
- rD
PEST replaced dematin 48-kDa isoform
- RBC
red blood cell
- TRIS
tris(hydroxymethyl)aminomethane.
References
- 1.Chishti AH, Faquin W, Wu CC, Branton D. Purification of erythrocyte dematin (protein 4.9) reveals an endogenous protein kinase that modulates actin-bundling activity. J Biol Chem. 1989;264:8985–8991. [PubMed] [Google Scholar]
- 2.Bennett V. The spectrin-actin junction of erythrocyte membrane skeletons. Biochim Biophys Acta. 1989;988:107–121. doi: 10.1016/0304-4157(89)90006-3. [DOI] [PubMed] [Google Scholar]
- 3.Vardar D, Chishti AH, Frank BS, Luna EJ, Noegel AA, Oh SW, Schleicher M, McKnight CJ. Villin-type headpiece domains show a wide range of F-actin-binding affinities. Cell Motil Cytoskeleton. 2002;52:9–21. doi: 10.1002/cm.10027. [DOI] [PubMed] [Google Scholar]
- 4.Roof DJ, Hayes A, Adamian M, Chishti A, Li T. Molecular characterization of abLIM, a novel actin-binding and double zinc finger protein. J Cell Biol. 1997;138:575–588. doi: 10.1083/jcb.138.3.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rana AP, Ruff P, Maalouf GJ, Speicher DW, Chishti AH. Cloning of human erythroid dematin reveals another member of the villin family. Proc Natl Acad Sci USA. 1993;90:6651–6655. doi: 10.1073/pnas.90.14.6651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barrientos T, Frank D, Kuwahara K, Bezprozvannaya S, Pipes GC, Bassel-Duby R, Richardson JA, Katus HA, Olson EN, Frey N. Two novel members of the ABLIM protein family, ABLIM-2 and -3, associate with STARS and directly bind F-actin. J Biol Chem. 2007;282:8393–8403. doi: 10.1074/jbc.M607549200. [DOI] [PubMed] [Google Scholar]
- 7.Faquin WC, Husain A, Hung J, Branton D. A immunoreactive form of erythrocyte protein 4.9 is present in non-erythroid cells. Eur J Cell Biol. 1988;46:168–175. [PubMed] [Google Scholar]
- 8.Khanna R, Chang SH, Andrabi S, Azam M, Kim A, Rivera A, Brugnara C, Low PS, Liu SC, Chishti AH. Headpiece domain of dematin is required for the stability of the erythrocyte membrane. Proc Natl Acad Sci USA. 2002;99:6637–6642. doi: 10.1073/pnas.052155999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Azim AC, Knoll JHM, Beggs AH, Chishti AH. Isoform cloning, actin binding, and chromosomal localization of human erythroid dematin, a member of the villin superfamily. J Biol Chem. 1995;270:17407–17413. doi: 10.1074/jbc.270.29.17407. [DOI] [PubMed] [Google Scholar]
- 10.Lutchman M, Pack S, Kim AC, Azim A, Emmert-Buck M, van Huffel C, Zhuang Z, Chishti AH. Loss of heterozygosity on 8p in prostate cancer implicates a role for dematin in tumor progression. Cancer Genet Cytogenet. 1999;115:65–69. doi: 10.1016/s0165-4608(99)00081-3. [DOI] [PubMed] [Google Scholar]
- 11.Siegel DL, Branton D. Partial purification and characterization of an actin-bundling protein, band 4.9, from human erythrocytes. J Cell Biol. 1985;100:775–785. doi: 10.1083/jcb.100.3.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chishti AH, Levin A, Branton D. Abolition of actin-bundling by phosphorylation of human erythrocyte protein 4.9. Nature. 1998;334:718–721. doi: 10.1038/334718a0. [DOI] [PubMed] [Google Scholar]
- 13.Frank BS, Vardar D, Chishti AH, McKnight CJ. The NMR structure of dematin headpiece reveals a dynamic loop that is conformationally altered upon phosphorylation at a distal site. J Biol Chem. 2004;279:7909–7916. doi: 10.1074/jbc.M310524200. [DOI] [PubMed] [Google Scholar]
- 14.Jiang ZG, McKnight CJ. A phosphorylation-induced conformation change in dematin headpiece. Structure. 2006;14:379–387. doi: 10.1016/j.str.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 15.Rechsteiner M, Rogers SW. PEST sequences and regulation by proteolysis. Trends Biochem Sci. 1996;21:267–271. [PubMed] [Google Scholar]
- 16.Rial VD, Ceccarelli EA. Removal of DnaK contamination during fusion protein purifications. Protein Expr Purif. 2002;25:503–507. doi: 10.1016/s1046-5928(02)00024-4. [DOI] [PubMed] [Google Scholar]
- 17.Doolittle RF. Redundancies in protein sequences. In: Fasman GD, editor. Prediction of protein structure and the principles of protein conformation. New York: Plenum Press; 1989. pp. 599–623. [Google Scholar]
- 18.Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::aid-prot50>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 19.Smirnov SL, Isern NG, Jiang ZG, Hoyt DW, McKnight CJ. The isolated sixth gelsolin repeat and headpiece domain of villin bundle F-actin in the presence of calcium and are linked by a 40-residue unstructured sequence. Biochemistry. 2007;46:7488–7496. doi: 10.1021/bi700110v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fink AL. Natively unfolded proteins. Curr Opin Struct Biol. 2005;15:35–41. doi: 10.1016/j.sbi.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 21.Uversky VN. What does it means to be natively unfolded? Eur J Biochem. 2002;269:2–12. doi: 10.1046/j.0014-2956.2001.02649.x. [DOI] [PubMed] [Google Scholar]
- 22.Uversky VN. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 2002;11:739–756. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanchez-Puig N, Veprintsev DB, Fersht AR. Human full-length Securin is a natively unfolded protein. Protein Sci. 2005;14:1410–1418. doi: 10.1110/ps.051368005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shojania S, O'Neil JD. HIV-1 Tat is a natively unfolded protethe solution conformation and dynamics of reduced HIV-1 Tat-(1-72) by NMR spectroscopy. J Biol Chem. 2006;281:8347–8356. doi: 10.1074/jbc.M510748200. [DOI] [PubMed] [Google Scholar]
- 25.Rechsteiner M. PEST sequences are signals for rapid intracellular proteolysis. Semin Cell Biol. 1990;1:433–440. [PubMed] [Google Scholar]
- 26.Marley J, Lu M, Bracken C. A method for efficient isotopic labeling of recombinant proteins. J Biomol NMR. 2001;20:71–75. doi: 10.1023/a:1011254402785. [DOI] [PubMed] [Google Scholar]
- 27.Pardee JD, Spudich JA. Purification of muscle actin. Methods Enzymol. 1982;85:164–181. doi: 10.1016/0076-6879(82)85020-9. [DOI] [PubMed] [Google Scholar]
- 28.Meng J, Vardar D, Wang Y, Guo H, Head JF, McKnight CJ. High-resolution crystal structures of villin headpiece and mutants with reduced F-actin binding activity. Biochemistry. 2005;44:11963–11973. doi: 10.1021/bi050850x. [DOI] [PubMed] [Google Scholar]
- 29.Harris JR. Negative staining-carbon film technique: new cellular and molecular applications. J Electron Microsc Tech. 1991;18:269–276. doi: 10.1002/jemt.1060180309. [DOI] [PubMed] [Google Scholar]
- 30.Greenfield N, Fasman GD. Computed circular dichroism spectra for the evaluation of protein conformation. Biochemistry. 1969;8:4108–4116. doi: 10.1021/bi00838a031. [DOI] [PubMed] [Google Scholar]
- 31.Piotto M, Saudek V, Sklenar V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J Biomol NMR. 1992;2:661–665. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- 32.Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, Sykes BD. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J Biomol NMR. 1995;6:135–140. doi: 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
- 33.Delaglio F, Grzesiek G, Vuister G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:227–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 34.Johnson BA, Blevins RA. NMRView: a computer program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]