


Abstract
The phototriggered cleavage of chemical bonds has found numerous applications in biology, particularly in the field of gene sequencing through photoinduced DNA strand scission. However, only a small number of modified nucleosides that are able to cleave DNA at selected positions have been reported in the literature. Herein, we show that a new photoactivable deoxyadenosine analogue, 3-nitro-3-deaza-2′-deoxyadenosine (d(3-NiA)), was able to induce DNA backbone breakage upon irradiation (λ > 320 nm). The d(3-NiA) nucleoside was chemically incorporated at desired positions into 40-mer oligonucleotides as a phosphoramidite monomer and subsequent hybridization studies confirmed that the resulting modified duplexes display a behaviour that is close to that of the related natural sequence. Enzymatic action of the Klenow fragment exonuclease free revealed the preferential incorporation of dAMP opposite the 3-NiA base. On the other hand, incorporation of the analogous 3-NiA triphosphate to a primer revealed high enzyme efficiency and selectivity for insertion opposite thymine. Furthermore, only the enzymatically synthesized base pair 3-NiA:T was a substrate for further extension by the enzyme. All the hybridization and enzymatic data indicate that this new photoactivable 3-NiA triphosphate can be considered as a photochemically cleavable dATP analogue.
INTRODUCTION
Selective DNA fragmentation is undeniably a useful technique for the structural and dynamic analysis of nucleic acids. For instance, DNA sequencing using microarrays, which require preliminary DNA fragmentation into oligomers of 20–40 nt in length, is a very promising technology in the field of medical diagnosis. DNA strand scission can involve either native restriction enzymes, synthetic nucleases (e.g. metallic complexes) (1–5), or photocleaving agents (6). The advantages of photocleaving agents are numerous: the cleavage reactions can be performed in reagent-free medium, they allow the spatial and temporal control of the reactions, and reaction by-products are easily removed. If many DNA-photocleaving agents have been developed so far, only a very few of them are nucleoside analogues that can be incorporated within DNA strands (7–10). These reported nucleoside analogues typically rely on the photochemistry of the O-nitrobenzyl group, which is attached at one C-position of the sugar backbone (11–16). Unfortunately, they often behave as universal nucleobases and then are not specifically recognized by enzymes (17–19). Thus, there is a significant incentive to develop new photoactive nucleoside derivatives that display only minor structural modifications in comparison with their natural analogues in order to maintain their specific recognition properties.
Previously, we reported the synthesis of 7-nitro-indole nucleoside (d(7-Ni)) (20,21) (Figure 1). Photochemical and cleavable properties of DNA strands incorporating d(7-Ni) have been examined (22). Upon irradiation in aerated solution, a chemical rearrangement involving the nitro group occurs, affording the corresponding deoxyribonolactone. Subsequently, mild alkaline or thermal treatment leads to DNA strand cleavage through a β-elimination reaction (Figure 2). DNA polymerase recognition of d(7-Ni) incorporated either in a DNA matrix or to a primer as its corresponding triphosphate has also been studied (23). It was shown that the modified base was not specifically recognized by the enzyme.
Figure 1.

Structure of the deoxynucleosides derived from 7-nitroindole (d(7-Ni)) and 3-nitro-3-deaza-2′-deoxyadenosine (d(3-NiA)).
Figure 2.

Illumination of DNA containing the 3-nitro-3-deaza-2′-deoxyadenosine nucleoside results in the formation of the alkali labile deoxyribonolactone lesion followed by backbone cleavage in alkaline conditions via β and δ-elimination reactions.
We recently designed and synthesized a second-generation photocleavable nucleoside, 3-nitro-3-deaza-2′-deoxyadenosine (d(3-NiA)), which conserves the base-pairing ability of the natural adenine and was therefore expected to be specifically recognized by polymerase enzymes as an adenine (Figure 1). Previously, we described the synthesis of d(3-NiA) and its incorporation in oligonucleotides as a phosphoramidite monomer (24,25). Photocleavage of DNA containing the modified 3-NiA base was shown to be quantitative and rapid, both in single- and double-strand DNA (22).
In the present paper, we report the thermodynamic properties of oligonucleotide duplexes that incorporate d(3-NiA) in the middle of their sequences. The enzymatic recognition by a DNA polymerase Klenow fragment exonuclease free of the 3-NiA unnatural base has also been studied. The synthesis of the analogous d(3-NiA)TP triphosphate as well as its enzymatic incorporation into DNA are described.
MATERIALS AND METHODS
Materials
All reagents were of the highest quality commercially available. Oligonucleotides were labelled using [γ-32P]ATP (specific activity 3000 Ci/mmol) (PerkinElmer) and T4 polynucleotide kinase purchased from MBI fermentas. The Klenow fragment of Escherichia coli DNA polymerase I deficient in 3′→5′ proofreading exonuclease activity (KF exo-) was obtained from Euromedex. Nucleotide triphosphates (dNTPs) were purchased from MBI fermentas. Reagents used for automated synthesis were obtained from PE Biosystems.
Oligonucleotide synthesis
Natural or modified d(3-NiA) oligonucleotides were synthesized using standard solid-phase β-cyanoethylphosphoramidite method on a Expedite DNA synthesizer (Perseptive Biosystem 8900). The 3-nitro-3-deaza-2′-deoxyadenosine nucleoside phosphoramidite d(3-NiA) was prepared as previously reported (20,22). The 40-nt-long fragments were used as templates in enzymatic reactions. During the automated synthesis, the standard cycles for 1 μmol DNA synthesis were modified since double amounts of phosphoramidites were used for coupling steps and after this step also double amount of capping reagent was added. For incorporation of the modified nucleoside d(3-NiA), standard coupling time was extended for 10 min and quantity of capping reagent was multiplied by five. After automated synthesis the oligomers were detritylated on column, cleaved from support by conc. aqueous ammonia 30% for 1 h at room temperature, then deprotected by heating at 55°C for 24 h.
These oligomers were purified by electrophoresis on a 20% polyacrylamide gel in the presence of 7-M urea. The concentration of oligodeoxynucleotides was determined by UV. Oligonucleotide sequences used for experiments have been reported in Table 1.
Table 1.
Oligonucleotide sequences
| No. | Sequence (5′→3′) |
|---|---|
| 1 | CGCAC (3-NiA) CACGC |
| 2 | GCGTGAGTGCG |
| 3 | GCGTGTGTGCG |
| 4 | GCGTGGGTGCG |
| 5 | GCGTGCGTGCG |
| 6 | CGCACACACGC |
| 7 | CGCACGCACGC |
| 8 | CATGCTGATGAATTCAGGCCT (3-NiA) GAGGAATCGCTGGTACCG |
| 9 | CGGTACCAGCGATTCCTC |
| 10 | CATGCTGATGAATTCAGGCCTTGAGGAATCGCTGGTACCG |
| 11 | CATGCTGATGAATTCAGGCCTCGAGGAATCGCTGGTACCG |
| 12 | CATGCTGATGAATTCAGGCCTGGAGGAATCGCTGGTACCG |
| 13 | CATGCTGATGAATTCAGGCCTAGAGGAATCGCTGGTACCG |
| 14 | CATGCTGATGAATTCAGGCCATGAGGAATCGCTGGTACCG |
| 15 | CATGCTGATGAATTCAGGCCCTGAGGAATCGCTGGTACCG |
| 16 | CATGCTGATGAATTCAGGCCGTGAGGAATCGCTGGTACCG |
| 17 | CGGTACCAGCGATTCCTCA |
Melting temperature experiments
Experiments were performed on 11-bp DNA duplexes containing d(3-NiA) paired with dG, dC, dA or dT. Equimolar solutions (3 μM) of unmodified or d(3-NiA) modified oligonucleotides were mixed with their complementary strands in buffer consisting of 10 mM sodium phosphate/1 mM EDTA/100 mM NaCl adjusted to pH 7. UV absorption spectra (at 260 nm) and melting experiments were recorded using a CARY 400 Scan UV-Visible spectrophotometer (Varian) equipped with a Peltier thermoelectric Cary temperature controller (Varian). Before each melting experiment, samples were heated at 80°C for 5 min and cooled down slowly to ensure that the oligonucleotides were in the duplex state. The absorbance was monitored at 260 nm from 5°C to 80°C at a heating rate of 0.3°C per minute Data were recorded every 1 min. Melting temperature (Tm) was determined from the derivative method using the “Cary Win UV-bio” (version 2) application software. Error in the determination of Tm has been estimated as ± 0.6°C (average of three independent measurements).
Synthesis
1-[3′-O-acetyl-5′-O-(4,4-dimethoxytrityl)-2′-deoxy-β-d-erythro-pentofuranosyl]-7-nitro-4-(2,4,6-trimethylphenoxy)-1H-imidazo[4,5-c]pyridine: The compound 2 (200 mg, 0.28 mmol) was co-evaporated twice with dry pyridine (1 ml) and then dissolved in dry pyridine (1 ml). To this solution, acetic anhydride (1 ml) was added and the mixture was stirred overnight at room temperature. Solvents were removed by evaporation and by co-evaporation with toluene and the oily residue was purified by flash chromatography (elution with AcOEt/cyclohexane 40/60 to 50/50). Title compound was obtained with quantitative yield (212 mg). Rf (AcOEt/cyclohexane 1/1): 0.48. 1H NMR (200 MHz, CDCl3): δ = 8.78 (s, 1 H), 8.62 (s, 1 H), 7.44–7.21 (m, 9 H), 6.97 (s, 2 H), 6.88–6.75 (m, 5 H), 5.49 (m, 1 H), 4.34 (m, 1 H), 3.79 (s, 6 H), 3.49 (m, 2 H), 2.86-2.69 (m, 2 H), 2.33 (s, 3 H), 2.14 (s, 3 H), 2.12 (s, 6 H). MS (DCI, NH3/isobutane): 759.9 [M + H]+.
1-[3′-O-acetyl-2′-deoxy-β-d-erythro-pentofuranosyl]-7-nitro-4-(2,4,6-trimethylphenoxy)-1H-imidazo[4,5-c]pyridine (3): The 5′-O-DMT-nucleoside (200 mg; 0.26 mmol) was dissolved in dichloromethane (10 ml) and methanol (200 μl) was added. To this solution, trichloroacetic acid (400 μl, 30% solution in dichloromethane) was added and the mixture was stirred for 45 min at room temperature. The solution was then washed with NaHCO3 saturated aqueous solution (8 ml) and dried with MgSO4. After evaporation of solvents, the oily residue was purified by flash chromatography (elution with AcOEt/cyclohexane 70/30) to afford the compound 3 (97 mg, 82%). Rf (AcOEt/cyclohexane 1/1): 0.25. 1H NMR (300 MHz, CDCl3): δ = 8.92 (s, 1 H), 8.75 (s, 1 H), 6.95 (s, 2 H), 6.82 (t, J = 6.2 Hz, 1 H), 5.45 (m, 1 H), 4.23 (m, 1 H), 3.97 (m, 2 H), 2.88 (m, 1 H), 2.77 (m, 1 H), 2.67 (m, 1 H), 2.32 (s, 3 H), 2.14 (s, 3 H), 2.10 (s, 6 H). 13C NMR (75 MHz, CDCl3): δ = 170.8, 158.7, 147.6, 144.3, 140.9, 135.8, 132.2, 130.6, 130.3, 130.0, 129.7, 89.4, 86.2, 76.7, 61.9, 41.2, 21.0 (2C), 16.5. MS (DCI, NH3/isobutane): 457.0 [M + H]+. Anal. Calcd for C22H24N4O7 (456.5): C 57.89, H 5.30, N 12.27. Found: C 57.50, H 5.36, N 11.86.
5′-Triphosphate of 1-[3′-O-acetyl-2′-deoxy-β-d-erythro-pentofuranosyl]-7-nitro-4-(2,4,6-trimethylphenoxy)-1H-imidazo[4,5-c]pyridine (5): The 3′-acetyl nucleoside 3 (30 mg, 66 μmol) was dried by co-evaporation with dry pyridine (twice) and then dissolved under argon in dry pyridine (66 μl) and dry dioxane (198 μl). To this solution, 2-chloro-4H-1,3,2-benzodioxaphosphorin-4-one 4 (86 μL of 1 M solution freshly prepared in dry dioxane, 86 μmol) was added and stirred at room temperature under argon for 10 min. Then tributylammonium pyrophosphate (212 μl of 0.5 M solution in dry DMF, 106 μmol) and tributylamine (86 μl, 352 μmol) were added simultaneously and stirred for 10 min. The reaction mixture was then treated with iodine (1.3 ml of 1% solution in aqueous pyridine solution: pyridine/water = 98/2 v/v, 192 μmol) for 15 min and then the excess of iodine was decomposed by addition of sodium hydrogen sulphite aqueous solution (0.5%). After stirring for 10 min, solvents were removed in vacuo; then the residue was dissolved in 1 ml of water. The crude product was purified by reverse-phase silica gel chromatography (LiChroprep RP 18 silica gel, 40–63 μm, elution with 0–80% MeOH/H2O). To combined fractions containing 5, NaHCO3 aqueous solution (0.2 M, 500 μl) was added and solvents were removed by evaporation in vacuo to afford 41 mg (47%) of 5. Rf (propan-1-ol/H2O/NH3 11/2/7): 0.65. 1H NMR (300 MHz, D2O): δ = 9.01 (s, 1 H), 8.70 (s, 1 H), 7.13 (s, 2 H), 6.93 (t, J = 6.3 Hz, 1 H), 5.58 (m, 1 H), 4.60 (m, 1 H), 4.37 (m, 2 H), 2.92 (m, 2 H), 2.37 (s, 3 H), 2.23 (s, 3 H), 2.10 (s, 6 H). 31P NMR (75 MHz, D2O): δ = –21.74 (t, Pβ), –10.91 (d, Pα), –5.80 (d, Pγ). MS (ESI): 784.9 [M–3H+ + 4Na+]+, 762.9 [M – 2H+ + 3Na+]+. UV 84 μM in H2O : λmax (ε) = 322 (4265).
5′-Triphosphate of 4-amino-1-[2′-deoxy-β-d-erythro-pentofuranosyl]-7-nitro-1H-imidazo[4,5-c]pyridine (5′triphosphate of 3-deaza-3-nitro-2′-deoxyadenosine) (1): The triphosphate 5 (4.5 μmol) was dissolved in water (200 μl) and concentrated NH4OH (30%) solution (1.2 ml) was added and the solution was heated at 60°C for 2 h. After evaporation of solvents, the residue was purified by reverse-phase silica gel chromatography (LiChroprep RP 18 silica gel, 40–63 μm, elution with H2O) to afford 1 (3.46 μmol, 77%). Rf (propan-1-ol/H2O/NH3 11/2/7): 0.46. 1H NMR (500 MHz, D2O): δ = 8.76 (s, 1 H), 8.70 (s, 1 H), 6.88 (m, 1 H) 5.58 (m, 1 H), 4.77 (m, 1 H), 4.29 (m, 3 H), 2.70 (m, 2 H). 31P NMR (121 MHz, D2O): δ = −22.18 (t, Pβ), –10.98 (d, Pα), –7.31 (d, Pγ). MS (ESI): 645.8 [M–3H+ + 4Na+]+, 623.8 [M – 2H+ + 3Na+]+. UV 58 μM in H2O: λmax (ε) = 373 (8900).
Polymerization assays with KF exo-
General preparation of primer/template duplexes
5′-32P-labelled primer were annealed to template oligonucleotides (two equivalents) in 50 mM Tris (pH 7.5), 10 mM MgCl2, 5 mM 2-mercaptoethanol by heating at 70°C for 5 min, then cooling to room temperature over 2 h.
Primer extension by KF exo- opposite d(3-NiA) present in template
Reactions were conducted either on modified (X = d(3-NiA), sequence 8) or unmodified (X = A, sequence 13) template (100 nM) hybridized to 5′-end labelled primer (sequence 9, 50 nM). Primer extension reaction catalysed by KF exo- (0.1 U) was carried out in buffer hybridization in presence of single dNTP (20 μM) or all four dNTPs (20 μM each) for 20 min at 30°C in a final volume of 10 μl. Reactions were quenched by adding 30 μl of formamide loading buffer (95% formamide, 20 mM EDTA pH 8, 0.1% bromophenol blue, 0.1% xylene cyanol) and heating at 70°C for 5 min. Aliquots (2 μl) were loaded on a 20% denaturing PAGE.
Insertion kinetics by Klenow exo- opposite d(3-NiA) or natural base
The 5′end-labeled 18-mer primer (sequence 9, 50 nM) was annealed to modified template containing d(3-NiA) (sequence 8, 0.1 μM) or unmodified template (sequences 10–16, 0.1 μM). Duplexes were incubated with various amounts of dNTP (10–1000 μM) and KF exo- (0.005–0.07 U per sample depending on the template and dNTP) for 2 min at 30°C in order to obtain <20% primer elongation. The reactions were quenched by adding 20 μl formamide dye mixture and heating at 70°C for 5 min. The reaction mixtures were resolved on 20% denaturing PAGE and the radioactivity was quantified by means of an Imager (Typhoon 9400) and the ImageQuant program. The initial velocities were plotted versus [dNTP] and fit to the Michaelis–Menten equation (using the program Kaleida-graph, Synergy Software) to determine kcat and Km. The data presented are averages of three independent determinations.
d(3-NiA) Triphosphate incorporation opposite d(3-NiA) or A, T, G and C followed by next nucleotide incorporation or full-length primer extension
Reactions were conducted on d(3-NiA) modified template (sequence 8, 0.1 μM) or one of the unmodified templates (sequences 10–16, 0.1 μM) annealed to 5′-end labelled primer (sequence 9 or 17, 50 nM). For the first step, d(3-NiA)MP incorporation to the 3′ primer terminus was carried out using KF exo- (0.1 U) in presence of d (3-NiA)TP (500 μM or 1000 μM, depending on the template used) for 5 min at 30°C. The reaction was stopped by heating at 70°C for 5 min. The second step of the reaction consisted either in incorporation of the next nucleotide opposite to the base in 5′ position of the d(3-NiA) in the template or in full-length primer extension. This was performed by KF exo- (0.1 U) in presence of either one dNTP (500 μM) or the four dNTPs (500 μM each). Reactions were incubated at 30°C for 20 min, then quenched with formamide loading buffer (30 μl) and analysed by 20% denaturing PAGE.
RESULTS
Preparation of the 3-deaza-3-nitro-2′-deoxyadenosine triphosphate 1
The 3-deaza-3-nitro-2′-deoxyadenosine triphosphate 1 was prepared from the trimethylphenoxy precursor nucleoside 2. The 5′-DMT nucleoside 2 (see Figure 3) was first converted into the 3′-protected nucleoside 3, which was then directly converted to the triphosphate 5 using the Ludwig–Eckstein method (26). Thus, the compound 3 was treated successively (1) with 2-chloro-4H-1,3,2-benzo-dioxaphosphorin-4-one (4), (2) with tributylammonium pyrophosphate and (3) with iode-aqueous pyridine. Purification of the crude mixture was carried out by reverse-phase chromatography. The triphosphate 5 was obtained with a 47% overall yield and characterized by 1H and 31P NMR and by ES-MS spectrometry. Finally the arylether substituent was replaced by –NH2 and the 3′-O-acetyl group was deprotected to afford the triphosphate 1 when 5 was treated with aqueous NH3 at 60°C for 2 h. The triphosphate 1 was obtained with 77% yield and characterized by 1H and 31P NMR and by electrospray mass ES-MS) spectrometry.
Figure 3.

Synthesis of the unnatural triphosphate (d(3-NiA)TP).
Hybridization properties
We evaluate the ability of the 3-nitro-3-deaza-2′-deoxyadenosine to pair with another base into a DNA duplex. Thermodynamic parameters were determined for a series of 11-mer duplexes containing the modified d(3-NiA) nucleoside in the middle of the sequence (sequence 1) and facing one of the four natural bases (sequences 2–5). For comparison, stability of duplexes containing the natural base pairs A:T or G:C (sequence 6: sequence 3 and sequence 7: sequence 5, respectively) or the mismatched bases A:C (sequence 6: sequence 5) were determined. The sigmoidal curves α = f (T) obtained from the standard UV melting curves for the different duplexes exhibited melting cooperativity and were analysed to derive the Tm values and the Van’t Hoff transition enthalpy ΔH, entropy ΔS and free energy ΔG°. The resulting data listed in Table 2 showed higher stability for the 3-NiA:T unnatural base pair duplex (Tm = 59°C) compared to the other duplexes in which 3-NiA was opposite to C, G or A (Tm = 46°C, 53°C and 49°C, respectively). Very similar Tm values were determined respectively for the matched duplexes A:T and 3-NiA:T (Tm = 59 and 60°C, respectively) and for the mismatched duplexes A:C and 3-NiA:C (Tm = 46°C for the both). The stabilization energies determined respectively for the matched duplexes A:T and 3-NiA:T were also similar (ΔS° = 231 and 236 cal/mol/K, ΔH° = 85 and 86 kcal/mol, respectively and ΔG° = 16 kcal/mol for both). On the contrary, a decrease of the Tm (ΔTm = 8°C) was measured when the natural base pair C:G was replaced by the unnatural pair 3-NiA:G. This corresponds to a loss of free energy of 4 kcal/mol. All these results indicate that the 3-NiA unnatural base behaves like an adenine.
Table 2.
Melting temperatures and thermodynamics parameters of 11-mer duplexes containing natural or unnatural base pairs (X:Y) in the middle of the sequence
| Duplex | X | Y | Tm (°C) | ΔS° (cal/mol/K) | ΔH° (kcal/mol) | ΔG° (kcal/mol) |
|---|---|---|---|---|---|---|
| 3′ CGCACXCACGC 5′ | Unnatural base pair | |||||
| 5′ GCGTGYGTGCG 3′ | ||||||
| 3-NiA | A | 49 | 85 | 36 | 10 | |
| 3-NiA | T | 60 | 236 | 86 | 16 | |
| 3-NiA | G | 53 | 182 | 67 | 13 | |
| 3-NiA | C | 46 | 154 | 57 | 11 | |
| Natural base pair | ||||||
| A | T | 59 | 231 | 85 | 16 | |
| G | C | 61 | 240 | 89 | 17 | |
| Mismatch | ||||||
| A | C | 46 | 185 | 67 | 12 | |
| Experiments were run in triplicate. See ‘Materials and Methods’ section for details. | ||||||
Replication: enzymatic recognition of the unnatural base 3-NiA by KF exo-
In general, DNA polymerase efficiency is defined by its ability to undergo base-pair synthesis (i.e. incorporation of a triphosphate opposite a natural or unnatural base in the template) and secondly by the aptitude to extend a primer ending by a natural or unnatural base pair. The efficiency and the selectivity of unnatural DNA synthesis involving the d(3-NiA) nucleoside by KF exo- were examined both in the context of base-pair synthesis and base-pair extension.
Insertion of dNMP opposite d(3-NiA)
Duplex containing the 3-NiA nucleobase was evaluated as a substrate for the exonuclease-deficient Klenow fragment of Escherichia coli DNA polymerase I (KF exo-). Studies were investigated using a 40-mer modified template containing the unnatural bases 3-NiA in the middle of the sequence (sequence 8) hybridized to the 18-mer primer (sequence 9). For comparison, a template containing the natural base A (sequence 13) hybridized to the same 18-mer primer was also examined. The resulting duplexes were subjected to primer extension in the presence of one single dNTP (Figure 4). DNA synthesis on template containing the natural base (sequence 13) led to the expected incorporation of dTMP opposite A (data not shown). When using the 3-NiA containing strand as a template (sequence 8), dTMP was incorporated as expected opposite 3-NiA. dAMP and guanosine monophosphate (GMP) were also incorporated opposite the unnatural base with quite different efficiency. Insertion of dCMP could not be detected in these conditions (Figure 4, lane 3).
Figure 4.

Single nucleotide incorporation opposite 3-NiA. Reactions were performed on 40-mer templates containing the unnatural base: 3-NiA, sequence 8 (lanes 1–5; 100 nM) primed with 32P-labeled 18-mer (sequence 9, 50 nM) in the presence of one single dNTP (20 μM) and KF exo- (0,1 U). Reactions were incubated for 20 min at 30°C before processing as described in ‘Materials and Methods’ section.
Kinetic parameters for insertion of dNMP opposite the modified sites were determined. The initial velocities were measured and plotted versus [dNTP] and fitted to the Michaelis–Menten equation to determine the Km and kcat constants. The specificity constants kcat/Km were also calculated. The steady-state kinetic constants for single nucleotide incorporation are reported in Table 3.
Table 3.
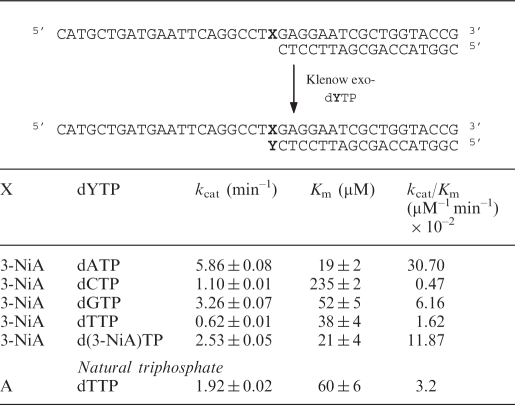
Steady-state kinetic parameters (kcat/Km) for incorporation by KF exo- of single nucleotides opposite the natural or unnatural bases present in the template (X = A or 3-NiA, respectively)
|
Opposite the modified 3-NiA nucleobase present in the template (sequence 8), dAMP was preferentially inserted. The kcat/Km value for dAMP incorporation was five times, 19 times and 65 times, respectively, higher than measured for dGMP, dTMP and dCMP incorporation. In addition the d(3-NiA) nucleoside was incorporated opposite 3-NiA in the template with a rather good efficiency since the specificity constants kcat/Km was only 2.6 times lower than for dAMP incorporation. Thus, the efficiency for nucleotide insertion opposite d(3-NiA) follows the order dAMP > d(3-NiA)MP > dGMP > dTMP > dCMP.
Full-length primer extension on DNA templates containing d(3-NiA)
Modified template containing the unnatural base 3-NiA (sequence 8) was annealed to the 18-mer primer (sequence 9) and subjected to extension in the presence of the four dNTPs. For comparison, measurements were also made by using a template containing A instead of 3-NiA (sequence 13, Figure 5). As expected KF exo- was able to fully extend the primer (sequence 9) annealed with the natural template (sequence 13, formation of 40-mer products, Figure 5, lane 2). Employing identical enzyme concentrations and dNTP quantities as used for the unmodified duplex, KF exo- synthesized full-length DNA starting from duplex containing d(3-NiA) in the template (Figure 5, lane 4).The additional major band seen in lane 4 results from single nucleotide extension of the primer through incorporation of a native triphosphate opposite the modified base. The accumulation of this intermediate suggests that a pause occurs after the first incorporation.
Figure 5.

Full-length extension of primer hybridized to modified or unmodified templates. Reactions were carried out at 30°C for 20 min using either a natural template (X = A, sequence 13, lanes 1, 2) or a modified template containing 3-NiA (X = 3-NiA, sequence 8, lanes 3 and 4) at a 100-nM concentration, primed with 32P-labelled 18-mer, sequence 9 (50 nM) in the presence of the four dNTPs (at a final concentration of 20 μM each). Reactions were catalysed by KF exo- (0.1 U).
Incorporation of d(3-NiA) triphosphate opposite the four natural bases A, C, G or T
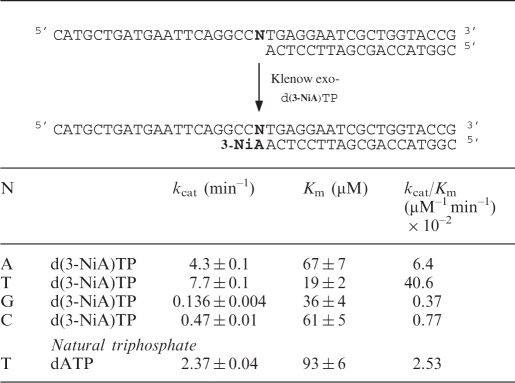
Efficiency of insertion of the triphosphate of d(3-NiA) against each natural base in the template has been measured. Experiments were run using the 19-mer primer (sequence 17) successively hybridized to the four templates (sequences 10, 14–16) varying by the nature of the base N at the initiation site of the polymerization. The kinetic parameters for insertion of the unnatural triphosphate opposite each natural base are reported in Table 4. The triphosphate of d(3-NiA) was preferentially incorporated opposite to thymine in the template (kcat/Km = 40.6 × 10–2 min−1 mM–1). The kcat/Km value was 6.3 times, 52 times and 110 times, respectively, higher than measured for incorporation opposite natural bases A, C and G. Furthermore, incorporation of d(3-NiA)TP opposite natural thymine was 16 times higher than incorporation of natural dATP opposite thymine (kcat/Km = 2.53 × 10–2 min–1 mM–1).
Table 4.
Efficiency of KF exo- incorporation of unnatural triphospate (d(3-NiA)TP) opposite natural bases N in the template
 |
Extension of the primer terminated by d(3-NiA)
The ability of KF exo- to extend a primer terminated by the unnatural 3-NiA base opposite the different natural bases N in the template was studied (Figure 6). Reactions were conducted on the 18-mer primer (sequence 9) annealed to the 40-mer templates in which base N19 was successively a thymine (sequence 10), an adenine (sequence 13), a cytosine (sequence 11) or a guanine (sequence 12). In a first step, primer extension was run in the presence of the triphosphate d(3-NiA)TP (Figure 6, lanes 2, 7, 12 and 17). Incorporation of one d(3-NiA) nucleoside was observed for all duplexes, permitting to obtain all possible unnatural base pairs 3-NiA: N. In a second step polymerization was continued on these extended primers in the presence either of dATP (the next complementary base in the template) (Figure 6, lanes 3, 8, 13 and 18) or in the presence of the four dNTPs (Figure 6, lanes 4, 9, 14 and 19). No extension could be observed for the duplexes in which d(3-NiA) was facing adenine, cytosine or guanine, neither in the presence of dATP (Figure 6, lanes 8, 13 and 18) nor in the presence of the mixed dNTPs (Figure 6, lanes 9, 14 and 19). Only the 3-NiA:T base pair was substrate for extension of the modified primer. In this case, formation of a full-length product was observed in the presence of the four natural triphosphates (Figure 6, lane 4). Incorporation of the modified monophosphate (d(3-NiA)MP) at the 3′ primer terminus has been proved for all extension reactions. Indeed, primers extended by one or two d(3-NiA)MP [products of lanes 7, 12, and 17 for one incorporation and lane 2 for extension by two d(3-NiA)MP; Figure 6] were first irradiated at λ > 320 nm for 60 min, in order to convert the modified momophosphate into deoxyribonolactone. Secondly, cleavage at the lactone site by piperidine treatment produced the initial 18-mer primer (Figure 6, lanes 5, 10, 15 and 20).
Figure 6.

Chain extension reaction catalysed by KF exo- in the presence of the modified triphosphate d(3-NiA)TP. Experiments were performed with different natural templates (100 nM) varying by the nature of the base N19 (N = T, sequence 3; N = C, sequence 4; N = G, sequence 5; N = A, sequence 6) hybridized to 18-mer primer, sequence 9 (50 nM). In a first step, chain extension reactions were conducted in the presence of d(3-NiA)TP (1 mM) and KF exo- (0.1 U) for 5 min at 30°C (lanes 2, 7, 12 and 17). In the second step, reactions were carried out using KF exo- (0.1 U) in the presence of dATP (lanes 3, 8, 13, 18) or in the presence of a mixture of the four dNTPs (lanes 4, 9, 14, 19) for 20 min at 30°C. One part of products of reactions corresponding to lanes 3, 8, 13 and 18 were illuminated at λ > 320 nm for 20 min and then treated with NaOH 1 M for 20 min at 70°C (lanes 5, 10, 15 and 20).
Influence of the downstream base on the extension of a primer terminated by the unnatural 3-NiA base
In this experiment, we investigated the influence of the downstream template base (N20) on the extension of the primer terminated by the 3-NiA unnatural base. Experiments were conducted on the same 18-mer primer (sequence 9) hybridized to different 40-mer templates (sequences 10, 14–16) varying only by the nature of base N20 (Figure 7). The primers were first elongated by one 3-NiA base as described in the previous experiment (Figure 7, lanes 2, 6, 10 and 14). It should be noted that the 18-mer primer annealed to template in which N20 was a guanine was only partially converted in the conditions used. In a second step, elongation was continued either in the presence of the triphosphate complementary to the next base in the template (Figure 7, dATP for T20: lane 3, dTTP for A20: lane 7, dGTP for C20: lane 11, dCTP for G20: lane 15) or in the presence of the four dNTPs (Figure 7, lanes 4, 8, 12 and 16). All the templates allowed elongation of the primer by one 3-NiA. For each duplex, primers terminated by 3-NiA were extended in the presence of a unique triphosphate (Figure 7, lanes 3, 7, 11 and 15). However, primer extension in the presence of dCTP only led to small amounts of reaction product. Incorporation of three guanines was observed for the duplex in which the template base N was a cytosine. Full-length extension (40-mer) was observed in the presence of the mixture of the four dNTPs for all duplexes (Figure 7, lanes 4, 8, 12 and 16).
Figure 7.

Effect of the downstream template base N20 on chain extension reaction catalysed by KF exo-. (i) 3-NiA Incorporation opposite T in the template. (ii) Incorporation of the complementary correct nucleotide opposite varying nucleotide in the template. Experiments were performed with different templates (100 nM) varying by the nature of the base N20 (N = T, sequence 10; N = A, sequence 14; N = C, sequence 15; N = G, sequence 16) hybridized to 18-mer primer (sequence 2, 50 nM). Addition of one d(3-NiA) to the primer was carried out using KF exo- (0.1 U) in presence of d(3-NiA)TP (500 μM) for 5 min at 30°C (lanes 2, 6, 10 and 14). The second step of reaction was performed using KF exo- (0.1 U) in the presence of one single dNTP (lanes 3, 7, 11 and 15) or in the presence of the mixture of the four dNTPs (lanes 4, 8, 12 and 16) for 20 min at 30°C.
DISCUSSION
In this article, we first studied the hybridization properties of DNA duplexes containing the 3-NiA unnatural base opposite each native base. Comparison of the thermodynamic parameters determined for the unnatural 3-NiA: N (N = A, C, G or T) base-pair duplexes and for the natural matched or mismatched base-pair duplexes clearly showed that 3-NiA behaves like the deoxyadenosine nucleoside.
We then investigated recognition by KF exo- of the unnatural nucleobase 3-NiA in the DNA template. The incorporation frequency opposite the unnatural base followed the order: dAMP>dGMP>dTMP>dCMP. It appears that d(3-NiA) was recognized by KF exo- rather like a pyrimidine since insertion of purines dAMP or dGMP opposite the unnatural base was favoured over dTMP and dCMP. The small preference for dAMP incorporation suggests that similarly to non-informative lesions, such as abasic sites (2′-deoxyribose, dR or 2′-deoxyribonolactone, dL), 3-NiA might obey the ‘A-rule’. Indeed, when the modified nucleosides are unable to fit within the polymerase active site, the enzyme inserts in most cases an adenine opposite the non-instructional nucleobase (27). Although 3-NiA is very similar to the natural adenine, we assume that the presence of the nitro group could disturb the polymerase binding and subsequently prevents its conformational change, which is necessary for the matched nucleotide insertion. Consequently, 3-NiA cannot be considered as a true photocleavable deoxyadenosine mimic. In addition, these primer extension experiments showed that the presence of d(3-NiA) in the template led to a decrease of the polymerase activity, although full-length products were obtained.
We also examined the incorporation of the d(3-NiA) triphosphate opposite each native base in the template. The measured kinetic parameters were significantly different between the four natural bases. Indeed, the modified d(3-NiA) triphosphate was preferentially inserted opposite thymine. The insertion frequency opposite each natural base followed the order T >> A > C ∼ G. If we refer to other studies in which the fidelity of KF exo- has been checked (28), it appeared that d(3-NiA)TP is inserted with a selectivity that is nearly as high as that those observed for dATP. In addition, the d(3-NiA) triphosphate was also efficiently inserted opposite itself in the template. This observation is in good agreement with a previous study from Kool, who reported that hydrophobic aromatic nucleosides are preferentially incorporated opposite themselves (29).
Extension of primers terminated by the unnatural base 3-NiA has been also monitored. We found that 3-NiA behaves as chain terminator when facing adenine, guanine or cytosine in the template. When facing thymine, the 3-NiA primer was elongated by one adenine in the presence of dAMP and full-length elongation occurred in the presence of the four native base triphosphates, indicating that only the terminal 3-NiA:T pair could be recognized by KF exo-. This elongation was not dependent on the nature of the base downstream to thymine in the template.
Several models have been proposed in order to explain how most DNA polymerases maintain an incredible degree of substrate fidelity during DNA synthesis. Replication fidelity has historically been interpreted with respect to the formation of proper hydrogen bonds between the nascent base pairs. According to this model, the incoming dNTP directly pairs opposite to its complementary base via direct hydrogen bonding, the enzyme catalysing the formation of the phosphodiester bond only if the two nucleobases are aligned correctly. However, several studies also indicate that hydrogen bonding should be not the unique factor involved in nucleotide selection by polymerase during DNA synthesis. It was proposed that solvatation, base stacking and steric (size and shape) factors should play a role in the selection process as well. In particular, Berdis et al.(30,31) highlighted the significant contribution of the stacking interactions between the π electrons-rich residues of both aromatic amino acids in the catalytic site of the enzyme and the incoming nucleobase. These non-covalent interactions were shown to facilitate the nucleotide binding and the conformational change step preceding the phosphoryl transfer during DNA synthesis, both steps being the most significant contributors towards catalysis fidelity. A complete kinetic scheme describing the polymerization of dNTP by the Klenow fragment of DNA polymerase I has been established by Kuchta (32).
With regards to the results obtained by Berdis, Kool and Kuchta, we suggest the hypothetic mechanism described explains the efficiency and selectivity of the d(3-NiA)TP incorporation by KF exo- opposite thymine in the template. The nucleobase moiety of the non-natural triphosphate d(3-NiA)TP contains an extended π-electron surface area (NO2 group) with reference to the natural dATP. This enhanced π electron surface area could be responsible of higher binding stabilization of the incoming nucleobase through π– π stacking interactions with the aromatic amino acids in the dNTP-binding site of the enzyme. Upon binding of d(3-NiA)TP, the polymerase/DNA complex then undergoes a conformational change to further align the incoming non-natural triphosphate into a favourable geometrical arrangement for subsequent phosphoryl transfer. Additionally, the hydrogen bonds formation in the nascent base pair, similar to which existing in the natural T:A Watson–Crick base pair, could facilitate this arrangement and consequently play a significant role in the selective incorporation of d(3-NiA)TP by KF exo- opposite thymine in the template. In this model, we propose that shape complementary, hydrogen bonding and an extended π-stacking all contribute to the efficiency and the selectivity of the d(3-NiA)TP insertion by KF exo- opposite thymine in the template.
In conclusion, the 3-NiA nucleoside was designed by analogy with the previously reported 7-nitroindole nucleoside to be a photocleavable precursor of the highly labile deoxyribonolactone lesion. In contrast with 7-Ni, the 3-NiA nucleoside is potentially able to establish two hydrogen bonds with thymine and was thus expected to be specifically recognized by enzymes as an adenine. Enzymatic experiments involving the 3-NiA nucleotide incorporated in a template indicated that the modified base was not recognized as an adenine. However, the selective recognition by KF exo- of the d(3-NiA)TP triphosphate as a dATP analogue has been demonstrated. Compared to the first-generation 7-Ni nucleoside, which was a universal nucleoside analogue, the 3-NiA was recognized by the DNA polymerase as an adenosine. In addition, hybridization properties of oligonucleotides that incorporate (3-NiA) are very similar to the corresponding native biopolymers. Further work also showed that d(3-NiA) is actually a highly interesting motif to generate DNA backbone cleavage at pre-selected positions. From these results, we propose that the d(3-Ni-A)TP may be a useful new fragmentation tool for DNA sequencing.
ACKNOWLEDGEMENTS
We are grateful to Dr. Jean-François Constant and Dr. Laurent Vial for critical reading of the manuscript. Funding for open access charge: Université Joseph Fourier.
Conflict of interest statement. None declared.
REFERENCES
- 1.Sigman DS. Chemical nucleases. Biochemistry. 1990;29:9097–9105. doi: 10.1021/bi00491a001. [DOI] [PubMed] [Google Scholar]
- 2.Pitié M, Bernardou J, Meunier B. Oxidation at carbon-1′ of DNA deoxyriboses by the Mn-TMPyP/KHSO5 system results from a cytochrome P-450-type hydroxylation reaction. J. Am. Chem. Soc. 1995;117:2935–2936. [Google Scholar]
- 3.Pratviel G, Bernadou J, Meunier B. Carbon-hydrogen bonds of DNA sugar units as targets for chemical nucleases and drugs. Angew. Chem. Int. Ed. Engl. 1995;34:746–769. [Google Scholar]
- 4.Sigman DS, Chen CH. Chemical nucleases: new reagents in molecular biology. Annu. Rev. Biochem. 1990;59:207–236. doi: 10.1146/annurev.bi.59.070190.001231. [DOI] [PubMed] [Google Scholar]
- 5.Pratviel G, Bernadou J, Meunier B. DNA and RNA cleavage by metal complexes. Adv. Inorg. Chem. 1998;45:251–312. [Google Scholar]
- 6.Armitage B. Photocleavage of nucleic acids. Chem. Rev. 1998;98:1171–1200. doi: 10.1021/cr960428+. [DOI] [PubMed] [Google Scholar]
- 7.Tronche C, Goodman BK, Greenberg MM. DNA damage induced via independent generation of the radical resulting from formal hydrogen atom abstraction from the C1′-position of a nucleotide. Chem. Biol. 1998;5:263–271. [PubMed] [Google Scholar]
- 8.Abramova TV, Silnikov VN. 4-Aminomethyl-3-Nitrobenzoic acid-A photocleavable linker for oligonucleotides containing combinatorial libraries. Nucleosides Nucleotides Nucleic Acids. 2005;24:1333–1343. doi: 10.1080/15257770500230509. [DOI] [PubMed] [Google Scholar]
- 9.Ruparel H, Bi L, Li Z, Bai X, Kim DH, Turro NJ, Ju J. Design and synthesis of 3′-O-allyl photocleavable fluorescent nucleotide as a reversible terminator for DNA sequencing by synthesis. Proc. Natl Acad. Sci. USA. 2005;102:5932–5937. doi: 10.1073/pnas.0501962102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.WU W, Stupi BP, Litosh VA, Mansouri D, Farley D, Morris S, Metzker S, Metzker ML. Termination of DNA synthesis by N6-alkylated, not 3′-O-alkylated, photocleavable 2′-deoxyadenosine triphosphates. Nucleic Acids Res. 2007;35:6339–6349. doi: 10.1093/nar/gkm689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ordoukhanian P, Taylor J-S. Design and synthesis of a versatile photocleavable DNA building block. Application to phototriggered hybridization. J. Am. Chem. Soc. 1995;117:9570–9571. [Google Scholar]
- 12.Zhang K, Taylor J-S. A caged ligatable DNA strand break. J. Am. Chem. Soc. 1999;121:11579–11580. [Google Scholar]
- 13.Ordoukhanian P, Taylor J-S. Caged single and double strand breaks. Bioconjug. Chem. 2000;11:94–103. doi: 10.1021/bc9900993. [DOI] [PubMed] [Google Scholar]
- 14.Zhang K, Taylor J-S. Phototriggered formation and repair of DNA contaianing a site specific single strand break of the type produced by ionizing radiation or AP-lyase activity. Biochemistry. 2001;40:153–159. doi: 10.1021/bi001781j. [DOI] [PubMed] [Google Scholar]
- 15.Lenox HJ, McCoy CP, Sheppard TL. Site-specific generation of deoxyribonolactone lesions in DNA oligonucleotides. Org. Lett. 2001;3:2415–2418. doi: 10.1021/ol016255e. [DOI] [PubMed] [Google Scholar]
- 16.Dussy A, Meyer C, Quennet E, Bickle TA, Giese B. New light-sensitive nucleosides for caged DNA strand breaks. Chem. Bio. Chem. 2002;3:54–60. doi: 10.1002/1439-7633(20020104)3:1<54::AID-CBIC54>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 17.Loakes D, Brown DM, Linde S, Hill F. 3-Nitropyrrole and 5-nitrindole as universal bases in primers for DNA sequencing and PCR. Nucleic Acids Res. 1995;23:2361–2366. doi: 10.1093/nar/23.13.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loakes D, Hill F, Linde S, Brown DM. Nitroindoles as universal bases. Nucleosides Nucleotides. 1995;14:1001–1003. [Google Scholar]
- 19.Pirrung MC, Zhao X, Harris SV. A universal, photocleavable DNA base: nitropiperonyl 2′-deoxyriboside. J. Org. Chem. 2001;66:2067–2071. doi: 10.1021/jo001594r. [DOI] [PubMed] [Google Scholar]
- 20.Kotera M, Bourdat A-G, Defrancq E, Lhomme J. A highly efficient synthesis of oligodeoxyribonucleotides containing the 2′-deoxyribonolactone lesion. J. Am. Chem. Soc. 1998;120:11810–11811. [Google Scholar]
- 21.Roupioz Y, Lhomme J, Kotera M. Chemistry of the 2-deoxyribonolactone lesion in oligonucleotides: cleavage kinetics and products analysis. J. Am. Chem. Soc. 2002:9129–9135. doi: 10.1021/ja025688p. [DOI] [PubMed] [Google Scholar]
- 22.Kotera M, Roupioz Y, Defrancq E, Bourdat A-G, Garcia J, Coulombeau C, Lhomme J. The 7-nitroindole nucleoside as a photochemical precursor of 2′-deoxyribonolactone: access to DNA fragments containing this oxydative abasic lesion. Chem. Eur. J. 2000;6:4163–4169. doi: 10.1002/1521-3765(20001117)6:22<4163::aid-chem4163>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 23.Crey-Desbiolles C, Berthet N, Kotera M, Dumy P. Hybridization properties and enzymatic replication of oligonucleotides containing the photocleavable 7-nitro-indole base analog. Nucleic Acids Res. 2005;33:1532–1543. doi: 10.1093/nar/gki293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crey-Desbiolles C, Lhomme J, Dumy P, Kotera M. 3-Nitro-3-deaza-2′-deoxyadenosine as a versatile photocleavable 2′-deoxyadenosine mimic. J. Am. Chem. Soc. 2004;126:9532–9533. doi: 10.1021/ja047976m. [DOI] [PubMed] [Google Scholar]
- 25.Crey-Desbiolles C, Kotera M. Synthesis of 3-deaza-3-nitro-2′-deoxyadenosine. Bioorg. Med. Chem. 2006;14:1935–1941. doi: 10.1016/j.bmc.2005.10.040. [DOI] [PubMed] [Google Scholar]
- 26.Ludwig J, Eckstein FJ. Rapid and efficient synthesis of nucleoside 5′-0-(1-thiotriphosphates), 5′-triphosphates and 2′,3′-cyclophosphorothioates Using 2-chloro-4H- 1,3,2-benzodioxaphosphorin-4-one. J. Org. Chem. 1989;54:631–635. [Google Scholar]
- 27.Taylor J-S. New structural and mechanistic insight into the A-rule and the instructional and non-instructional behavior of DNA photoproducts and other lesions. Mutat. Res. 2002;510:55–70. doi: 10.1016/s0027-5107(02)00252-x. [DOI] [PubMed] [Google Scholar]
- 28.Caroll SS, Cowart M, Benkovic SJ. A mutant of DNA polymerase I (Klenow Fragment) with reduced fidelity. Biochemistry. 1991;30:804–813. doi: 10.1021/bi00217a034. [DOI] [PubMed] [Google Scholar]
- 29.Kool ET, Morales JC, Guckian KM. Mimicking the structure and function of DNA: insights into DNA stability and replication. Angew. Chem. Int. Ed. Engl. 2000;39:990–1009. doi: 10.1002/(sici)1521-3773(20000317)39:6<990::aid-anie990>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 30.Zhang K, Lee I, Berdis AJ. The use of nonnatural nucleotides to probe the contributions of shape complementarity and π-electron surface area during DNA polymerization. Biochemistry. 2005;44:13101–13110. doi: 10.1021/bi050585f. [DOI] [PubMed] [Google Scholar]
- 31.Sherriff A, Motea E, Lee I, Berdis AJ. Mechanism and dynamics of translesion DNA synthesis catalyzed by the Escherichia coli Klenow Fragment. Biochemistry. 2008;47:8527–8537. doi: 10.1021/bi800324r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuchta RD, Mizrahi V, Benkovic PA, Johnson KA, Benkovic SJ. Kinetic mechanism of DNA polymerase I (Klenow) Biochemistry. 1987;26:8410–8417. doi: 10.1021/bi00399a057. [DOI] [PubMed] [Google Scholar]