

Abstract
IpaH proteins are E3 ubiquitin ligases delivered by the type III secretion apparatus into host cells upon infection of humans by the Gram-negative pathogen Shigella flexneri. These proteins comprise a variable leucine-rich repeat—containing N-terminal domain and a conserved C-terminal domain harboring an invariant cysteine residue that is crucial for activity. IpaH homologs are encoded by diverse animal and plant pathogens. Here we demonstrate that the IpaH C-terminal domain carries the catalytic activity for ubiquitin transfer and that the N-terminal domain carries the substrate specificity. The structure of the IpaH C-terminal domain, determined to 2.65-Å resolution, represents an all-helical fold bearing no resemblance to previously defined E3 ubiquitin ligases. The conserved and essential cysteine residue lies on a flexible, surface-exposed loop surrounded by conserved acidic residues, two of which are crucial for IpaH activity.
Type III secretion (T3S) systems are used by numerous Gram-negative pathogenic bacteria to deliver effector proteins into the cells of their human, animal or plant hosts. T3S systems comprise the T3S apparatus (T3SA) that form syringe-like structures that span the bacterial envelope and extend like a needle from the bacterial surface, translocators that transit through the T3SA and form a pore within the target cell membrane, effectors that transit through the T3SA and the pore into the cytosol of host cells, and specific chaperones and transcription regulators for secretion and transcription of these effectors1,2. Some effectors target the actin cytoskeleton to promote entry or inhibit phagocytosis of the bacterium, whereas other effectors interfere with the host’s innate immune responses1.
The ubiquitin pathway is a common target of bacterial effectors. This pathway involves one ubiquitin-activating enzyme (E1), a limited number of ubiquitin-conjugating enzymes (E2s) and many ubiquitin-ligating enzymes (E3s). The C-terminal glycine residue of 76-residue ubiquitin is first charged via a thioester linkage onto a cysteine residue of E1 and then transferred to a cysteine residue of an E2. E3s recruit an E2 or a subset of E2s for ubiquitin transfer to specific substrates. Two classes of E3s are differentiated on the basis of their mechanism of action and on sequence or structural similarities. RING (really interesting new gene) and U-box (a modified RING motif) domain—containing E3s act as adaptor-like molecules by bringing a ubiquitinated E2 and the substrate into sufficiently close proximity to promote the ubiquitination of the substrate. In contrast, HECT (homologous to E6-associated protein C terminus) domain—containing E3s possess an essential cysteine residue that acts as an acceptor for the ubiquitin carried by the E2 before its transfer to the substrate. The N- and C-terminal domains of HECT E3s are usually involved in substrate binding and catalytic activity, respectively3.
Several T3S effectors are E3 ligases that modulate cellular ubiquitination. For example, the effector AvrPtoB from Pseudomonas syringae pv. tomato belongs to the U-box—containing family and targets the kinase Fen in Arabidopsis thaliana4-6. The seven effectors of the GALA family from the plant pathogen Ralstonia solanaceaurum contain F-box motifs that bind the host Skp1 proteins and function as SCF (Skp1, Cullin, F-Box) E3s7. The effector SopA from Salmonella enterica possesses a HECT-like E3 ubiquitin-ligase activity8 and is organized into two N-terminal subdomains, which resemble the bilobed architecture of HECT domain E3s, and a leucine-rich repeat (LRR)—containing C-terminal domain thought to be involved in substrate recognition9. The sequence similarity between SopA and HECT E3s is restricted to residues surrounding the catalytic cysteine. Finally, the proposed T4S effector LubX from Legionella pneumophila contains multiple U-boxes and ubiquitinates Clk1 (ref. 10).
Bacteria of the genus Shigella are responsible for bacillary dysentery in humans. Their virulence is dependent upon a T3S system encoded by a 200-kb virulence plasmid11. Among the Shigella flexneri T3S effectors are members of the IpaH family, which comprises nine proteins encoded by both the chromosome and the virulence plasmid. IpaH proteins are characterized by an approximately 250-residue N-terminal domain that is variable and contains six to eight LRR segments and an approximately 300-residue C-terminal domain that is virtually identical in all IpaH proteins12. IpaH homologs are encoded by numerous pathogens containing genes for a T3S apparatus, including Salmonella enterica, Yersinia pestis and Yersinia pseudotuberculosis, Edwardsiella ictulari, Rhizobium spp., Bradyrhizobium japonica and various Pseudomonas spp13. IpaH9.8 promotes degradation of the mitogen-activated protein kinase kinase (MAPKK) Ste7 in yeast and ubiquitinates this protein in vitro; the homologous protein SspHI from S. enterica ubiquitinates the mammalian protein kinase PKN1 in vitro13. The two-domain architecture of IpaH family members and the presence of a cysteine residue in their conserved C-terminal domains are reminiscent of HECT domain E3s; however, the C-terminal domain of IpaH family members does not show sequence similarity with HECT domain E3s. To better understand the function of this family of E3 ubiquitin ligases, we decided to define the catalytic domain of these enzymes, determine its structure and investigate the functional role of conserved residues by using in vivo and in vitro assays. We present evidence that the IpaH C-terminal domain (IpaH-CTD) is an E3 ubiquitin ligase that adopts a new all-helical fold in which the conserved and essential cysteine residue is located in a surface-exposed flexible loop.
RESULTS
E3 ligase activity of the IpaH C-terminal domain
The region of the C-terminal domain that is identical in all nine IpaH proteins (IpaH-CTD) corresponds to residues 254–540 in IpaH9.8 and residues 285–571 in IpaH1.4, another IpaH protein encoded by the Shigella virulence plasmid (Fig. 1a). To better understand the ubiquitin-ligase activity of IpaH proteins, we purified the C-terminal fragments of IpaH9.8 and IpaH1.4 as GST-IpaH9.8208–545 and His6-IpaH1.4265–575 fusions and tested them in vitro for autoubiquitination activity in the presence of ATP, ubiquitin, E1 and the E2 UBE2D2 (Fig. 1b). Both truncated IpaH proteins were highly active; we detected molecular species ~7 kDa larger than corresponding IpaH fragments within 2 min of the ubiquitination reaction, and larger species accumulated thereafter. In accordance with E3 ligase activity, no such species were detected even after 2 h of incubation if the reaction lacked ubiquitin, E1 or E2 (Fig. 1c,d).
Figure 1.

The C-terminal domain of IpaH proteins shows E3 ubiquitin-ligase activity. (a) Schematic representation of IpaH9.8 and IpaH1.4 C-terminal fragments (not to scale). The IpaH-CTD is shown in gray, and its position in each IpaH protein is indicated. (b) Immunoblot analysis using anti-IpaH antibodies of reactions performed in the presence of ATP, ubiquitin, E1, UBE2D2 and GST-IpaH9.8208–545 or His6-IpaH1.4265–575. The positions of native GST-IpaH9.8208–545 and His6-IpaH1.4265–575 are indicated on the right. (c,d) Immunoblot analysis with anti-IpaH (c) and anti-ubiquitin (d) antibodies in the presence or absence of ATP, ubiquitin, E1, UBE2D2 and His6-IpaH1.4265–575. The reactions were performed at 25 °C for 2 h (except for T0, which represents the degree of ubiquitination at t = 0 min). The stars mark the 70-kDa species corresponding to the dimer of IpaH1.4265–575 possibly formed through a disulfide bond between cysteine residues in IpaH1.4265–575 monomers.
In contrast to the C-terminal domain, the full-length IpaH9.81–545 protein was inactive in autoubiquitination assays even after 1 h of incubation, unless the substrate Ste7 protein was added to the reaction (Fig. 2a). In IpaH9.81–545 autoubiquitination reactions, in the presence of Ste7, we detected species larger than GST-IpaH9.81–545 by anti-IpaH antibodies after 5 min (Fig. 2b). Using immunoblot analysis of the IpaH9.81–545 autoubiquitination reactions using anti-Ste7 antibodies (Fig. 2b), we detected species larger than Ste7 after 5 min of reaction. The formation of ubiquitinated forms of IpaH and Ste7 detected during this reaction were also confirmed using anti-ubiquitin antibodies (Fig. 2b). Combined, these data demonstrate that IpaH9.8 requires the presence of Ste7 protein to show similar autoubiquitination activity to its truncated C-terminal domain. This suggests that the N-terminal domain may be inhibitory for the autoubiquitination activity of the full-length protein and that substrate binding releases this inhibition.
Figure 2.
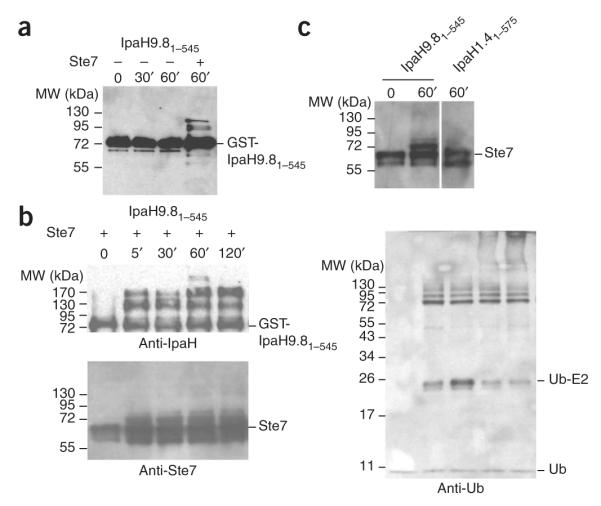
E3 ubiquitin ligase activity of full-length IpaH proteins. (a) Immunoblot analysis using anti-IpaH antibodies of reactions performed in the presence of ATP, ubiquitin, E1, UBE2D2 and GST-IpaH9.81–545. The reaction mixture performed in the presence of a complex containing Ste7, Ste11-4 and Kss1 is indicated. (b) Immunoblot analysis using anti-IpaH (above left), anti-Ste7 (below left) and anti-ubiquitin (right) antibodies of reactions performed in the presence of ATP, ubiquitin, E1, UBE2D2, GST-IpaH9.81–545 and a complex containing Ste7, Ste11-4 and Kss1. (c) Immunoblot analysis using anti-Ste7 antibodies of reactions performed in the presence of ATP, ubiquitin, E1, UBE2D2 and GST-IpaH9.81–545 or His6-IpaH1.41–575 and a complex containing Ste7, Ste11-4 and Kss1. Reactions were incubated for the times indicated. Samples were treated with DTT before loading.
IpaH homologs differ in their N-terminal regulatory regions (Fig. 1a). To test whether the N-terminal domains mediate substrate specificity, we examined whether IpaH1.4 could also ubiquitinate Ste7 in vitro. The IpaH1.4 N-terminal domain shares 38% sequence identity with the N-terminal domain of IpaH9.8. In contrast to GST-IpaH9.81–545, His6-IpaH1.41–575 was unable to ubiquitinate Ste7 (Fig. 2c).
Structure of the IpaH C-terminal domain
The IpaH-CTD has E3 ligase activity, but its sequence does not resemble that of any known E3. To obtain insights into the structure of the IpaH-CTD, we determined the crystal structure of IpaH1.4265–575 to 2.65-Å resolution using SAD from selenium (Se-SAD; Supplementary Fig. 1 online) using selenium-enriched protein. The final model was refined to an Rwork and Rfree of 22.8% and 28.4%, respectively, and contains 6,192 atoms, with three molecules (A, B and C) in the asymmetric unit. In each molecule, 18–20 residues at the N terminus and 10 residues at the C terminus are disordered. Additional disordered regions included 3 to 9 residues located between helices α3 and α4 and 8 to 12 residues located between helices α8 and α9 in molecules A, B and C, as well as 8 residues located between helices α5 and α6 in molecule B. The final refined model showed excellent stereochemistry, with 99.4% of the residues within the most favored and additional allowed regions of the Ramachandran plot. IpaH1.4 residues 284–565 visualized in this structure correspond almost exactly to the IpaH-CTD and are identical in IpaH1.4 and IpaH9.8 (Figs. 1a, 3 and 4).
Figure 3.
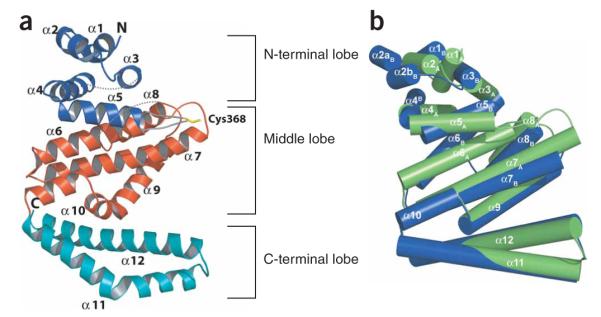
Overall structure of the IpaH1.4 C-terminal domain. (a) Ribbon representation of the IpaH-CTD in molecule A; the N-terminal, middle and C-terminal lobes are colored in blue, red and cyan, respectively. The loop between helices α5 and α6 is colored in gray with Cys368 in yellow. The unstructured loops between residues 329 to 334 and 449 to 462 are indicated as dashed lines, and the N and C termini are labeled. (b) Superimposition of molecules A and B present in the asymmetric unit. Each IpaH polypeptide is shown by cylindrical helices, with molecule A in green and molecule B in blue, and positions of each helix in molecules A and B are labeled. In molecule B, residue 303 was not modeled owing to poor electron density, and helix α2 is split in two parts (α2aB and α2bB).
Figure 4.

Multiple sequence alignment of IpaH homologs. The alignment is restricted to the C-terminal domain identical in all IpaH proteins (IpaH-CTD), which spans residues 254 to 540 (indicated by arrow) in IpaH9.8 and residues 285 to 571 in IpaH1.4. A set of nine representative proteins was selected from the alignment of 25 IpaH homologs13 including (1) IpaH9.8 and (2) IpaH1.4 from S. flexneri, (3) SspH1 from Salmonella typhimurium, (4) Y4fR from Rhizobium spp., (5) Blr1904 from Bradyrhizobium japonicum, (6) PP1072 and (7) PP2212 from Pseudomonas putida, (8) Pfl14099 from Pseudomonas fluorescens and (9) PSPTO4093 from P. syringae pv. tomato. Sequences of the C-terminal domain of the ~600-residue proteins (rows 1–5) and ~1,500-residue proteins (rows 6–9) are shown in black and blue characters, respectively. Identical residues within sequences of the first and second groups are highlighted in green and blue, respectively. Residues that are identical in all or most sequences are highlighted in red and purple, respectively. Secondary-structure elements derived from the structure of the IpaH1.4 C-terminal domain are shown above the sequence alignment and labeled. The broken lines correspond to the disordered regions in the structure. The α-helices that are part of N-terminal, middle and C-terminal lobes are colored blue, red and cyan, respectively. Residues selected for mutagenesis are marked with asterisks.
The overall IpaH-CTD structure contains 12 α-helices arranged into three distinct lobes: the N-terminal lobe is formed by helices α1 to α5, the middle lobe is formed by helices α6 to α10, and the C-terminal lobe comprises a two-helix (α11 and α12) coiled coil (Figs. 3a and 4 and the stereo view in Supplementary Fig. 1). The N-terminal and middle lobes are closely packed, whereas the C-terminal lobe extends from the rest of the structure, with a large cavity between the C-terminal lobe and the two others (Fig. 3a and Supplementary Fig. 1). Comparison of the three IpaH molecules in the asymmetric unit indicates that the three lobes in each molecule show considerable conformational flexibility, in part demonstrated by higher B-factors for molecules A and B. Molecules A and C superimpose well (r.m.s. deviation of 0.6Å), whereas molecule B presents a rotation of ~25° of the N-terminal lobe and helix α6 relative to the rest of the structure in molecules A and C (Fig. 3b). The observed conformational diversity points to an intrinsic flexibility of the N-terminal lobe.
We detected no noteworthy similarities between the structures of IpaH-CTD and major types of E3s, including HECT domain E3s (PDB 1ND7, PDB 2ONI and PDB 1D5F), SopA (PDB 2QZA and PDB 2QYU) and RING-finger proteins such as AvrPtoB (PDB 2FD4) or any other proteins in the Protein Data Bank by using Secondary Structure Matching (SSM; http://www.ebi.ac.uk/msd-srv/ssm/ssmstart.html) and Dali (http://www.ebi.ac.uk/dali/) servers. Accordingly, the IpaH-CTD structure represents a previously undescribed fold and defines a new class of E3s.
Role of the invariant cysteine residue in IpaH activity
We previously showed that replacement of the IpaH9.8 invariant cysteine residue with alanine dramatically impaired its activity both in vitro and in vivo13. In the IpaH1.4-CTD structure, this cysteine corresponds to Cys368 and is part of an 8-residue loop connecting helices α5 and α6 (Figs. 3a and 4). Cys368 is located at the border between the N-terminal and middle lobes and is exposed to the large cavity formed by the C-terminal lobe and the rest of the molecule (Fig. 5a,b). In molecules A and C, the loop protrudes from the IpaH-CTD surface, with its side chains fully exposed to the solvent (Fig. 5c). This conformation is stabilized mainly by intermolecular contacts resulting from close packing of neighboring IpaH-CTD molecules in the crystal lattice, which brings Cys368 residues from molecules A and C into proximity (data not shown). Although the interaction between the two cysteine residues is weak and noncovalent, as evidenced by their 3.34-Å separation, it is sufficient for ordering of the loops in the two molecules. An equivalent packing arrangement is observed between molecule B and its symmetry mate in the crystal lattice; however, the corresponding loop in molecule B seemed disordered between residues 363 to 372 and could not be modeled. Intramolecular interactions of the cysteine-containing loop are limited to contacts at or near the loop between helices α7 and α8, where a hydrogen bond is formed between OE2 of Glu369 and the hydrogen of the backbone N of Glu429 belonging to helix α8, as seen in molecules A and C (Fig. 5c). These features, together with the high temperature factors of corresponding residues, indicate that the loop harboring the cysteine residue has considerable conformational flexibility.
Figure 5.

Environment of the conserved cysteine residue. (a) Ribbon diagram of IpaH-CTD (molecule A). The nine invariant residues are shown in a stick representation, labeled and colored in red. The N and C termini are labeled. (b) Overall surface of IpaH-CTD. Positions of surface-exposed invariant residues are colored red, whereas residues conserved only among a subset of IpaH proteins are colored purple. Cys368 and Asp370 are the only residues from a that appear in this view. The surface-exposed regions for the six residues mutated to alanine or serine are labeled. (c) Close-up of the region of the IpaH-CTD structure around the cysteine-containing loop in stereo. Residues in this loop as well as three nearby residues subjected to mutagenesis are shown in a stick representation and labeled. Carbon atoms in this loop are colored cyan and other residues are represented with their carbon atoms uncolored. All nitrogen, oxygen and sulfur atoms are colored blue, red and yellow, respectively.
IpaH activity was eliminated when we mutated the invariant cysteine to alanine13. We also analyzed the effect of mutating this residue to serine in IpaH9.8, as this substitution is less invasive than the previously described alanine substitution. We tested the C337S IpaH9.8 variant for its ability to rescue growth of sst2Δ yeast cells (Fig. 6a). These cells are unable to dampen the pheromone-response pathway and cannot grow in the presence of pheromone14. By promoting ubiquitination and degradation of Ste7, IpaH9.8 interrupts the pheromone-response pathway and permits growth of sst2Δ cells in the presence of pheromone13. Following induction of their expression by galactose, wild-type IpaH9.8 and the C337S variant were produced in similar amounts, as monitored by immunoblotting (data not shown). As with the C337A mutation13, the C337S mutant failed to rescue sst2Δ cells for growth in the presence of pheromone (Fig. 6a), indicating that the presence of a thiol group at this position is essential for IpaH9.8 activity.
Figure 6.

Functional analysis of conserved residues. (a) Growth on glucose- or galactose-containing plates of serial dilutions of sst2Δ yeast cells harboring plasmids encoding indicated proteins (WT, wild-type IpaH9.8-Flag; C337A; IpaH9.8 in which Cys337 is replaced by alanine (the same nomenclature is used for the other variants)). Below, plates containing α-factor are shown. (b) Immunoblot analysis with anti-IpaH (above) and anti-His (below) antibodies of eluates from glutathione beads added to GST-IpaH9.8208–545 or its C337A, C337S, D397A, D339A and R340A variants incubated with His6-UBE2D2. (c) Immunoblot analysis using anti-IpaH antibodies of reactions performed in the presence of ATP, ubiquitin, E1, UBE2D2, GST-IpaH9.8208–545 (WT, above) or its C337A (middle) and C337S (below) variants. Samples were incubated at 25 °C for the indicated time and treated (+) or not treated (-) with DTT before loading. Arrows indicate the DTT-sensitive species. (d) Immunoblot analysis using anti-IpaH antibodies of reactions performed in the presence of ATP, ubiquitin, E1, UBE2D2, GST-IpaH9.8208–545 or the GST-IpaH9.8208–545 variants carrying the indicated replacement. Samples were incubated at 25 °C for 30 min (except for WT To, which represents the degree of ubiquitination of the wild-type protein at t = 0 min) and treated with DTT before loading.
To test whether replacement of cyteine by alanine or serine affected the binding of IpaH9.8 to the E2, we tested the ability of C337A and C337S GST-IpaH9.8208–545 variants to associate with UBE2D2 in GST pull-down experiments (Fig. 6b). The amounts of UBE2D2 co-precipitated by both C337A and C337S variants were similar to that obtained with the wild-type GST-IpaH9.8208–545, indicating that these variants were not impaired in their ability to bind UBE2D2.
In HECT and SopA E3 ligases, the essential cysteine residue forms an E3-ubiquitin thioester intermediate, which enables the transfer of ubiquitin onto the substrate. This intermediate can be visualized on a gel as ubiquitinated species that are sensitive to reducing agents such as DTT. Thus, we examined the possibility that GST-IpaH9.8208–545 formed a DTT-sensitive ubiquitinated species during autoubiquitination reactions (Fig. 6c). We carried out these assays using GSTIpaH9.8208–545 and the C337A and C337S GST-IpaH9.8208–545 variants and a ubiquitin molecule in which all lysine residues were replaced by arginine (KO ubiquitin, Boston Biochem), such that it could not give rise to polyubiquitinated chains.
In the reaction carried out with GST-IpaH9.8208–545, a molecular species larger than GST-IpaH9.8208–545 that would correspond to GST-IpaH9.8208–545 linked to one ubiquitin molecule was detected after 10 s of incubation (Fig. 6c). This band was considerably more intense in the sample that had not been treated with DTT, suggesting that this species represents a mixture of DTT-sensitive and DTT-resistant ubiquitinated GST-IpaH9.8208–545. Upon prolonged incubation times, we detected larger species that were also sensitive to DTT treatment; these species are likely to correspond to thioester intermediates of already ubiquitinated IpaH molecules. Concomitantly, large DTT-resistant species accumulated and, after 2 h incubation, the same species were present in the DTT-treated and untreated samples. No DTT-sensitive species were detected in reactions performed with the C337A and C337S GST-IpaH9.8208–545 variants, confirming that formation of DTT-sensitive Ub-IpaH species is dependent upon the cysteine residue in the IpaH-CTD. However, we detected large-molecular-weight species in reactions carried out with both variant proteins after 20 min of incubation. The formation of these ubiquitinated species occurred more slowly than with wild-type GST-IpaH9.8208–545 and did not lead to the accumulation of multiubiquitinated IpaH species, even after 2 h of incubation. As the only cysteine residue of IpaH9.8208–545 had been replaced by alanine or serine, the formation of these species was not mediated by the formation of a thioester ubiquitin-IpaH intermediate, but probably by a direct transfer of ubiquitin from E2 onto IpaH lysine residues.
Analysis of residues within the proposed catalytic site
Elucidation of the role of the invariant cysteine residue led us to analyze the potential role of the other eight invariant residues in the IpaH-CTD, that is, Trp305, Asp370, Leu400, Arg409, Ala417, Leu434, Leu444 and Leu446 in IpaH1.4 (Fig. 4). In the IpaH1.4 C-terminal domain structure, the conserved tryptophan, alanine and leucine residues are buried in the core of the structure and are probably required to maintain the integrity of this domain (Fig. 5a,b). Asp370 is located 8 Å from and is part of the same loop as Cys368. Arg409, along with Arg405 and Glu477, participate in a network of surface salt bridges between helices α7 and α9, far from the cysteine-containing loop (data not shown). In addition, several residues located near Cys368 are conserved in a subset of IpaH homologs; these residues include Glu369 and Arg371 (from the loop between α5 and α6), Arg418 (belonging to the C-terminal part of α7) and Asp428 (from the loop between α7 and α8) (Figs. 4 and 5b). The invariant Asp370 together with Glu369 and Asp428 form a negatively charged surface above the plane of the cysteine-containing loop, whereas Arg371 and Arg418 contribute to a positively charged patch below it (Fig. 5b,c).
To investigate the role of invariant and conserved charged residues in IpaH function, we substituted the corresponding residues in IpaH9.8 (that is, the invariant Asp339 and the conserved Glu338, Arg340, Arg387 and Asp397 residues) individually to alanine. These IpaH9.8 variants, which were produced in yeast in similar amounts compared to wild-type IpaH9.8 (data not shown), were tested for their ability to rescue growth of sst2Δ yeast cells (Fig. 6a). D339A and D397A variants did not rescue the growth of sst2Δ cells. By contrast, E338A and R387A variants were still active in vivo. We observed a lower growth rate for sst2Δ yeast cells expressing the R340A variant. These results identified Asp339 and Asp397 as crucial residues for IpaH9.8 activity.
To investigate whether the growth phenotypes observed in sst2Δ cells expressing IpaH9.8 variants were the result of a decreased E3 activity of these proteins, we introduced the same substitutions into GST-IpaH9.8208–545 and tested the resulting proteins for their ability to bind UBE2D2 in GST pull-down experiments and for their autoubiquitination activity (Fig. 6b,d). Proteins carrying the substitutions D339A, D397A and R340A, which failed to rescue or only partially rescued growth of sst2Δ cells, were still able to bind UBE2D2 (Fig. 6b). The D397A variant showed lower autoubiquitination activity compared to wild-type GST-IpaH9.8208–545, whereas the E338A, R340A and R387A variants produced mono- and polyubiquitinated species similarly to wild-type GST-IpaH9.8208–545. Thus, the D339A and R340A substitutions did not abolish the autoubiquitination activity, suggesting that the in vitro assay is less sensitive than the in vivo assay to detect low or intermediate activities. Nonetheless, there is good agreement between the in vitro autoubiquitination activity of GST-IpaH9.8208–545 variants and the ability of full-length IpaH9.8 variants to rescue the growth of sst2Δ yeast cells.
IpaH interactions with E2
To explore the specificity of the IpaH-CTD toward E2s, we tested 28 purified human E2s for their ability to support IpaH1.4265–575 autoubiquitination activity (Fig. 7a). In addition to UBE2D2, the E2s UBE2D1, UBE2D3 and UBE2D4 were able to support polyubiquitination of IpaH1.4265–575. Molecular species larger than IpaH1.4265–575 were also detected in the reactions performed with 11 other E2s (Fig. 7a), although these reactions did not lead to formation of polyubiquitinated species as in the case of the four E2s mentioned above.
Figure 7.

IpaH interactions with E2 conjugating enzymes. (a) Immunoblot analysis using anti-IpaH antibodies of reactions performed in the presence of ATP, ubiquitin, E1, IpaH1.4265–575 and one of the indicated human E2 enzymes. The reactions were performed at 25 °C for 40 min. The double asterisk indicates the E2s supporting the strong autoubiquitination reaction. Single asterisk indicates the E2s supporting formation of only a single modified species. A star indicates the (70 kDa) species that is present in all lanes, including the one that did not contain any E2 (below, third lane). This band would correspond to a dimer of IpaH1.4265–575, presumably formed through a disulfide bond between cysteine residues in IpaH265–575 monomers (see also Fig. 1). The E2 nomenclature is in accordance with that used by the Human Genome Organization (http://www.genenames.org/genefamily/ube2.php). The reaction with UBE2D3 E2 was done in duplicate (above, lanes 5 and 7). (b) Immunoblot analysis using anti-IpaH antibodies showing IpaH1.4265–575 autoubiquitination in the presence of wild type (WT) and R5A, F62A and K63A UBE2D2 variants. Reactions were performed at 25 °C for 5 min. (c) Determination of the dissociation constants of IpaH1.4265–575 interactions with UBE2D2 using fluorescence polarization. The change in fluorescence polarization of fluorescein-labeled UBE2D2 (WT) or its indicated variants are plotted as a function of IpaH1.4265–575 concentration with error bars indicating s.d.
The interaction of UBE2D2 with the E6AP HECT domain and the cNOT4 RING domain E3s involves the N-terminal helix and loops 4 and 7 of UBE2D2, which insert into a hydrophobic groove on the E3 surface15-17. UBE2D2 residues implicated in E3 binding include primarily the conserved phenylalanine residue (Phe62), which provides the main contact with both HECT domain and RING domain E3s. The corresponding phenylalanine residue (Phe63) in UBE2L3 (also known as UbcH7) was also found to be crucial for the interaction of UBE2L3 with SopA9. In addition, the interaction of UBE2D2 with cNOT4 was abrogated by the alanine substitution of Arg5 and Lys63 residues, which belong to the UBE2D2 N-terminal helix and loop 4, respectively16. To gain insight on the interaction of IpaH with UBE2D2, we examined the effect of alanine substitutions of Arg5, Phe62 and Lys63 in UBE2D2 on the autoubiquitination activity of His6-IpaH1.4265–575. All three variant E2s supported the formation of polyubiquitinated IpaH species similarly to the wild-type UBE2D2 (Fig. 7b). To quantify the IpaH-UBE2D2 interaction, we labeled the wild-type and variant UBE2D2 with fluorescein-5-maleimide and monitored the change in fluorescence polarization upon titration of IpaH1.4265–575 (Fig. 7c). Kd values for IpaH1.4265–575 with the wild-type UBE2D2 and its R5A, F62A and K63A variants were 317 μM, 151 μM, 176 μM and 226.9 μM, respectively. The Kd obtained for the interaction of UBE2D2 with IpaH1.4265–575 (317 μM) is higher yet comparable to the Kd (160 μM) reported for the interaction of this E2 with E6AP18. The Kd values obtained for the E2 variants confirmed that these replacements did not prevent the interaction of UBE2D2 with IpaH, in contrast to what has been described for E6AP and SopA.
DISCUSSION
We report a functional and structural characterization of the IpaH C-terminal domain, an ~300-residue domain conserved in a large family of known or presumptive T3S effectors13. This domain is endowed with the catalytic activity of this family of E3s and its structure does not show similarities to other eukaryotic or bacterial E3s (Fig. 8). Accordingly, the IpaH-CTD structure represents a previously uncharacterized fold and defines a new class of E3s.
Figure 8.

Structures of different classes of E3 ligases in which a cysteine residue is essential for activity. Helices are shown in red, strands in cyan and loops in gray. The positions of the N and C termini of each protein are indicated by N and C. The WWP1 HECT domain (PDB 1ND7) is used to represent the HECT domain fold. The catalytic cysteine (α- and β-carbon and γ-sulfur atoms) is shown as a space-filling model and colored yellow.
Autoubiquitination activity is common among E3s19 and this activity was used to demonstrate that the IpaH-CTD harbors the catalytic activity. Using variants of IpaH9.8 and its C-terminal domain, we found a good correlation between the activity of the full-length protein in yeast and the autoubiquitination activity of the C-terminal domain in vitro. Nevertheless, no ubiquitinated forms of IpaH9.8 were detected upon its expression in yeast, suggesting that this E3 may not autoubiquitinate in vivo or that ubiquitinated IpaH molecules are quickly degraded by the proteasome. The inability to detect ubiquitinated IpaH molecules may also be due to activity of deubiquitinating enzymes (DUBs)20. It is intriguing that the in vitro autoubiquitination activity of full-length IpaH9.8 is dependent upon the presence of the substrate Ste7. These results suggest that, in the absence of the substrate, the N-terminal domain of IpaH9.8 inhibits the autoubiquitination activity. As the full-length IpaH9.8 ubiquitinates ubiquitin in vitro13, it seems unlikely that the N-terminal domain interferes with the binding of the E2 on the C-terminal domain. IpaH1.4 was not able to promote ubiquitination of the IpaH9.8 substrate Ste7 in vitro, suggesting that the N-terminal LRR-containing domain of IpaH mediates the substrate specificity. Accordingly, each member of the IpaH family may have its own target(s).
The IpaH-CTD contains a single invariant cysteine residue that is essential to the E3 activity. In HECT domains and SopA E3s, ubiquitin is transiently linked by a thioester linkage to the catalytic cysteine residue located approximately 30 residues from the C terminus of the E3; in these E3s, leucine and threonine residues are located 6 residues and 1 residue, respectively, upstream from the catalytic cysteine. In IpaH proteins, the invariant cysteine is located approximately in the middle of the IpaH-CTD sequence and is not surrounded by conserved residues similar to those of eukaryotic E3s. Nevertheless, the cysteine residue of the IpaH-CTD is required for the activity in vivo and robust autoubiquitination in vitro. In addition, we detected DTT-sensitive ubiquitinated forms of wild-type IpaH but not of the cysteine to serine or alanine variants. These data strongly suggest that IpaH enzymes use this cysteine residue as an acceptor of ubiquitin from E2s and then transfer the ubiquitin to their target, as described for HECT domain E3s. Detection of the IpaH-Ub thioester intermediate proved challenging, owing to the high activity of the IpaH catalytic domain. Analogous difficulties were encountered upon characterization of the intermediate of some HECT domain E3s21.
Replacement of either of the two conserved residues Asp339 or Asp397 with alanine abrogated IpaH9.8 activity in vivo. These residues, corresponding to Asp370 and Asp428 in IpaH1.4, are located on the surface of the protein and their replacement with alanine is not predicted to affect the overall structure of the protein. GST pull-down experiments demonstrated that these mutations do not affect the ability of the IpaH-CTD to bind UBE2D2. Thus, these residues may have an analogous role to the charged residues surrounding the catalytic cysteine in E2s, in which they maintain an electrostatic environment that favors catalysis22,23. However, in the IpaH-CTD structure, the carboxyl groups of these aspartate residues are located at least 7 Å from the sulfhydryl group of the cysteine, suggesting that large conformational changes must occur during catalysis. It is also possible that these residues do more than just promote an acidic microenvironment, because replacement of the neighboring acidic residue (Glu338) led to only a modest decrease in IpaH9.8 activity. Another possible role for these two acidic residues is the recognition of the acceptor lysine residue(s) of the substrate for ubiquitination.
The conformational heterogeneity revealed by the conformations of the three molecules in the IpaH crystal is consistent with the predicted mechanism for HECT domain E3s; in the latter enzymes, the conformational flexibility enables the shuttling of ubiquitin from the E2 to the catalytic cysteine residue of the E3 and then to the target. The transfer of ubiquitin from the catalytic cysteine of E2 to the HECT domain is accompanied by large structural changes that involve E2 binding to one end of the N-terminal lobe of the HECT domain, followed by a swiveling of the small lobe of the HECT domain relative to the large lobe, bringing the E2 close to the catalytic cysteine in the C-terminal lobe of the HECT domain24. Conformational flexibility is also observed in SopA, for which different conformations of the C-terminal lobe were described in the multiple molecules in the asymmetric unit. For the IpaH-CTD, transfer of ubiquitin to the substrate might be enabled by the intrinsic mobility of the α5-α6 loop. Rotation around the swivel formed by the α6-α7 loop and helix α6 would bring the substrate-bound N-terminal domain close to the active site loop, thereby facilitating the transfer of ubiquitin from IpaH to the substrate.
Of the 28 human E2s that we tested, UBE2D2 and its closest homologs supported the highest activity in the IpaH autoubiquitination reaction. Although these proteins have been studied extensively, the novelty of the IpaH-CTD structure did not allow us to identify the binding surface of the E2 onto IpaH. UBE2D2 variants described as severely impaired in binding to HECT domain and SopA E3s still supported the IpaH autoubiquitination reaction and demonstrated even tighter binding to IpaH-CTD. Further mutagenesis analysis and structural information on the IpaH-E2 complex will be necessary to gain a detailed knowledge on the IpaH-E2 interaction.
IpaH homologs are classified in two groups according to the size of their N-terminal domain and the overall sequence similarity of their C-terminal domain (Fig. 4). Proteins of the first group are present in Enterobacteriaceae, whereas proteins of the second are present in Pseudomonas spp. Proteins of the two groups share little sequence similarity in their far C-terminal region; this region corresponds exactly to the third lobe (composed of helices α11 and α12), which is separated from the rest of the domain by a large cavity. This observation suggests that this third lobe might be endowed with a function that is different between these two classes of E3s, possibly binding to different E2s.
T3S effectors endowed with ubiquitin-ligase activity identified so far bear F-box, U-box or RING motifs (GALA family, LubX and AvrPtoB) or show a general architecture reminiscent of the HECT domain (SopA). In contrast, members of the IpaH family represent a new class of E3s, both in sequence and in structure. This finding raises questions on the origin of the structural diversity among E3 ubiquitin ligases and suggests that bacterial and eukaryotic genomes might contain other E3s that remain to be identified.
METHODS
Materials
We used yeast strain BY4741 sst2::kanMX MATa leu2Δ met15Δ ura3Δ (Research Genetics) to monitor sensitivity to α-factor. E1, ubiquitin and KO ubiquitin were purchased from Boston Biochem, anti-Ste7 from Santa Cruz Biotechnology, M2 anti-Flag antibodies and the mating pheromone α-factor from Sigma. Polyclonal IpaH antibodies have been described25. His6-tagged UBE2D2 (UbcH5b) was prepared as described26. GST-IpaH9.81–545, GST-IpaH9.8208–545 and all derivatives thereof were prepared as described25. Complexes containing Ste11-4, Ste7 and Kss1 were prepared as described27 and eluted from Flag M2 agarose—affinity gel (Sigma) using a Flag peptide.
Plasmids
The ipaH9.8 3’ region (codons 208 to 575) was amplified by PCR and cloned as a BamHI-XhoI fragment into pGEX6P1 to create pJR009. Site-directed mutagenesis of pJR009 was performed using the Stratagene Quick Change II kit (Invitrogen) to create pJR010 (C337A), pJR011 (C337S), pJR012 (E338A), pJR013 (D339A), pJR014 (R340A), pJR015 (R387A) and pJR016 (D397A). The ipaH9.8 entire coding sequence was cloned into pDONR201 (Invitrogen) and used for site-directed mutagenesis as described above. The destination vector pGAL1-CFLAG was used to create plasmids pMT4330 (wild type), pMT4331 (C337A), pMT4332 (C337S), pMT4333 (E338A), pMT4334 (D339A), pMT4335 (R340A), pMT4336 (R387A) and pMT4337 (D397A) for expression of Flag-tagged IpaH9.8 in yeast as described previously28. Plasmid pRT7 encoding GST-IpaH9.8 has been described25. In the case of the His6-tag constructs, the DNA fragments encoding full-length IpaH1.4 and the C-terminal domains of IpaH1.4 (residues 265 to 575) and IpaH9.8 (residues 208 to 545) were amplified from S. flexneri genomic DNA and cloned into the T7 expression vector in fusion with a DNA fragment encoding an N-terminal His6 tag followed by a TEV protease recognition and cleavage site29.
Protein purification and crystallization
IpaH-CTDs were expressed and purified as previously described29. Briefly, the expression plasmid for each polypeptide was transformed into Escherichia coli BL21-Gold (DE3) (Stratagene), which harbors an extra plasmid (pMgk) encoding three rare tRNAs (AGG and AGA for arginine, and ATA for isoleucine). These E. coli cells were then cultured in 1 liter LB medium supplemented with ampicillin (100 μg ml−1) and kanamycin (50 μg ml−1), and incubated at 37 °C with shaking until the culture reached an optical density at 600 nm (OD600) of 0.6–0.8. At this point the culture was induced with 0.4 mM IPTG and allowed to grow overnight at 15 °C. Cells were harvested by centrifugation, disrupted by sonication, and the insoluble material was removed by centrifugation. IpaH C-terminal fragments were purified using nickel—nitrilotriacetic acid (Ni-NTA) affinity chromatography and dialyzed and stored in a buffer containing 10 mM HEPES, pH 7.5, 500 mM NaCl and 0.5 mM tris-(2-carboxyethyl) phosphine (TCEP). Selenomethionine-enriched proteins were prepared after growth of bacteria in SeMet high-yield media (Shanghai Medicilon).
Crystallization was performed using proteins concentrated to 10.0 mg ml−1. Initial crystals were obtained by sitting drop vapor diffusion at room temperature (20–24 °C) using an optimized sparse matrix crystallization screen30. A single crystal from an optimized crystallization mixture containing 2M ammonium sulfate and 1% (w/v) PEG MME 2000 was cryoprotected by transferring to a mixture of 3M ammonium sulfate, 5% (w/v) PEG MME 2000, 0.5 M NaCl, 10mM HEPES, pH 7.5, and flash frozen in liquid nitrogen.
Data collection, structure determination and refinement
The structure of IpaH1.4265–575 was determined using the SAD method. Diffraction data collected at 100° K using synchrotron radiation on beamline 19-ID (Structural Biology Centre, Advanced Photon Source, Argonne National Laboratories) at the peak wavelength of the selenium K absorption edge were integrated and scaled using HKL2000 (ref. 31). Data collection and refinement statistics for this structure are summarized in Table 1. Using diffraction data between 50Å and 3.4Å, the positions of 15 of the 21 anomalous scatterers were found using SHELXD32 followed by heavy-atom refinement and maximum likelihood—based phasing as implemented in the autoSHARP program suite33,34. Phase improvement by density modification generated an interpretable experimental SAD map at 2.65 Å, allowing an initial model to be built using ARP/wARP35. The model was subsequently improved through alternate cycles of manual building using COOT36 and restrained refinement against a maximum-likelihood target with 5% of the reflections randomly excluded as an Rfree test set. All refinement steps were performed using REFMAC37 in the CCP4 program suite38. In total, 155 out of a possible 933 residues residing mostly at the N and C termini as well as flexible loop regions 328–337 and 447–460 were omitted owing to poor electron density. The final model comprising three molecules of IpaH1.4-CTD and solvent molecules refined to an Rwork of 22.8% and Rfree of 28.4%, including TLS parameterization39,40. Data collection, phasing and structure-refinement statistics are summarized in Table 1. The Ramachandran plot generated by PROCHECK41 showed excellent stereochemistry overall, with 99.4% of the residues in the most favored and additional allowed regions with no disallowed residues.
Table 1.
Data collection, phasing and refinement statistics
| IpaH-CTD SeMet | |
|---|---|
| Data collection | |
| Space group | I422 |
| Cell dimensions | |
| a = b, c (Å) | 128.9, 282.8 |
| Peak | |
| Wavelength | 0.97845 |
| Resolution (Å) | 49.21-2.65 |
| Rmerge | 7.3 (47.9) |
| I / σI | 35.3 (3.8) |
| Completeness (%) | 99.0 (99.5) |
| Redundancy | 8.2 (8.1) |
| Refinement | |
| Resolution (Å) | 50.0-2.65 |
| No. reflections | 34,706 |
| Rwork / Rfree | 22.8/28.4 |
| No. atoms | |
| Protein | 6,268 |
| Ligand/ion | 35 |
| Water | 24 |
| B-factors | |
| Protein | 61.2 |
| Ligand/ion | 54.7 |
| Water | 38.7 |
| r.m.s. deviations | |
| Bond lengths (Å) | 0.016 |
| Bond angles (°) | 1.586 |
Values in parentheses are for the highest-resolution shell.
In vitro assays
Ubiquitination reactions were performed in a 20-μl reaction mixture containing buffer A (50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 10 mM ATP, 10 mM MgCl2, 0.5 mM DTT), 4 μg of His6-tagged ubiquitin, 0.13 μg of E1 and 2 μg of E2 in the presence or absence of 2 μg of GST-IpaH9.81–545,GST-IpaH9.8208–545 or His6-IpaH1.4265–575. Reactions were incubated at 25 °C for the indicated period of time and stopped by the addition of an equal volume of 2x Laemmli sample buffer (0.125 M Tris-HCl, pH 6.8, 20% (v/v) glycerol, 4% (w/v) SDS, 0.004% (w/v) bromophenol blue) with or without 100 mM DTT. Reaction mixtures were separated by SDS-PAGE, transferred onto a nitrocellulose membrane and probed with specific antibodies.
The collection of human E2 expression constructs was received as a gift from the S. Dhe-Paganon laboratory at the Structural Genomics Consortium (http://thesgc.com/sgc-webpages/sgc-toronto.php). The constructs provided a T7 promoter—driven expression of the N-terminal poly-histidine fusion for each human E2 enzyme. The constructs were then transformed into E. coli BL21-Gold (DE3) (Stratagene), expressed, and purified using Ni-NTA affinity chromatography as described above.
GST pull-down assay
The GST-IpaH9.8208–545 and its C337A, C337S, D339A, R340A and D397A variants were incubated with His6-UBE2D2 in equimolar ratio for 2 h in PBS buffer (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.5) at 4 °C. We then added glutathione beads and precipitated the bound proteins by centrifugation. After extensive washing, the bound proteins were separated by SDS-PAGE and visualized by western blotting using anti-His and anti-IpaH antibodies.
Fluorescence polarization assay
Plasmids encoding UBE2D2 R5A, F62A and K63A variants were generated using the Quick Change Site-Directed Mutagenesis II kit (Stratagene) using the plasmid encoding His6-UBE2D2 as a template. E2s were incubated with fluorescein-5-maleimide (Molecular Probes) at a 1:25 molar ratio at 4 °C for 12 h. The completeness of labeling was confirmed by MS. Free fluorescent dye was removed by gel-filtration chromatography followed by dialysis. A starting concentration of the labeled E2 variants of 1–2 nM was selected according to the extent of conjugated fluorophore. Binding assays with IpaH1.4265–575 were performed in the buffer containing 25 mM HEPES Na, pH 7.5, 0.15 M NaCl, 0.5 mM TCEP. Fluorescence anisotropy was measured at 25 °C using a ‘Synergy 2’ fluorescence polarization instrument (Biotek) with the excitation wavelength set at 485 nm and emission wavelength set at 528 nm. Increasing amounts of IpaH1.4265–575 were added to aliquots of labeled E2s. Data from four measurements were averaged and fitted to a single-site binding model using nonlinear regression with GrafPad Prism 4 (GrafPad Software).
Supplementary Material
ACKNOWLEDGMENTS
We wish to thank the staff at the Argonne National Laboratory beam line 19-ID for assistance with data collection and A. Edwards for critical reading of the manuscript. We also wish to thank S. Dhe-Paganon and G. Avvakumov at the Structural Genomics Consortium, Toronto, for providing the collection of expression constructs for human E2-conjugating enzymes. We thank D. Briant for insight to E3 ubiquitination assays. This work was supported by US National Institutes of Health Grants GM62414-01, by the Ontario Research and Development Challenge Fund and by a grant from the Canadian Institutes of Health Research Grant. M.T. is supported by grants from the Canadian Institutes of Health Research (MT012466 and MOP-57795), the National Cancer Institute of Canada, the Royal Society and the Scottish Universities Life Sciences Alliance.
Footnotes
Accession codes. Protein Data Bank: IpaH1.4 C-terminal domain structure coordinates and factors have been deposited under accession code 3CKD.
Note: Supplementary information is available on the Nature Structural & Molecular Biology website.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/
References
- 1.Bhavsar AP, Guttman JA, Finlay BB. Manipulation of host-cell pathways by bacterial pathogens. Nature. 2007;449:827–834. doi: 10.1038/nature06247. [DOI] [PubMed] [Google Scholar]
- 2.Galán JE, Cossart P. Host-pathogen interactions: a diversity of themes, a variety of molecular machines. Curr. Opin. Microbiol. 2005;8:1–3. doi: 10.1016/j.mib.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 3.Ardley HC, Robinson PA. E3 ubiquitin ligases. Essays Biochem. 2005;41:15–30. doi: 10.1042/EB0410015. [DOI] [PubMed] [Google Scholar]
- 4.Mattoo S, Lee YM, Dixon JE. Interactions of bacterial effector proteins with host proteins. Curr. Opin. Immunol. 2007;19:392–401. doi: 10.1016/j.coi.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 5.Abramovitch RB, Janjusevic R, Stebbins CE, Martin GB. Type III effector AvrPtoB requires intrinsic E3 ubiquitin ligase activity to suppress plant cell death and immunity. Proc. Natl. Acad. Sci. USA. 2006;103:2851–2856. doi: 10.1073/pnas.0507892103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosebrock TR, et al. A bacterial E3 ubiquitin ligase targets a host protein kinase to disrupt plant immunity. Nature. 2007;448:370–374. doi: 10.1038/nature05966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Angot A, et al. Ralstonia solanacearum requires F-box-like domain-containing type III effectors to promote disease on several host plants. Proc. Natl. Acad. Sci. USA. 2006;103:14620–14625. doi: 10.1073/pnas.0509393103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Y, Higashide WM, McCormick BA, Chen J, Zhou D. The inflammation-associated Salmonella SopA is a HECT-like E3 ubiquitin ligase. Mol. Microbiol. 2006;62:786–793. doi: 10.1111/j.1365-2958.2006.05407.x. [DOI] [PubMed] [Google Scholar]
- 9.Diao J, Zhang Y, Huibregtse JM, Zhou D, Chen J. Crystal structure of SopA, a Salmonella effector protein mimicking a eukaryotic ubiquitin ligase. Nat. Struct. Mol. Biol. 2008;15:65–70. doi: 10.1038/nsmb1346. [DOI] [PubMed] [Google Scholar]
- 10.Kubori T, Hyakutake A, Nagai H. Legionella translocates an E3 ubiquitin ligase that has multiple U-boxes with distinct functions. Mol. Microbiol. 2008;67:1307–1319. doi: 10.1111/j.1365-2958.2008.06124.x. [DOI] [PubMed] [Google Scholar]
- 11.Buchrieser C, et al. The virulence plasmid pWR100 and the repertoire of proteins secreted by the type III secretion apparatus of Shigella flexneri. Mol. Microbiol. 2000;38:760–771. doi: 10.1046/j.1365-2958.2000.02179.x. [DOI] [PubMed] [Google Scholar]
- 12.Ashida H, Toyotome T, Nagai T, Sasakawa C. Shigella chromosomal IpaH proteins are secreted via the type III secretion system and act as effectors. Mol. Microbiol. 2007;63:680–693. doi: 10.1111/j.1365-2958.2006.05547.x. [DOI] [PubMed] [Google Scholar]
- 13.Rohde JR, Breitkreutz A, Chenal A, Sansonetti PJ, Parsot C. Type III secretion effectors of the IpaH family are E3 ubiquitin ligases. Cell Host Microbe. 2007;1:77–83. doi: 10.1016/j.chom.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 14.Dohlman HG, Song J, Ma D, Courchesne WE, Thorner J. Sst2, a negative regulator of pheromone signaling in the yeast Saccharomyces cerevisiae: expression, localization, and genetic interaction and physical association with Gpa1 (the G-protein alpha subunit) Mol. Cell. Biol. 1996;16:5194–5209. doi: 10.1128/mcb.16.9.5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Houben K, et al. Solution structure of the ubiquitin-conjugating enzyme UbcH5B. J. Mol. Biol. 2004;344:513–526. doi: 10.1016/j.jmb.2004.09.054. [DOI] [PubMed] [Google Scholar]
- 16.Dominguez C, et al. Structural model of the UbcH5B/CNOT4 complex revealed by combining NMR, mutagenesis, and docking approaches. Structure. 2004;12:633–644. doi: 10.1016/j.str.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 17.Winkler GS, et al. An altered-specificity ubiquitin-conjugating enzyme/ubiquitin-protein ligase pair. J. Mol. Biol. 2004;337:157–165. doi: 10.1016/j.jmb.2004.01.031. [DOI] [PubMed] [Google Scholar]
- 18.Eletr ZM, Kuhlman B. Sequence determinants of E2—E6AP binding affinity and specificity. J. Mol. Biol. 2007;369:419–428. doi: 10.1016/j.jmb.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beaudenon S, Dastur A, Huibregtse JM. Expression and assay of HECT domain ligases. Methods Enzymol. 2005;398:112–125. doi: 10.1016/S0076-6879(05)98011-7. [DOI] [PubMed] [Google Scholar]
- 20.Sun SC. Deubiquitylation and regulation of the immune response. Nat. Rev. Immunol. 2008;8:501–511. doi: 10.1038/nri2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang M, Pickart CM. Different HECT domain ubiquitin ligases employ distinct mechanisms of polyubiquitin chain synthesis. EMBO J. 2005;24:4324–4333. doi: 10.1038/sj.emboj.7600895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu PY, et al. A conserved catalytic residue in the ubiquitin-conjugating enzyme family. EMBO J. 2003;22:5241–5250. doi: 10.1093/emboj/cdg501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tolbert BS, et al. The active site cysteine of ubiquitin-conjugating enzymes has a significantly elevated pKa: functional implications. Biochemistry. 2005;44:16385–16391. doi: 10.1021/bi0514459. [DOI] [PubMed] [Google Scholar]
- 24.Verdecia MA, et al. Conformational flexibility underlies ubiquitin ligation mediated by the WWP1 HECT domain E3 ligase. Mol. Cell. 2003;11:249–259. doi: 10.1016/s1097-2765(02)00774-8. [DOI] [PubMed] [Google Scholar]
- 25.Mavris M, et al. Regulation of transcription by the activity of the Shigella flexneri type III secretion apparatus. Mol. Microbiol. 2002;43:1543–1553. doi: 10.1046/j.1365-2958.2002.02836.x. [DOI] [PubMed] [Google Scholar]
- 26.Kim DW, et al. The Shigella flexneri effector OspG interferes with innate immune responses by targeting ubiquitin-conjugating enzymes. Proc. Natl. Acad. Sci. USA. 2005;102:14046–14051. doi: 10.1073/pnas.0504466102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Breitkreutz A, Boucher L, Tyers M. MAPK specificity in the yeast pheromone response independent of transcriptional activation. Curr. Biol. 2001;11:1266–1271. doi: 10.1016/s0960-9822(01)00370-0. [DOI] [PubMed] [Google Scholar]
- 28.Ho Y, et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- 29.Zhang RG, et al. Structure of Thermotoga maritima stationary phase survival protein SurE: a novel acid phosphatase. Structure. 2001;9:1095–1106. doi: 10.1016/s0969-2126(01)00675-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kimber MS, et al. Data mining crystallization databases: knowledge-based approaches to optimize protein crystal screens. Proteins. 2003;51:562–568. doi: 10.1002/prot.10340. [DOI] [PubMed] [Google Scholar]
- 31.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 32.Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr. D Biol. Crystallogr. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 33.Bricogne G, Vonrhein C, Flensburg C, Schiltz M, Paciorek W. Generation, representation and flow of phase information in structure determination: recent developments in and around SHARP 2.0. Acta Crystallogr. D Biol. Crystallogr. 2003;59:2023–2030. doi: 10.1107/s0907444903017694. [DOI] [PubMed] [Google Scholar]
- 34.de la Fortelle E, Bricogne G. Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Methods Enzymol. 1997;276:472–494. doi: 10.1016/S0076-6879(97)76073-7. [DOI] [PubMed] [Google Scholar]
- 35.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 36.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 37.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 38.Collaborative Computational Project, Number 4 The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 39.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 40.Winn MD, Murshudov GN, Papiz MZ. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 2003;374:300–321. doi: 10.1016/S0076-6879(03)74014-2. [DOI] [PubMed] [Google Scholar]
- 41.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.