

Abstract
The Fragile Histidine Triad gene (human FHIT, mouse Fhit) has been shown to act as a tumor suppressor gene. Nit1 and Fhit form a fusion protein, encoded by the NitFhit gene in flies and worms, suggesting that mammalian Nit1 and Fhit proteins, which are encoded by genes on different chromosomes in mammals, may function in the same signal pathway(s). A previous study showed that Nit1 deficiency in knockout mice confers a cancer prone phenotype, as does Fhit deficiency. We have now assessed the tumor susceptibility of Fhit−/−Nit1−/− mice and observed that double knockout mice develop more spontaneous and carcinogen-induced tumors than Fhit−/− mice, suggesting that the extent of tumor susceptibility due to Nit1 and Fhit deficiency is additive, and that Nit1 and Fhit affect distinct signal pathways in mammals. Nit1, like Fhit, is present in cytoplasm and mitochondria but not nuclei. Because Fhit deficiency affects responses to replicative and oxidative stress, we sought evidence for Nit1 function in response to such stresses in tissues and cultured cells: when treated with hydroxyurea, the normal kidney-derived double-deficient cells appear not to activate the pChk2 pathway and when treated with H2O2, show little evidence of DNA damage, compared with wild type and Fhit−/− cells. The relevance of Nit1 deficiency to human cancers was examined in human esophageal cancer tissues, and loss of Nit1 expression was observed in 48% of esophageal adenocarcinomas.
Keywords: Nit1, Fhit, NMBA tumor induction, Tumor suppressor
Introduction
Galperin and Koonin (2004) proposed prioritizing ‘conserved hypothetical’ proteins for characterization, based on criteria such as wide phyletic spread, information about protein structure and expression. Authors gave a ‘top 10’ list of attractive targets, for which biochemical activity but not biological function could be predicted, with the expectation that characterization of these conserved proteins would reveal new aspects of biology. The fourth entry in the ‘top 10’ table was Nit1, a gene conserved from bacteria, through yeast and plants to insects, invertebrates, vertebrates and mammals; it is a predicted amidase in mammals and Nit1 mRNA is expressed in most mammalian tissues, though it is not an essential gene in mice (Semba et al., 2006). Our interest in Nit1 began with the finding that in flies and worms, it is fused to the N-terminus of the Fhit protein (Pekarsky et al., 1998). Fhit proteins are encoded by fragile genes, located at common chromosome fragile sites, in mouse and human genomes (Matsuyama et al., 2003). We had speculated that, due to the ‘Rosetta stone’ hypothesis, ie, genes fused in one organism are likely to be involved in the same signal or metabolic pathway in organisms in which they map to separate locations (Marcotte et al., 1999), Fhit and Nit1 would function in the same pathway in mammals. We had also speculated that Nit1, an enzyme of unknown function in mouse and man, might physically interact with Fhit, though physical interaction between Fhit and Nit1 or Nit2, another member of the nitrilase family (Pace and Brenner, 2001), has not been confirmed in co-immunoprecipitation experiments (Lin et al., 2007).
Fhit expression is reduced or absent in many human cancers (Huebner and Croce, 2003) and over-expression of Fhit in cancer cells suppresses cell growth and induces apoptosis. Fhit-deficient mice are more susceptible to carcinogen induction of tumors than wild type mice (Fong et al., 2000), and Fhit gene therapy inhibits tumor induction and progression (Ishii et al., 2001). Nit1 is a member of the nitrilase superfamily, which includes 13 branches (Pace and Brenner, 2001). Members of the nitrilase family play important roles in plant and bacteria by catalyzing the reactions involving amides, amines, nitriles and other nitrogen containing compounds. Little is known about the function of mammalian Nit1 and its substrates. But very recently, the substrate for Nit2, another predicted amidase of the Nitrilase branch 10 that shares 55% homology (Lin et al., 2007) with Nit1, has been identified (Cooper et al., 2009). Although the substrate specificities of Nit1 and Nit2 were unknown, they were assumed to be amidases based on sequence homology to known amidases and to the presence of a Cys-Glu-Lys triad in the putative active site (Barglow et al., 2008). Nit1 knockout in mouse tissues/cells results in increased cell proliferation, enhanced survival of cells exposed to DNA-damaging agents and an increased incidence of NMBA-induced tumors (Semba et al., 2006). Conversely, Nit1 over-expression leads to decreased cancer cell viability and increased caspase-3-dependent apoptosis, suggesting that Nit1 is a tumor suppressor that stimulates apoptosis in cancer cells. Evidence that Nit2 is also a tumor suppressor has been reported (Lin et al., 2007). Cooper et al identified Nit2 as ω-amidase that catalyzes the deamidation of α-ketoglutaramate (αKGM) and α-ketosuccinamate (αKSM) to α-ketoglutarate and oxaloacetate, respectively (Cooper et al., 2009). αKGM and αKSM are generated in vivo by transamination of glutamine and asparagine, respectively. ω-Amidase is important in nitrogen and sulfur metabolism. The natural substrates of Nit1 remain unknown.
To investigate roles for Nit1 in Fhit pathways, we generated Nit1 knockout mice and observed that Nit1 absence increases susceptibility to forestomach tumor induction, as does Fhit loss, (Semba et al., 2006). Nit1-deficient kidney cells established from Nit1 knockout (Nit1−/−) mice are resistant to UV induced- and Nit1 over-expression-induced apoptosis in normal and cancer cells (Semba et al., 2006), again similarly to Fhit-deficient cells.
The aim of the current study was to compare the tumor susceptibility of Fhit−/− mice with that of Fhit−/−Nit1−/− double knockout (DKO) mice after oral delivery of N–nitrosomethylbenzylamine (NMBA). We anticipated that the tumor incidence would be similar between these two mouse strains if Fhit and Nit1 function in the same pathway(s). We observed that DKO mice develop significantly more spontaneous and NMBA-induced tumors than Fhit−/− mice, suggesting that Fhit and Nit1 affect additive tumor suppressor pathways.
MATERIALS AND METHODS
Mouse breeding, genotyping and NMBA treatment
Fhit−/− and Nit1−/− mice were generated as previously described (Fong et al., 2000; Semba et al., 2006) and maintained on a mixed 129/SvJ X C57BL/6J background. Fhit−/− mice were crossed with Nit1−/− mice; F1 generation mice were inter-crossed and progeny genotyped by PCR amplification, using specific primers and mouse tail DNA templates, as illustrated in Fig 1A. Genotyping to identify wild type (wt) and knockout Fhit alleles was performed as described previously (Fong et al., 2000). Genotyping primers for wt and knockout Nit1 alleles were reported previously (Semba et al., 2006).
Figure 1. Identification and characterization of the Fhit−/−Nit1−/− double knockout mouse strain.
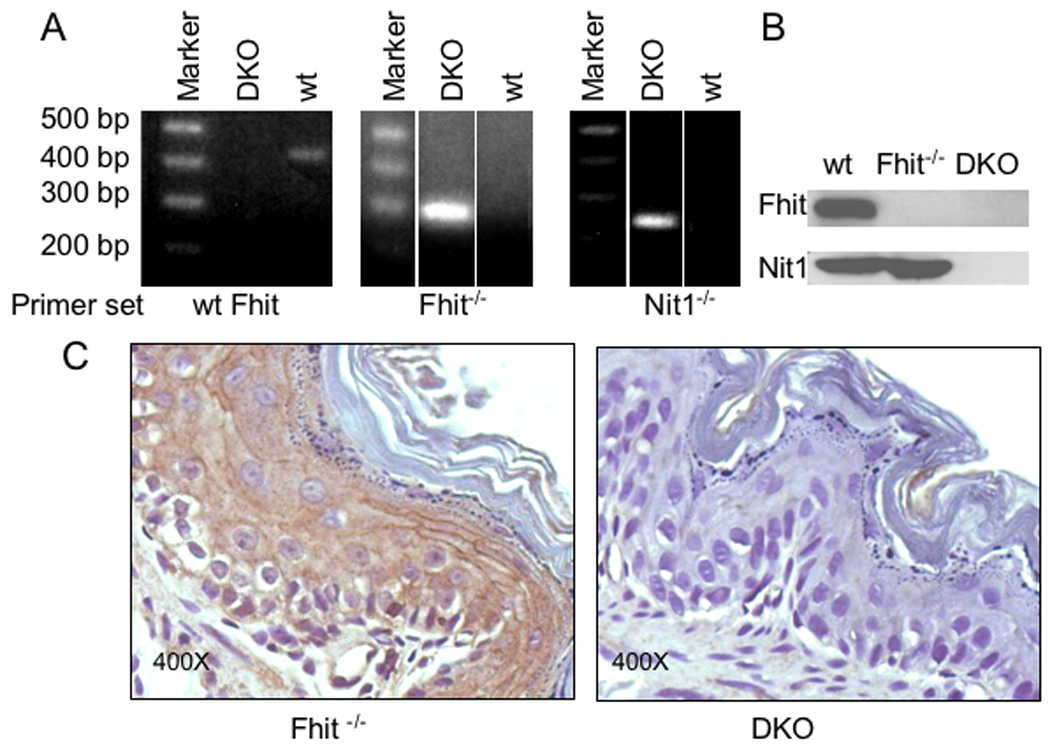
A. PCR genotyping of Fhit and Nit1 knock out mice, using DNA isolated from tail clips of pre-weanling mice. The left two panels show PCR genotyping results for the Fhit amplicon of ~400 bp for wt and ~270 bp for the KO allele. The right panel shows the Nit1 KO allele of 220 bp. PCR was performed with primers specific for wt and knockout alleles. B. Western blots for cell lysates of wt, Fhit−/− and DKO cells with rabbit anti-Fhit and anti-Nit1 sera confirmed absence of expression of Fhit and Nit1 in DKO cells and of Fhit in Fhit−/− cells. C. Immunohistochemical staining of Nit1 in both Fhit−/− and DKO mouse tissues. There is no expression of Nit1 in forestomach tissue from DKO mice.
NMBA was purchased from Midwest Research Institute (Kansas City, MO). A single dose of NMBA (2 mg/kg body weight) was administered by oral gavage to Fhit−/− and DKO mice. Untreated littermates served as controls. Mice were observed at least once per week and sacrificed 15 or 25 weeks post treatment to determine tumor incidence in esophagus, forestomach, glandular stomach, liver, kidney, intestine and spleen. Mouse studies were performed following protocols approved by The Ohio State University Institutional Animal Care and Use Committee.
At autopsy, esophagus and whole stomach were removed and opened longitudinally. Liver, spleen, kidney and intestine were inspected for tumors. The number of visible tumors in esophagus and forestomach was recorded. The incidence of spontaneous lesions in untreated aged DKO mice was similarly determined by visual inspection of internal organs at necropsy, and by histopathological examination of fixed, hematoxylin and eosin (H&E) stained sections of kidney, liver and gastrointestinal tract tissues by a pathologist (WF); cohorts of mice were examined at ages from 16 to 26 months.
Histopathological analysis
Tissues were fixed in buffered formalin, processed and embedded in paraffin. Esophagus and forestomach sections were stained with H&E for histological assessment and unstained sections were placed on treated slides for immunohistochemical analyses. Papillomas, dysplasias and cancers were enumerated in esophagus and forestomach sections by a pathologist (HO).
Establishment of kidney cell lines and hydroxyurea treatment
Establishment of wt and Fhit−/− kidney cells was described previously (Ottey et al., 2004). DKO kidney cells were established as follows: kidneys of baby mice were dissected and cultured with minimal essential medium (Sigma) with 20% fetal bovine serum and 100 µg/ml gentamycin (Sigma) for the first 3 generations. After that, cells were subcultured in the same medium with 10% fetal bovine serum. Hydroxyurea (HU, Sigma) stock solution at 200 mM was prepared immediately before treatment. Wt, Fhit−/− and DKO kidney cells were treated with 2 mM HU for various time periods as indicated.
Immunoblot analysis
Cells were lysed in RIPA buffer (25 mM Tris•HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS). Samples were run on SDS-PAGE Ready Gel Precast Gels (Bio-Rad, Hercules, CA) and protein was transferred to nitrocellulose membrane. Immunoblots were performed with rabbit anti-Fhit (1:5000) (Fong et al., 2000), rabbit anti-Nit1 (1:2000) (Semba et al., 2006), mouse anti-cyclin D1 (1:500, SC-2004, Santa Cruz Bio., Santa Cruz, CA), mouse anti-Gapdh (1:10,000, CB-1001, Calbiochem, La Jolla, CA), rabbit anti-γH2AX (1:5000, A300-081A, Bethyl Lab, Montgomery, TX), rabbit anti-pChk2 (phospho T68) (1:500, AB38461, Abcam, Cambridge, MA). Detection was by SuperSignal Pico (Thermo Scientific, Rockford, IL).
Immunofluorescence analysis
To determine subcellular location of Nit1 protein, wt kidney cells were seeded at 1×105 cells/well in LAB-TEK 8-well plate and cultured for 48 h. Cells were incubated with culture medium containing 500 nM Mitotracker (Invitrogen, Carlsbad, CA) for 30 min at 37°C and washed with PBS, fixed for 15 min with 3.7% buffered formaldehyde solution, and permeabilized with 0.2% TritonX-100 for 5 min. After blocking with 5% BSA for 1 h at 37°C, cells were washed with PBS and incubated with rabbit anti-Nit1 polyclonal serum (1:200) overnight at 37°C. Negative controls were prepared by replacing mitotracker or primary antiserum with 5% BSA in PBS. Fluorescein isothiocyanate (FITC)-conjugated anti-rabbit serum was used as secondary serum at a dilution of 1:500. Nuclei were counterstained with 4’, 6-Diamidino-2-phenyindole (DAPI). Images were captured with a ZEISS AxioCam and Axio Vision system (Zeiss, Thornwood, New York).
Subcellular fractionation
To confirm Nit1 subcellular localization, cellular protein fractionation was performed as described (Wu et al., 2005) with modifications. To prepare whole-cell extracts, cells were lysed in Solution A (50 mM Tris-HCl, pH 7.5, 420 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40, 0.34 M sucrose, 10% glycerol, 1 mM Na3VO4, 10 mM NaF, 10 mM β-glycerophosphate, 1 mM PMSF and 1 × Protease Inhibitor Cocktail (Roche, Indianapolis, IN)). After incubation on ice for 30 min, lysates were centrifuged at 3500 rpm at 4°C for 10 min. Supernatant was used for protein assay. For nuclear extract preparation, cells were lysed in Buffer B (10 mM HEPES at pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 0.1% Triton X-100, protease and phosphatase inhibitors as above), homogenized with 10 strokes of syringe with 28.5 G needle. Then cytoplasmic proteins were separated from nuclei by centrifugation at 3500 rpm for 5 min. Isolated nuclei were washed twice with Solution B followed by centrifugation at 3500 rpm for 5 min. Complete lysis of nuclei was performed in Solution A. To assess the level of Nit1 protein in mitochondria, a Mitochondria and Cytosol Fractionation kit (Biovision, Mountain View, CA) was used, following the manufacturer’s protocol.
Cytogenetic analysis
Wt, Fhit−/− and DKO kidney cells were cultured in T25 flasks. One day later, cells were treated with aphidicolin followed by the fixation of chromosomes. Karyotype analyses were performed in the Cytogenetics shared facility.
Immunohistochemical analysis
Consecutive 4 µm tissue sections were cut from paraffin blocks and placed on polylysine-coated slides for immunohistochemistry (IHC) analysis. Slides were de-paraffinized and subject to microwave antigen retrieval in 0.01M sodium citrate buffer (pH6.0) for 40 min. Endogenous peroxidase was blocked by incubation in 3% H2O2 for 10 min at room temperature. Sections were washed three times with PBS, each for 5 min, and then blocked by incubation with PBS containing 5% milk for 30 min at room temperature. Slides were incubated overnight at 4°C with primary antisera at optimal dilutions in PBS. Antisera for IHC were used to detect Ki67 (M7240, Dako North America, Inc., Carpinteria, CA), cyclin D1 (RM-9104, Thermo Fisher Scientific, Fremont, CA), pChk2 (AB38461, Abcam), and γH2AX (IHC-00059, Bethyl Lab). After primary antisera incubation, slides were rinsed in PBS and incubated in biotinylated anti-rabbit second antibody (1:300) in PBS with 5% milk for 60 min followed by incubation with streptavidin-HRP complex at 1:300 in water for 30 min. After washing with PBS, specimens were incubated in enhanced 3, 3’-diaminobenzidine tetrahydrochloride for 5 min. Finally, sections were counterstained with hematoxylin, dehydrated through graded alcohols, and cleared in xylene. Negative controls were prepared by replacing the primary antisera with non-specific antiserum.
Tissue microarray of human esophagus tissues
Archival files from the Ohio State University Department of Pathology were searched for esophageal adenocarcinoma resection specimens. Cases were reviewed for representative areas of esophageal adenocarcinoma, Barrett’s esophagus negative and positive for dysplasia, normal glands, and normal squamous epithelium. 2 mm cores were punched out of the areas of interest from formalin-fixed, paraffin embedded blocks in duplicate and transferred to the recipient TMA blocks using a precision instrument (Beecher Instruments, Silver Springs, MD, USA). The paraffin-embedded tissues were then cut in 4 µm slices and placed on a positively charged slide. Slides with specimens were then placed in a 60 °C oven for 1 h, cooled, and deparaffinized and rehydrated through xylenes and graded ethanol solutions to water. All slides were quenched for 5 min in a 3% H2O2 solution in water to block for endogenous peroxidase. Antigen retrieval was performed by placing slides in buffer (pH 6.1) for 25 min at 94°C using a vegetable steamer and cooling for 15 min in solution. Slides were then placed on a Dako Autostainer, immunostaining system, for use with IHC. Fhit was diluted 1:7000 and Nit was diluted 1:5000 and incubated for 60 min and 30 min respectively. Next slides were blocked for endogenous biotin with a biotin blocking system. Secondary antibody used was goat anti-rabbit (Vector, BA-1000) at a dilution of 1:200 and incubated for 30 min. The detection system used was Vectastain Elite (Vector, PK-6100) for 30 min. Staining was visualized with 3, 3’-diaminobenzidine chromogen. Slides were then counterstained in Richard Allen hematoxylin, dehydrated through graded ethanol solutions and coverslipped.
Statistical Analysis
The P values for tumor incidence differences and for histopathological analyses were calculated by Fisher’s exact test. Tumor multiplicity (no. of tumors/mouse) difference analysis was calculated by Student t-test. The frequency of Fhit and Nit1 deficiency in esophageal cores on TMAs was assessed and correlations determined by Fisher’s exact test. All statistical analyses were 2-tailed.
Detection of 8-hydroxyguanosine
Wt, Fhit−/−, and DKO cells (10,000 cells/well, LAB-TEC 8 well) were plated and after 48 h treated with or without 0.5 mM H2O2 for 5 h. Cells were fixed for 15 min with 3.7% buffered formaldehyde and permeabilized with 0.5% TritonX-100 for 10 min. Then, after blocking with 5% milk and normal goat serum for 1 h, slides were incubated with goat anti-8-hydroxyguanosine (8-OHdG) polyclonal antiserum (Chemicon) overnight at 4°C (1:200) after blocking with 5% milk for 30 min. Negative controls were prepared by replacing primary antiserum with PBS. Texas red conjugated anti-goat serum (Santa Cruz Biotechnology) was used as secondary antiserum (1:500). Cells were counterstained with DAPI. Images were captured with Zeiss Axio Cam and Axio Visionsystem (×400).
RESULTS
Genotyping and characterization of the mouse strains
Genotyping, illustrated in Figure 1A, identified cohorts of Fhit−/− and DKO mice. Effectiveness of the gene knockout procedures for Fhit and Nit1 alleles was confirmed at the protein level by assessment of expression of the respective proteins in kidney cells, a tissue that expresses the highest level of both proteins in wt cells (Pekarsky et al., 1998); the kidney-derived cell cultures from each of the three genotypes of mice, wt, Fhit−/− and DKO, were cultured and lysed for immunoblot analysis. Immunoblot results confirmed that DKO kidney cells did not express Fhit or Nit1, while Fhit−/− cells expressed Nit1 only (Fig. 1B); Fig. 1C shows Nit1 expression in mouse tissue by IHC staining of both Fhit−/− and DKO mice tissues. Nit1 was expressed as coarse particles in cytoplasmic regions of Fhit−/− forestomach, but not in DKO mouse forestomach (Fig. 1C).
As reported previously, Fhit−/− and Nit1−/− mice are healthy, fertile and long-lived (Fong et al., 2000; Semba et al., 2006) as are DKO mice. However, The DKO strain exhibited more spontaneous lesions than either Fhit−/− or Nit1−/− mice. DKO mice were aged 18 to 26 months and assessed for appearance of spontaneous lesions in internal organs, by visual inspection during necropsy; tissues were then fixed and processed for histopathological assessment of tissue sections. At 16 months, 4/4 mice did not show abnormalities; at 18–23 months, 3 of 6 mice showed forestomach tumors; at 26 months, 4 of 5 mice showed enlarged livers, with one enlarged liver showing dysplastic or adenomatous changes and the other 3 showing mild inflammation. Three aged mice, one at 23 months and two at 26 months, showed apparent blockage in the region of the rectal end of the colon; these apparent blockages, on histopathologic examination were diagnosed as large, secretion-filled hydrocystomas or cystadenomas of some perianal apocrine gland (data not shown; detailed description of spontaneous abnormalities in the DKO mice awaits assessment of another cohort of mice when they reach ~2 years of age). These glandular lesions were somewhat reminiscent of the sebaceous gland tumors observed in Fhit−/− and Msh2−/− mouse strains (de Wind et al., 1998; Fong et al., 2000). In earlier studies of aged Fhit-deficient and Nit1-deficient mice, we have not observed liver or anal gland abnormalities or spontaneous forestomach tumors (Fong et al., 2000; Semba et al., 2006; Zanesi et al., 2001).
Increased susceptibility to carcinogen induction of gastrointestinal tumors in DKO mice
In pilot experiments to determine optimal carcinogen treatment regimen for detecting differences in susceptibility of the two mouse strains, we treated small numbers of Fhit−/− and DKO mice, at ~8 weeks of age, with a single NMBA dose and sacrificed mice at 15 or 25 weeks after NMBA treatment, and visually inspected tissues for lesions and tumors, to define endpoints for our larger cohort treatment schedule. In these pilot experiments it appeared that the DKO mice developed more tumors after carcinogen treatment. Tumors were observed only in forestomach, as shown previously for Fhit and Nit1 KO mice; photographs of representative forestomachs are shown in Figure 2, which depicts one lesion in an untreated DKO forestomach and 4 tumors in a DKO forestomach of an NMBA-treated mouse. Under the same experimental conditions, the Fhit−/− forestomachs showed no visible tumors, though the NMBA-exposed Fhit−/− forestomachs were thickened throughout. As summarized in Table 1, 15 weeks post induction, 75% of Fhit−/− mice and 100% of DKO mice developed forestomach tumors. The average tumors per mouse were 1.75 for Fhit−/− versus 2.25 for DKO. Twenty-five weeks post induction, most treated mice of both genotypes exhibited tumors and average tumors per mouse were 3.09 for Fhit−/− and 5.03 for DKO forestomachs (P<0.01); ~19% of untreated DKO mice developed spontaneous tumors while none of untreated Fhit−/− mice had tumors (P<0.1). No tumors were found in glandular stomach, liver, kidney, intestine or spleen. Histopathological analysis of fixed and H&E stained tissues for esophagi and forestomachs of the NMBA-treated and untreated mice gave the following results: 1) papillomas were characterized by exophytic tumors of squamous cells with hyperkeratinization; the basal layer of papilloma was well preserved, and cellular and nuclear atypism was slight. 2) Dysplasia included amorphous cells extending from the basal cell layer, except for a thin superficial layer. 3) When invasion in the basement membrane and severe cellular atypia were present, the lesion was considered invasive carcinoma (Nishida et al., 2005). Examples of specific types of lesions in mouse esophagus and forestomach are shown in Figure 3 and results are enumerated for specific mouse strains in Table 2. Papillomas were observed in all forestomachs and esophagi, untreated and NMBA-treated, but dysplasia was observed only in NMBA-treated Fhit−/− and in both treated and untreated DKO mice. Under the conditions examined here, 1 NMBA dose and analysis at 25 weeks post-treatment, carcinoma was observed only in NMBA-treated DKO forestomachs. The incidence of dysplasia in untreated DKO mice was significantly higher than in untreated Fhit−/− mice. In esophagus, papilloma was observed in all groups and dysplasia was found in NMBA-treated Fhit−/− mice and both DKO groups. Carcinomas were not observed in esophagus, in accordance with previous results in Fhit and Nit1-deficent mice (Fong et al., 2000; Semba et al., 2006). The results of histopathological analysis were consistent with visual observations of lesions in fresh tissues at necropsy, confirming that DKO mice were more susceptible to spontaneous and carcinogen-induced tumors, especially carcinomas, than Fhit−/− mice after only 1 carcinogen dose.
Figure 2. Gross appearance of mouse forestomach and glandular stomach from untreated and NMBA treated mice.
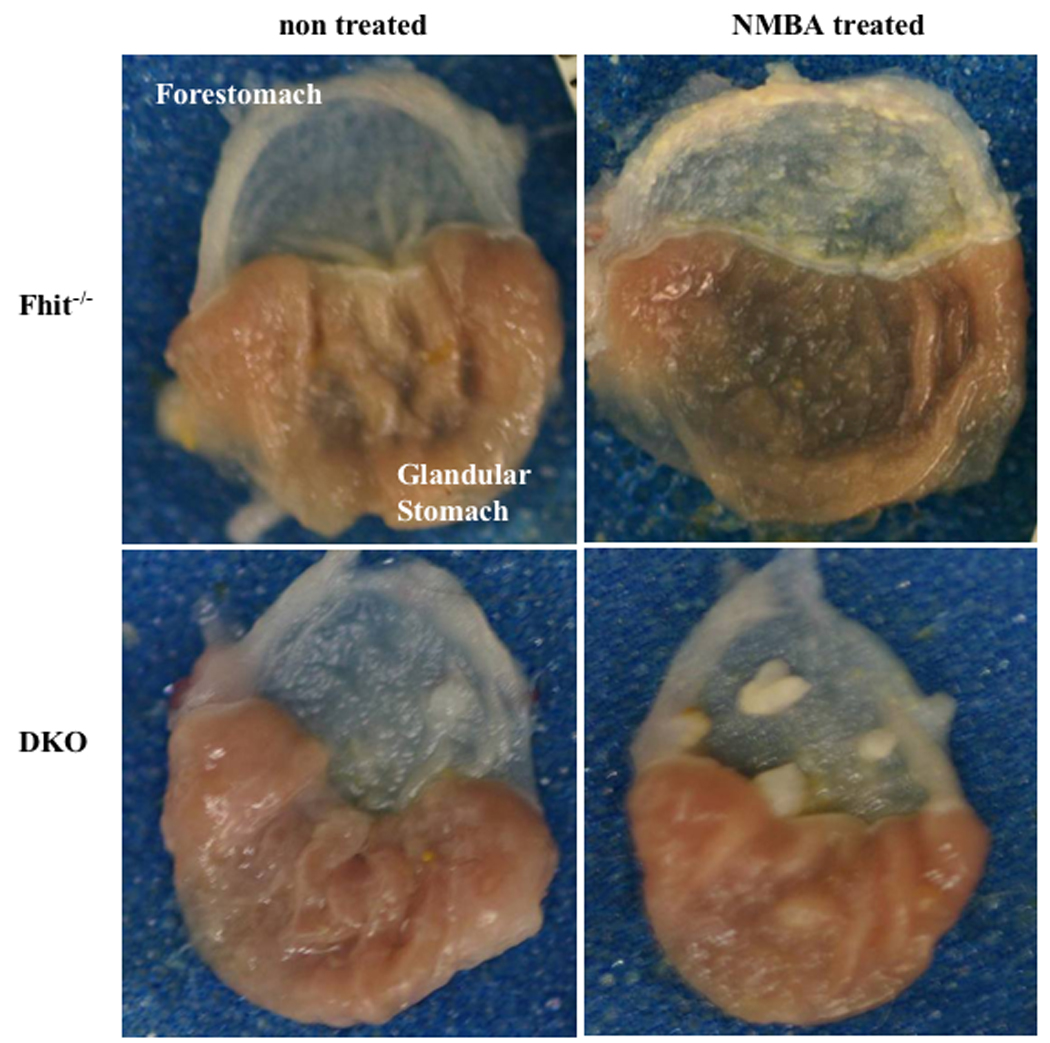
Fhit−/− and DKO mice at 6–8 weeks of age received a single dose of NMBA or were left untreated. Representative photographs are shown with forestomachs oriented to the top. Tumors were not observed in untreated Fhit−/− mice; one spontaneous tumor was found in an untreated DKO forestomach. Tumors were induced in both NMBA treated groups.
Table 1.
Tumor incidence in Fhit−/− and DKO mice
| Genotype | Treatment (weeks) |
number of tumors |
Tumor bearing mice |
Total mice |
Tumors/ mouse |
||||
|---|---|---|---|---|---|---|---|---|---|
| ≤0.5mm | ~1mm | ≥2mm | Total | ||||||
| Fhit−/− | 15 | 0 | 0 | 0 | 0 | 0(0) | 2 | 0 | |
| NMBA, | 15 | 5 | 2 | 0 | 7 | 3(75) | 4 | 1,75+1.3 | |
| 25 | 0 | 0 | 0 | 0 | 0(0) | 12 | 0 | ||
| NMBA, | 25 | 43 | 21 | 7 | 71 | 22(95.7) | 23 | 3.09±2.37 | |
| Fhit−/−Nit1−/−(DKO) | 15 | 1 | 0 | 0 | 1 | 1(25) | 4 | 0.25±0.5 | |
| NMBA, | 15 | 10 | 3 | 0 | 13 | 6(100) | 6 | 2.25±0.96 | |
| 25 | 0 | 5 | 1 | 6 | 3(18.8) | 16 | 0.313±0.7 | ||
| NMBA, | 25 | 65 | 74 | 28 | 167 | 31(96.9) | 32 | 5.03+3.02 | |
Upon dissection, esophagus, stomach, liver, kidney, spleen and intestine were examined and tumors were only found in esophagus and forestomach. Tumors were measured and enumerated by visual inspection. Numbers in parentheses denote percentages of tumor bearing mice. Tumors per mouse was expressed as means and standard deviation and pairs of the mean values were compared by Student t-test for: Fhit−/− vs NMBA-treated Fhit−/− 25 week (P<0.001); DKO untreated vs DKO treated 25 week (P<0.001); untreated groups (P<0.10) and 25 week treated groups (P<0.01).
Figure 3. Histopathological characterization of esophagus and forestomach tissues.
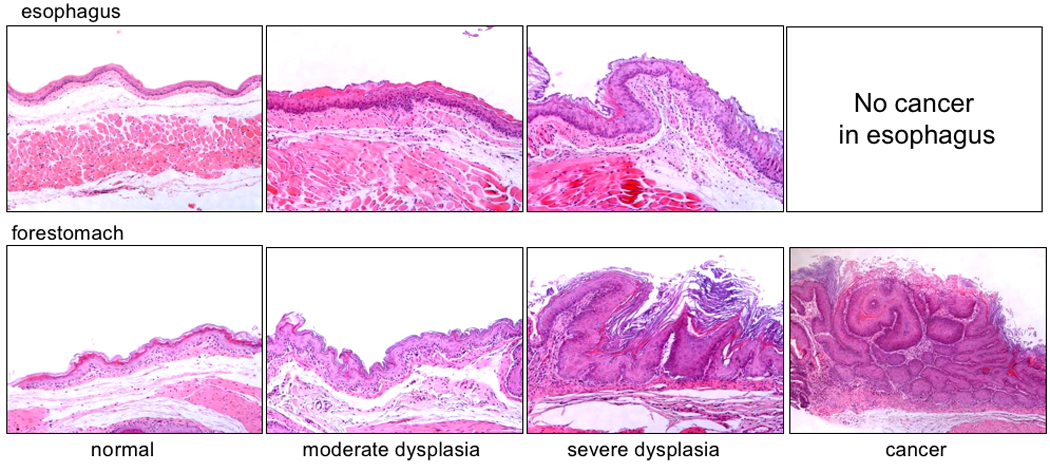
Tissues were fixed in 10% buffered formalin phosphate, processed and paraffin embedded; 4 µm sections were stained with H&E and observed by a pathologist (HO). Normal, moderate dysplasia and severe dysplasia were found in esophageal and forestomach tissues of NMBA-treated DKO mice (representative photos shown, X200); cancers were observed only in forestomach.
Table 2.
Histopathological analysis of esophageal and forestomach lesions
| Incidence (% of mice with) |
% of Mice with lesions |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Papilloma | Dysplasia | Carcinoma | ||||||||||
| Fore stomach | ||||||||||||
| Fhit−/− | (n=7) | 72 | 0 | 0 | 72 | |||||||
| Fhit−/− | NMBA | (n=18) | 80 | * | 40 | * | 0 | * | 100 | |||
| DKO | (n=ll) | 89 | 45 | 0 | 89 | |||||||
| DKO | NMBA | (n=26) | 100 | 72 | 28 | 100 | ||||||
| Esophagus | ||||||||||||
| Fhit−/− | (n=7) | 13 | 0 | 0 | 13 | |||||||
| Fhit−/− | NMBA | (n=18) | 28 | 45 | 0 | 50 | ||||||
| DKO | (n=ll) | 23 | 33 | 0 | 56 | |||||||
| DKO | NMBA | (n=26) | 35 | 62 | 0 | 70 | ||||||
P<0.05
Nit1 protein is distributed in cytosol and mitochondria
To develop clues to mechanisms through which double loss of Fhit and Nit1 proteins may contribute to tumor development, we began by assessing Nit1 subcellular localization. Firstly, mouse kidney cells expressing endogenous Nit1 were fractionated into mitochondrial, nuclear and cytosol subfractions and presence of Nit1 protein in the fractions was detected by immunblot analysis of the subfraction lysates. As shown in the immnoblots in Figure 4A, Nit1 protein was detected in cytosol and mitochondria, but not in nucleus. GAPDH served as marker for the cytosol fraction, Cox IV for mitochondria and Parp1 for nuclear fraction. Secondly, we examined Nit1 subcellular localization by immunofluorescence analysis (Fig. 4B). Nit1 protein was detected by use of FITC-conjugated anti-rabbit serum, and mitochondria were labeled with red-fluorescent Mitotracker. The yellow color in the merged panel indicates subcellular areas where Nit1 and mitochondria are co-localized. The results of immunofluorescence analysis, which illustrates the presence of Nit1 in mitochondria, are in accord with the subcelllular fractionation results and confirm that Nit1 protein is present in the cytosol and mitochondria, as is Fhit protein (Zanesi et al., 2001).
Figure 4. Subcellular localization of Nit1.
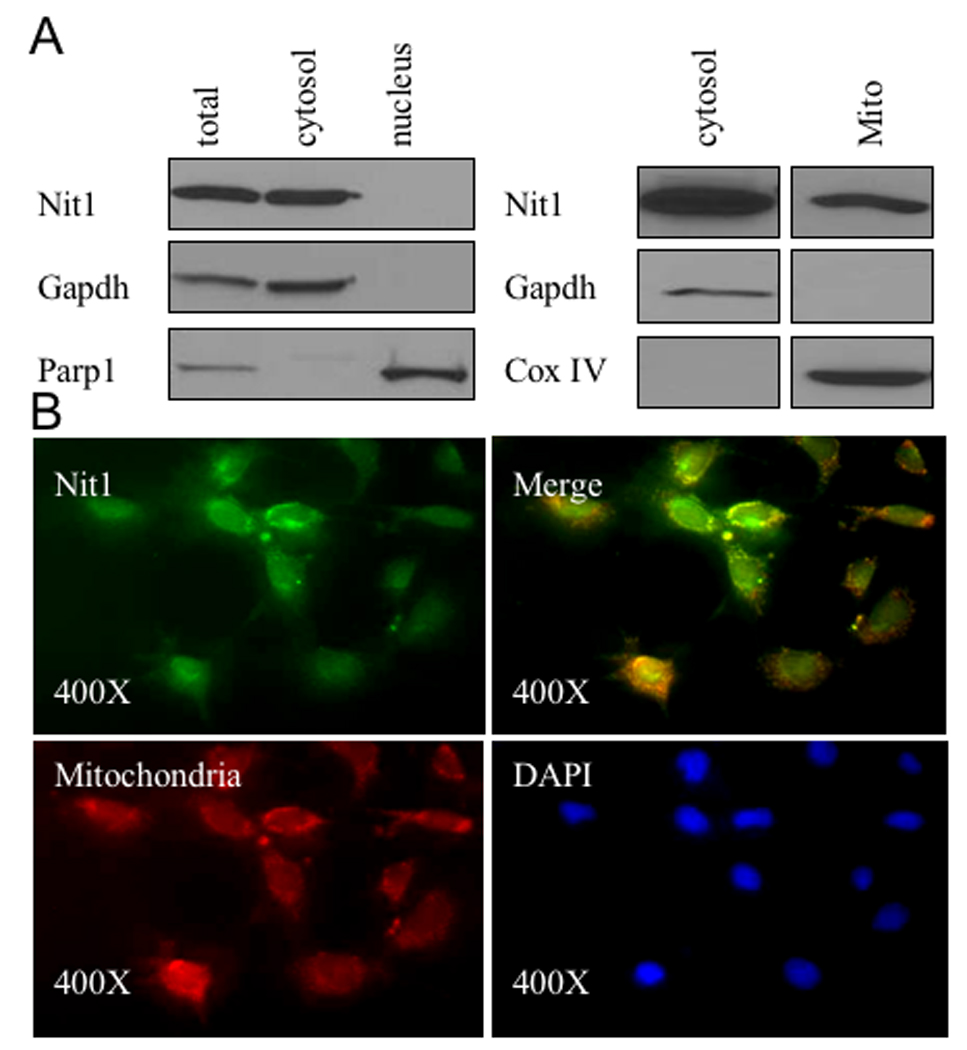
A. Western blot analysis of Nit1 in whole cell lysate of wt kidney cells, cytosol, mitochondria and nucleus. Cell fractions were run on 12% Ready Gel and immunoblotted with anti-Nit1 serum. Gapdh served as positive control for cytosol, Parp1 for nucleus and Cox IV for mitochondria. Nit1 is detected in cytosol and mitochondria, but not in nucleus. B. Immunofluorescence analysis of Nit1 subcellular localization. Wt kidney cells were incubated with rabbit anti-Nit1 serum followed by incubation with FITC (green)-conjugated anti-rabbit serum. Nuclei were counterstained with DAPI (blue). Mitochondria were stained in red. Nit1 green fluorescence (x 400) was detected in cytosol and mitochondria (see merged green and red, appearing yellow in some cells), but not in nucleus.
DKO kidney cells are pseudotetraploid
Assessment of chromosome number, using the established kidney cell lines (wt at 10th subculture, Fhit−/− at 21st subculture, and DKO at 4th subculture) for the three mouse genotypes, showed that the number of chromosomes in wt, Fhit−/− and DKO kidney cells were 40, 44 and 71, respectively.
The DNA damage response is altered by Fhit and Nit1 deficiency
We have reported previously that Fhit is involved in the DNA damage response through modulation of checkpoint proteins (Ishii et al., 2008; Okumura et al., in press). To determine if Nit1 may also have a role in responses to DNA damage, we examined the expression of γH2AX and pChk2 in wt, Fhit−/− and DKO non-neoplastic tissues of treated and non NMBA-treated mice; pChk2 and γH2AX were expressed in NMBA-treated Fhit−/−, non-treated and treated DKO forestomach tissues, but not in untreated Fhit−/− tissue (Fig. 5A), suggesting that the untreated DKO tissues express an activated checkpoint in the non-neoplastic DKO forestomach tissue, consistent with a preneoplastic condition (Bartkova et al., 2005; Gorgoulis et al., 2005). We also treated wt, Fhit−/− and DKO kidney cells with HU, a reagent known to induce DNA damage and DNA damage response checkpoint. As shown in Figure 5B, wt cells showed induction of γH2AX at 4 and 48 h after HU, while Fhit−/− and DKO cells showed minimal induction only at 48 h. The expression of pChk2 was slightly increased after HU treatment in wt cells, but was apparently not activated after HU in Fhit−/− and DKO cells. To examine effects of oxidative stress, cells of the three genotypes were treated with 0.5 mM H2O2 and examined for presence of 8-OHdG in DNA by staining with antiserum against 8-OHdG (Fig. 5C). Immunofluorescence results of this staining are shown in Figure 5C, left panel, which illustrates that wt cells show the highest level of 8-OHdG and DKO cells the lowest, with quantification of 8-OHdG levels shown in Figure 5C, right panel; >95% (P<0.01) of wt cells were positive for 8-OHdG, while only 14% of DKO cells and ~55% of Fhit−/− cells were positive. Thus, knockout of both Fhit and Nit1 resulted in near elimination of oxidative damage to DNA under the conditions of oxidative stress tested.
Figure 5. Expression of DNA damage response proteins in tissues of NMBA-treated KO mice and in KO-derived ‘normal’ kidney cells.
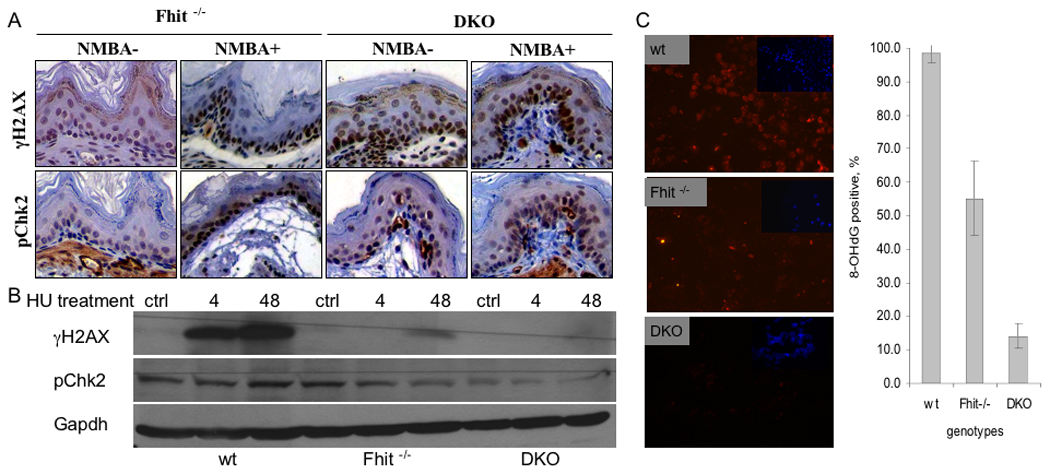
A. Examples of immunohistochemical staining for γH2AX and pChk2. The strongest expression of γH2AX was in NMBA treated Fhit−/− forestomach tissue. B Western blots of γH2AX and pChk2. Wt, Fhit−/− and DKO cells were treated with HU for 0 (ctrl), 4 or 48 h; γH2AX expression was the highest in wt cells followed by Fhit−/− cells. C. Left panel, cultured cells were treated with 8-OH-dG after exposure to 0.5 mM H2O2 for 5 h; DNA damage in wt, Fhit−/−, and DKO kidney cells was assessed by immunofluorescence detection of 8-OHdG (red); nuclei were counterstained with DAPI (blue, inset) (200x). Similar staining of cells that were untreated by H2O2 served as negative controls; fewer than 10% of cells showed 8-OHdG staining in the negative control cells (not shown). Right panel, bar graph illustrates frequency of 8-OHdG positive cells in the three cell strains; error bars indicate standard deviation of frequencies in three fields of stained cells (P<0.01 by Student t-test).
Proliferative and apoptotic markers in DKO tissues
We previously reported that Cyclin D1 was up-regulated in tumor tissues in Nit1-deficient mice (Semba et al., 2006). In this experiment, we compared expression of Cyclin D1, Ki67 and Bcl-2 in forestomachs of NMBA treated DKO and untreated wt mice. As shown in Figure 6, Cyclin D1 was strongly expressed in DKO tissues compared with wt. The expression of Ki67 in DKO mice was also higher than in wt forestomach, while Bcl-2 expression was undetectable or very low in the hyperplastic DKO tissue.
Figure 6. Expression of proliferation and apoptosis associated proteins in DKO forestomach.
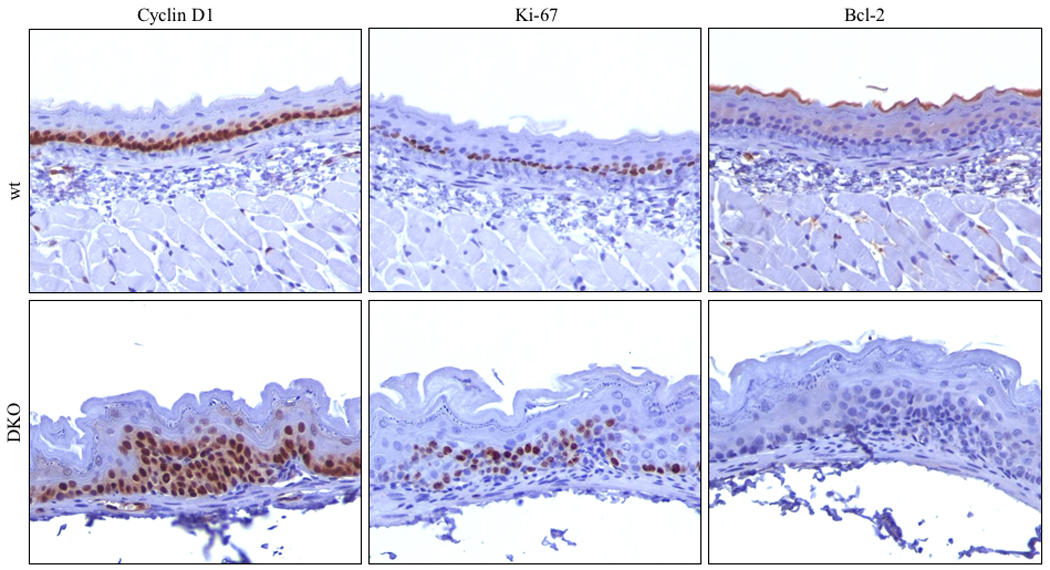
Immunohistochemical staining of Cyclin D1, Ki67, and Bcl-2 expression in wt and DKO forestomach tissues; Note the increase of Cyclin D1 and Ki67 and the decrease of Bcl-2 expression in DKO forestomach cells.
Nit1 expression in human esophageal lesions
To determine if Nit1, like Fhit, may have a role in human cancer development, we assessed expression of Nit1 in esophageal tissues of human cancer patients by immunohistochemical analyses of Nit1 and Fhit expression. The TMAs used in the analysis contained tissue cores from biopsies of Barrett esophagus cases and thus included tissue cores representing squamous epithelium, glandular epithelium, glandular epithelium with dysplasia and adenocarcinoma. Glandular cells were positive for Nit1 and Fhit. In contrast, the Nit1 expression level was reduced in 48% of adenocarcinomas and Fhit expression was reduced in 64% (see Fig. 7 for representative photographs); Fhit and Nit1 loss was not correlated (p=0.35) in these cancers, as indicated in the data summary in Table 3.
Figure 7. Assessment of Nit1 expression in human esophageal cancer tissues.
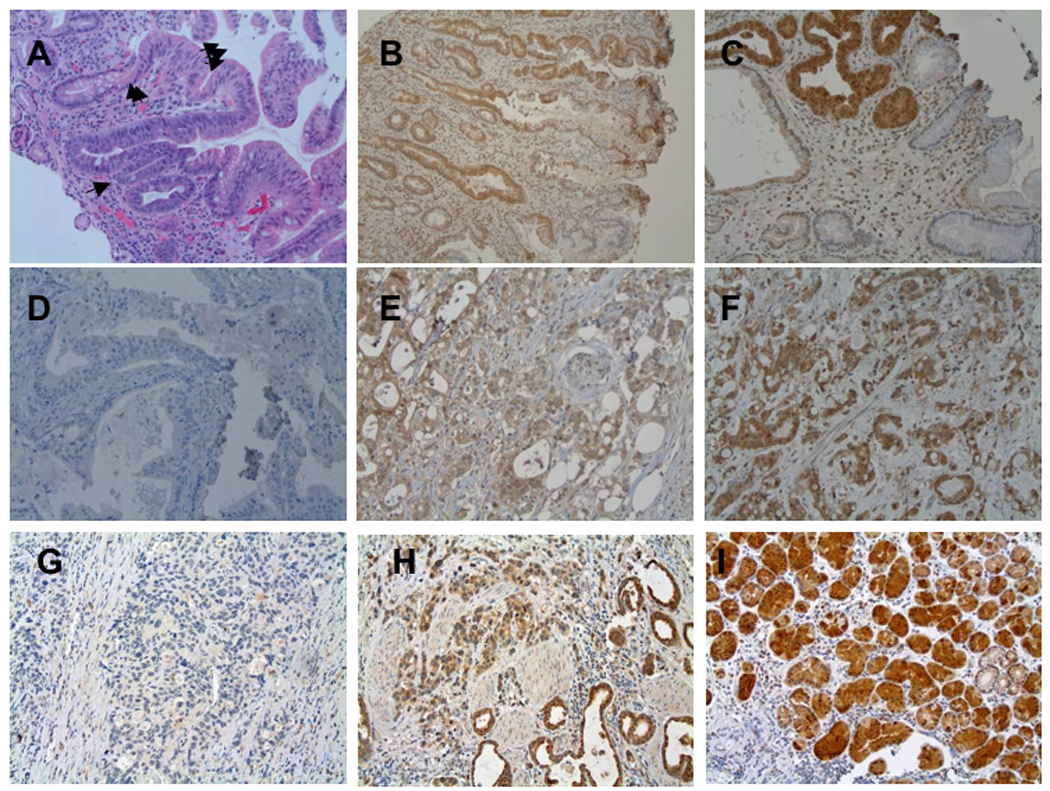
A. H&E of a section exhibiting Barrett glandular type tissue in esophagus, 1 arrow: basal compartment, 2 arrows: neck, 3 arrows: surface. B. Nit1 IHC staining of Barrett esophagi; slight Nit1 staining in basal compartment and negative in neck. C. Strong Nit1 staining in dysplasia and negative in Barrett esophagi without dysplasia. D-F. Nit1 staining of tumors, negative (D), slight (E), and strong (F). G–I: Fhit IHC staining. G: no Fhit staining (G); weak staining (H); strong staining (I).
Table 3.
Loss of Fhit and Nit1 protein in human esophageal tumors
| IHC staining | Nit1 negative | Nit1 positive |
|---|---|---|
| Fhit negative | 31 (36.0%) | 29 (33.7%) |
| Fhit positive | 10(11.6%) | 16(18.8%) |
Number and percentage of cancers expressing Fhit and Nit1 - Loss of both proteins was not significantly correlated (P=0.35, Fisher Exact test).
DISCUSSION
It was a surprise to some investigators that the human genome appears to encode only ~30,000 genes, not considering alternative isoforms and the many recently recognized conserved non-protein coding RNAs encoded in mammalian genomes. Another form of protein diversity might include enzymes that have evolved functions in addition to their catalytic activities. Fhit tumor suppressor function, which does not require that the protein be catalytically active (Okumura et al., in press; Siprashvili et al., 1997), is an example, and Nit1 tumor suppressor function is likely another, since over-expression of a recombinant Nit1 catalytic mutant showed tumor suppressor activity (Semba et al., 2006).
Nit1 suppressor function
We previously reported that Nit1 and Fhit-deficient mice showed similar susceptibility to NMBA induction of forestomach tumors (Semba et al., 2006). In this study we find that DKO mice were more susceptible to NMBA induction of such tumors and developed more spontaneous tumors than Fhit−/− mice, suggesting that loss of Nit1 and Fhit in gastrointestinal tissue leads to increased susceptibility to tumor development than the singly deficient mice. The results shown in Table 1, concerning visible tumors during necropsy, suggests that the average tumor burden per mouse is not quite 2-fold higher in the DKO mice in comparison to Fhit−/− tumor burden for each comparable group. However, histopathological results summarized in Table 2 suggests that the difference between the single and double deficient mice is greater than 2-fold since only the DKO mice, under the selected tumor induction protocol developed carcinomas of the forestomach. Thus, the DKO mice were much more likely to develop malignant tumors of the forestomach. Overall the results suggest that, though Fhit and Nit1 absence may affect some similar signal pathways involved in tumor development, they also affect independent, additive pathways.
Comparison of Nit1 and Fhit proteins
Both Nit1 and Fhit are cytosolic as well as mitochondrial proteins; and for both we have not detected presence in the nucleus. DKO kidney cells were aneuploid, suggesting a greater degree of chromosome instability, while wt and Fhit−/− cells, that had been subcultured more frequently, were diploid or pseudodiploid, respectively. The preliminary comparison of the response of DKO and Fhit−/− cells to stressful agents, suggested that the abnormal response of DKO cells to HU, a DNA damage inducer, and H2O2, an oxidative stress agent, was more extreme then the response of Fhit−/− cells. We have previously shown that Fhit-deficient human esophageal cancer cells, when exposed to a DNA damaging agent, exhibit weak checkpoint activation; results shown in Figure 5B suggest that the DKO cells show almost no response to HU treatment. Perhaps more surprisingly, the DKO cells show almost no evidence of oxidative damage to DNA after exposure to 0.5 mM H2O2, while Fhit−/− cells show a 2-fold reduction in 8-OHdG residues in nuclear DNA. We had previously shown that Fhit protein is chaperoned to mitochondria by the Hsp60/10 complex, where it binds and stabilizes Fdxr and thus participates in production of reactive oxygen species (ROS) after exposure to H2O2 (Pichiorri et al., 2009; Trapasso et al., 2008). Conversely, Fhit-deficient cells show reduced Fdxr levels and produce less ROS (Pichiorri et al., 2009; Trapasso et al., 2008). Fhit-deficient cells thus escape ROS-induced apoptosis, possibly carrying a mutational burden. The preliminary assessment of the lack of production of oxidative damage in H2O2-exposed DKO cells suggests that DKO cells would be even more likely to escape ROS-induced apoptosis, possibly contributing to tumor susceptibility through this escape. The absence of 8-OHdG in nuclei of DKO cells suggests that Nit1, like Fhit, participates in production of ROS in response to oxidative stress, though this must be investigated in greater detail through investigation of the function of Nit1 in mitochondria. Another similarity between Nit1 and Fhit protein is the high level of expression in glandular tissue, as seen in Figure 7, and the effect of absence of Fhit or Nit1 on tumor susceptibility of glandular tissue, such as sebaceous tumors in Fhit-deficient mice (Fong et al., 2000) and the anal gland cystadenomas in DKO mice.
However, due to location at a chromosome fragile site, the Fhit gene product is much more likely to be lost in cancers, and in fact is more frequently reduced in expression in human esophageal adenocarcinomas than is Nit1 protein, as shown in Table 3. Fhit and Nit1 are not coordinately lost in these tumors, as Fhit and Wwox protein are in tumors (Guler et al., 2004; Guler et al., 2005); Fhit and Wwox are both encoded at fragile sites and thus are highly susceptible to similar intrinsic and extrinsic DNA damaging agents, while Nit1 is not encoded at a fragile site.
Comparison to Nit2
Only branch 1 of the nitrilase superfamily branches has retained nitrilase catalytic activity. Genes in the other branches encode amidases or proteins with related enzymatic activities. Nit1 and Nit2, of branch 10 are found in most mammalian tissues, and share ~55% sequence homology (Barglow et al., 2008). They are considered amidases based on sequence and presence of a Cys-Glu-Lys triad in the putative active site. Nit1 knockout in mouse tissues/cells results in increased cell proliferation, enhanced survival of cells exposed to DNA-damaging agents and increased incidence of NMBA-induced tumors (Semba et al., 2006). Nit1 over-expression leads to decreased cancer cell viability and increased apoptosis, even after over-expression of a Nit1 protein with the Cys residue of the catalytic site mutated to Ala (Semba et al., 2006). These findings suggested that Nit1 is a tumor suppressor protein that stimulates apoptosis in cancer cells. Evidence was presented recently that Nit2 protein can also act as a tumor suppressor, even when its catalytic site is mutated (Lin et al., 2007).
Recent identification of Nit2 as ω-amidase that catalyzes the deamidation of αKGM and αKSM to α-ketoglutarate and oxaloacetate, respectively, and the idea that Nit2, as ω-amidase, may metabolize toxic substrates to useful products (Cooper et al., 2009), is intriguing in view of the apparent liver abnormalities of aged DKO mice. The concentration of αKGM is increased in the cerebrospinal fluid of patients with liver disease (Vergara et al., 1974); perhaps the thus far unknown Nit1 substrate could have a role in the liver abnormalities of the DKO mice.
ACKNOWLEDGEMENTS
We acknowledge the expert assistance of personnel in the Histology Facility of the Department of Molecular Virology, Immunology and Medical Genetics, the Pathology and Cytogenetics Facilities of the Department of Pathology, and the shared Facilities of the Ohio State University Comprehensive Cancer Center.
Funded by
Contract grant sponsor: National Institutes of Health; Contract grant numbers: CA77738 and CA132453.
REFERENCES
- Barglow K, Saikatendu K, Bracey M, Huey R, Morris G, Olson A, Stevens R, Cravatt B. Functional proteomic and structural insights into molecular recognition in the nitrilase family enzymes. Biochem (Wash) 2008;47:13514–13523. doi: 10.1021/bi801786y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland J, Lukas C, Orntoft T, Lukas J, Bartek J. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- Cooper AJ, Krasnikov B, Nostramo R, Nieves E, Callaway M, Chien CH, Pinto JT. Ubiquitously expressed w-amidase is a potential neuroprotectant identical to tumor suppressor Nit2. J Neurochem. 2009;108:S100. [Google Scholar]
- de Wind N, Dekker M, van Rossum A, van dV, te Riele H. Mouse models for hereditary nonpolyposis colorectal cancer. Cancer Res. 1998;58:248–255. [PubMed] [Google Scholar]
- Fong L, Fidanza V, Zanesi N, Lock L, Siracusa L, Mancini R, Siprashvili Z, Ottey M, Martin S, Druck T, McCue P, Croce C, Huebner K. Muir-Torre-like syndrome in Fhit-deficient mice. Proc Natl Acad Sci U S A. 2000;97:4742–4747. doi: 10.1073/pnas.080063497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galperin M, Koonin E. ‘Conserved hypothetical’ proteins: prioritization of targets for experimental study. Nucleic Acids Res. 2004;32:5452–5463. doi: 10.1093/nar/gkh885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgoulis V, Vassiliou L, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio R, Jr, Kastrinakis N, Levy B, Kletsas D, Yoneta A, Herlyn M, Kittas C, Halazonetis T. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- Guler G, Uner A, Guler N, Han SY, Iliopoulos D, Hauck WW, McCue P, Huebner K. The fragile genes FHIT and WWOX are inactivated coordinately in invasive breast carcinoma. Cancer. 2004;100:1605–1614. doi: 10.1002/cncr.20137. [DOI] [PubMed] [Google Scholar]
- Guler G, Uner A, Guler N, Han SY, Iliopoulos D, McCue P, Huebner K. Concordant loss of fragile gene expression early in breast cancer development. Pathol Int. 2005;55:471–478. doi: 10.1111/j.1440-1827.2005.01855.x. [DOI] [PubMed] [Google Scholar]
- Huebner K, Croce CM. Cancer and the FRA3B/FHIT fragile locus: it’s a HIT. Br J Cancer. 2003;88:1501–1506. doi: 10.1038/sj.bjc.6600937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii H, Dumon K, Vecchione A, Fong L, Baffa R, Huebner K, Croce C. Potential cancer therapy with the fragile histidine triad gene: review of the preclinical studies. JAMA. 2001;286:2441–2449. doi: 10.1001/jama.286.19.2441. [DOI] [PubMed] [Google Scholar]
- Ishii H, Mimori K, Ishikawa K, Okumura H, Pichiorri F, Druck T, Inoue H, Vecchione A, Saito T, Mori M, Huebner K. Fhit-deficient hematopoietic stem cells survive hydroquinone exposure carrying precancerous changes. Cancer Res. 2008;68:3662–3670. doi: 10.1158/0008-5472.CAN-07-5687. [DOI] [PubMed] [Google Scholar]
- Lin C, Chung M, Chen W, Chien C. Growth inhibitory effect of the human NIT2 gene and its allelic imbalance in cancers. FEBS J. 2007;274:2946–2956. doi: 10.1111/j.1742-4658.2007.05828.x. [DOI] [PubMed] [Google Scholar]
- Marcotte E, Pellegrini M, Ng H, Rice D, Yeates T, Eisenberg D. Detecting protein function and protein-protein interactions from genome sequences. Science. 1999;285:751–753. doi: 10.1126/science.285.5428.751. [DOI] [PubMed] [Google Scholar]
- Matsuyama A, Shiraishi T, Trapasso F, Kuroki T, Alder H, Mori M, Huebner K, Croce C. Fragile site orthologs FHIT/FRA3B and Fhit/Fra14A2: evolutionarily conserved but highly recombinogenic. Proc Natl Acad Sci U S A. 2003;100:14988–14993. doi: 10.1073/pnas.2336256100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida K, Mine S, Utsunomiya T, Inoue H, Okamoto M, Udagawa H, Hanai T, Mori M. Global analysis of altered gene expressions during the process of esophageal squamous cell carcinogenesis in the rat: a study combined with a laser microdissection and a cDNA microarray. Cancer Res. 2005;65:401–409. [PubMed] [Google Scholar]
- Okumura H, Ishii H, Pichiorri F, Croce CM, Mori M, Huebner K. The fragile gene product, Fhit, in oxidative and replicative stress responses. Cancer Sci. doi: 10.1111/j.1349-7006.2009.01168.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottey M, Han S, Druck T, Barnoski B, McCorkell K, Croce C, Raventos-Suarez C, Fairchild C, Wang Y, Huebner K. Fhit-deficient normal and cancer cells are mitomycin C and UVC resistant. Br J Cancer. 2004;91:1669–1677. doi: 10.1038/sj.bjc.6602058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace H, Brenner C. The nitrilase superfamily: classification, structure and function. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-1-reviews0001. REVIEWS0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekarsky Y, Campiglio M, Siprashvili Z, Druck T, Sedkov Y, Tillib S, Draganescu A, Wermuth P, Rothman J, Huebner K, Buchberg A, Mazo A, Brenner C, Croce C. Nitrilase and Fhit homologs are encoded as fusion proteins in Drosophila melanogaster and Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1998;95:8744–8749. doi: 10.1073/pnas.95.15.8744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichiorri F, Okumura H, Nakamura T, Garrison P, Gasparini P, Suh S, Druck T, McCorkell K, Barnes L, Croce C, Huebner K. Correlation of fragile histidine triad (Fhit) protein structural features with effector interactions and biological functions. J Biol Chem. 2009;284:1040–1049. doi: 10.1074/jbc.M806638200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semba S, Han S, Qin H, McCorkell K, Iliopoulos D, Pekarsky Y, Druck T, Trapasso F, Croce C, Huebner K. Biological functions of mammalian Nit1, the counterpart of the invertebrate NitFhit Rosetta stone protein, a possible tumor suppressor. J Biol Chem. 2006;281:28244–25283. doi: 10.1074/jbc.M603590200. [DOI] [PubMed] [Google Scholar]
- Siprashvili Z, Sozzi G, Barnes L, McCue P, Robinson A, Eryomin V, Sard L, Tagliabue E, Greco A, Fusetti L, Schwartz G, Pierotti M, Croce C, Huebner K. Replacement of Fhit in cancer cells suppresses tumorigenicity. Proc Natl Acad Sci U S A. 1997;94:13771–13776. doi: 10.1073/pnas.94.25.13771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapasso F, Pichiorri F, Gaspari M, Palumbo T, Aqeilan RI, Gaudio E, Okumura H, Iuliano R, Di LevaG, Fabbri M, Birk DE, Raso C, Green-Church K, Spagnoli LG, Venuta S, Huebner K, Croce CM. Fhit interaction with ferredoxin reductase triggers generation of reactive oxygen species and apoptosis of cancer cells. J Biol Chem. 2008;283:13736–13744. doi: 10.1074/jbc.M709062200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Vergara F, Plum F, Duffy TE. Alpha-ketoglutaramate: increased concentrations in the cerebrospinal fluid of patients in hepatic coma. Science. 1974;183:81–83. doi: 10.1126/science.183.4120.81. [DOI] [PubMed] [Google Scholar]
- Wu X, Shell S, Zou Y. Interaction and colocalization of Rad9/Rad1/Hus1 checkpoint complex with replication protein A in human cells. Oncogene. 2005;24:4728–4735. doi: 10.1038/sj.onc.1208674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanesi N, Fidanza V, Fong L, Mancini R, Druck T, Valtieri M, Rüdiger T, McCue P, Croce C, Huebner K. The tumor spectrum in FHIT-deficient mice. Proc Natl Acad Sci U S A. 2001;98:10250–10255. doi: 10.1073/pnas.191345898. [DOI] [PMC free article] [PubMed] [Google Scholar]