

Conspectus
Activity and selectivity are typically the first considerations when designing a drug. However, absorption, distribution, metabolism, excretion, and toxicity (ADMET) are equally important considerations. Peptides can provide a combination of potent binding and exquisite selectivity, as evidenced by their pervasive use as enzymes, hormones, and signaling agents within living systems. In particular, peptidic turn motifs are key elements of molecular recognition. They may be found at the exposed surfaces of globular proteins, where they are available for binding interactions with other peptides and small molecules.
However, despite these advantages, peptides often make poor drugs. The amide backbone is subject to rapid enzymatic proteolysis, resulting in short half-lives. Furthermore, the ability of the amide backbone to hydrogen-bond with water restricts its ability to cross membranes and, consequentially, results in poor oral bioavailability. Accordingly, the development of non-peptidic scaffolds that mimic peptidic turn motifs represents a promising means of converting peptidic agents into more drugable molecules. In this Account, we describe the design and synthesis of β-turn mimetics that use a β-D-glucose scaffold—the first use of a sugar scaffold for this purpose.
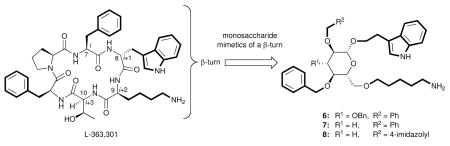
Somatostatin (SRIF) is a small-protein (14 amino acid residues) human hormone; a shorter (6 amino acid residues) synthetic peptide, L-363-301, is a fully peptidal agonist. These two cyclic peptides share the β-turn motif comprising Phe7-Trp8-Lys9-Thr10(D-Trp8 in the case of L-363,301), of which the Trp and Lys residues in the i+1 and i+2 positions respectively are critical for binding. In 1988, we initiated a program that tested and validated the then-novel proposition that the β-D-glucose scaffold can mimic the β-turn in L-363,301. The β-D-glucose scaffold proved to be an attractive mimic of a β-turn in part because it permits the convenient attachment of amino acid side chains via facile etherification reactions, rather than carbon–carbon bond formations; it is also an inexpensive starting material with well-defined stereochemistry. From the beginning, biological assays were used alongside physical measurements to assess the relevance of the design. Our first two synthetic targets, compounds 6 and 7, bound the SRIF receptors on benchmark (AtT-20) cells, albeit weakly, consistent with the objective of the design. Subsequently, a better ligand (8) and two congeners were found to be agonists at the SRIF receptors, providing convincing evidence that the peptide backbone is not required for receptor binding or signal transduction.
The unexpectedly high level of receptor affinity of selected analogs—as well as the fortuitous discovery that our peptidomimetics were active against several chemically distinct receptors—led us tohypothesize that these monosaccharides could access multiple potential binding modes. Our later studies of this sugar scaffold confirmed this property, which we termed pseudosymmetry, whereby multiple similar but non-identical motifs are displayed within a single analog. We propose the presence of pseudosymmetry to be an element of privilege and an advantage for lead discovery.
Introduction
Peptides play a fundamental role in biology. However, their use as therapeutics has been limited by their poor pharmacokinetic properties.1 Peptides are subject to proteolysis, which results in short biological half-lives even after parenteral administration. Furthermore, peptides have poor bioavailability because of their poor cellular transport properties. Rapid proteolysis of peptides can be overcome in several ways, including the use of amide surrogates, retro enantiomeric peptides,2 or simplified cyclic peptides. Additionally, D-amino acids may be incorporated.3 Stein noted that the cellular transport of organic compounds correlated inversely with their ability to hydrogen bond with water, and concluded that solvation impedes transport because the desolvation process during the extraction of a molecule into a lipid bilayer from an aqueous phase requires energy.1c Similarly, Diamond and Wright found that 1,2-cyclohexanediol crosses cell membranes more readily than 1,3-diols, a result attributed to the fact that the former can form an intramolecular hydrogen bond, thus reducing solvation and, in turn, decreasing the energy required for desolvation.1d More recently, we reported that β-strands in which the peptide backbone is replaced by a pyrrolinone scaffold show enhanced transport kinetics, presumably because of the reduced energetic cost of desolvation.4,5
The poor metabolic stability and transport properties associated with the backbone of potential peptidal therapeutic agents led us to investigate the development of peptidomimetics.6 The design of peptidomimetics was anticipated by Farmer, who proposed, but did not explore, the then novel idea of replacing a peptide scaffold with a cyclohexane ring and attaching relevant amino acid side chains to this construct.7 Subsequently, Bélanger and Dufresne implemented this proposal, describing non-peptidic mimic 1 (Figure 1) of the enkephalins (see 4 and 5, Figure 2) that incorporated the bicyclo [2.2.2]-octane scaffold.8 Later, Olson and collaborators reported peptidomimetic 2 (Figure 1), which was designed to mimic the tripeptide thyroid releasing hormone (TRH).9 Although 2 did not bind the endocrine receptor for which it was designed, it showed oral activity in animal models of cognitive dysfunction.
Figure 1.

Early designed peptidomimetics.
Figure 2.

Morphine (3) and enkephalins (4, 5).
Replacing the amide backbone with a nonpeptidal scaffold devoid of the capacity to hydrogen bond with the receptor can succeed only if the hydrogen bonds between the amide backbone of the peptide ligand and that of the receptor are not required for binding or signal transduction.10 The enkephalins (4 and 5) are the endogenous peptide ligands for the morphine receptor.11 The activity of the non-peptidic natural product morphine (3, Figure 2) provides compelling evidence that an amide backbone is not always required for binding of peptidal receptor ligands, or, indeed, for signal transduction by peptide hormones or neurotransmitters.7
In contrast, ligands and inhibitors of proteolytic enzymes often take advantage of critical backbone hydrogen bonds. The importance of these interactions has been well established by X-ray crystallography,12 emphasizing the functional role of the amide backbone in enzymatic catalysis. For example, backbone hydrogen bonds contribute to the optimal strict alignment of proteolytic enzymes with their substrates and facilitate the efficient catalysis of the resulting reactions. This does not preclude a role for induced fit, and, indeed, Kern et al. demonstrated in NMR studies that rapid motion of one residue of cyclophilin A is required during catalysis.12 In contrast with proteolytic enzymes, guanine nucleotide-binding protein coupled receptors (GPCRs) interact with ligands through helical bundles in which hydrogen bonding opportunities with ligands are largely unavailable.13
Our peptidomimetic program, initiated in 1988, employed monosaccharide scaffolds for the attachment of amino acid-mimicking side chains via ether linkages.14 Supported by extensive biological studies, our research has provided strong support for Farmer’s speculations.7
Somatostatin (SRIF)
Somatostatin (somatotropin release inhibiting factor, SRIF, see Figure 3) is a peptide hormone that was isolated, characterized and synthesized by Guillemin, Rivier, Vale and coworkers at the Salk Institute.15 SRIF is a cyclic tetradecapeptide which inhibits the release of several physiologically important substances, including growth hormone, glucagon, insulin, and gastric acid. SRIF is produced and released throughout the central nervous system and in major peripheral organs, such as the stomach and pancreas.16 Within the nervous system, SRIF acts as a neuromodulator, with physiological effects on neuroendocrine, motor, and cognitive functions. At the periphery, SRIF is a modulator of endocrine and exocrine functions and also regulates the differentiation and proliferation of normal and tumor cells.17
Figure 3.

Somatostatin (SRIF), L-363,301 (a potent agonist), and first-generation monosaccharide peptidomimetics (6–8).
Somatostatin is present in two forms, SRIF-14 and SRIF-28, which contain 14 and 28 (SRIF-28) amino acids, respectively.18 Both are formed by selective cleavage of prosomatostatin, a common precursor, and bind to SRIF receptors that are coupled to heterotrimeric guanine nucleotide-binding proteins. The SRIF receptors thus belong to the GPCR family.13 Five human SRIF receptor subtypes (hSSTR1–5) have been cloned and expressed, and the localization of specific receptor subtypes within the body has facilitated the elucidation of their individual functions.18,19 It is worth noting that the lack of specificity of SRIF for the receptor subtypes does not result in undesirable physiological side effects since SRIF is released in close proximity to the intended receptors and rapidly metabolized.
Since peptides normally exist in solution as an equilibrium mixture of conformers, knowing the bioactive conformation is invaluable when seeking to simplify the hormone or design a peptidomimetic. Building on SARs established at the Salk Institute,20 Merck scientists proposed a bioactive conformation of SRIF which has served as the basis for subsequent research related to this hormone.21 The fact that D-Trp8-SRIF is ten-fold more potent suggested that Trp8 might occupy the i+1 position of a β-turn.20 This hypothesis was based on theoretical calculations by Ramachandran establishing that a D-amino acid in the i+1 position of a β-turn stabilizes the motif,22 and subsequently validated by Veber and collaborators.21 This information, together with extensive SARs elucidated at both the Salk Institute23 and Merck,24 revealed that the residues required for binding and signal transduction are contained in the tetrapeptide sequence Phe7-Trp8-Lys9-Thr10 that defines the β-turn motif. Arison, Hirschmann, Veber, and their collaborators recognized that the bioactive conformation, especially in the D-Trp8 series (possessing a type II′ β-turn), places the indole side chain of Trp8 in close proximity to the side chain of Lys9.21,25 Extensive NMR studies in combination with the SAR data led the Merck team to propose that the bioactive conformation of SRIF resembles more closely the solution conformation of the D-Trp diastereomer.25
Constrained SRIF Analogs
The importance of SRIF in physiology has suggested a potential therapeutic role for a longer acting analog or peptidomimetic of this hormone.26 Having established the bioactive conformation of SRIF, the Merck team set out to develop a lower molecular weight peptide in hopes of improving both stability toward proteases and oral bioavailability.1b Veber and associates designed and synthesized cyclic hexapeptides, known at the time to be metabolically stable, that incorporated the critical β-turn.21 Due to limits in molecular modeling capabilities, the choice of the dipeptide Phe-Pro linkage in L-363,301 (Figure 3) was the result of a systematic variation of the dipeptide linker unit. Importantly, the side chains of the critical Phe7-D-Trp8-Lys9-Thr10 β-turn of L-363,301 displayed the same orientation as in D-Trp8-SRIF. Later, the more potent cyclic hexapeptide MK-678 (Figure 4) was prepared.27 The Phea residue in both analogs was found to be important and thought to mimic the hydrophobic region defined by amino acids Asn5 and Thr12 of SRIF. Clinical evaluation of MK-678 confirmed the hoped-for longer duration of action (>4 hours) when administered orally.1a However, the oral bioavailability of MK-678 is only 1–3%. In contrast, Sandoz discovered and successfully developed the SRIF octapeptide analog octreotide (Figure 4) for cancer chemotherapy and the treatment of acromegaly.28 Octreotide has to be administered parenterally, but provides adequate plasma half-lives.
Figure 4.

MK-678 and octreotide.
The observation that even small, metabolically stable peptides such as MK-678 lack adequate oral bioavailability suggested to Hirschmann in the 1980s that the amide backbone may be at least partially responsible for the poor transport properties. Therefore, the search for metabolically stable SRIF receptor ligands with good oral bioavailability continued. The discovery by Freidinger, Veber, and coworkers that a modified retro-enantio cyclic analog of MK-678 (9, Figure 5) possessed biological activity29 strengthened the proposition that the amide backbone of the cyclic hexapeptides and SRIF is not necessary for binding or signal transduction. Taken together, these facts suggested to us that the side chains of the β-turn of L-363,301, if attached to an appropriate non-peptidic scaffold, might bind the SRIF receptors. This proposition resulted in the initiation of a research project for the development of peptidomimetics of SRIF at the University of Pennsylvania.
Figure 5.

Retro-enantio cyclic analog of MK-678 (9).
The main objective of the SRIF peptidomimetic program was the discovery of molecules with improved pharmacokinetic properties.14a Using the NMR-based solution conformation of cyclic hexapeptide L-363,301 as a guide, we eventually turned to the use of β-D-glucose as a scaffold. The resulting glucoside 6 (Figure 3) appeared to overlay well with L-363,301, with the side chains of 6 at C2, C1 and C6 mimicking the Phe7, D-Trp8 and Lys9 residues of L-363,301 at the i, i+1 and i+2 positions of the β-turn, respectively. Thr10, the fourth amino acid of this β-turn, was not believed to be required for binding and, therefore, was not incorporated into the design of 6. The choice of the sugar scaffold offered several advantages over hydrocarbon scaffolds as suggested by Farmer,7 such as a well-defined conformation, stereochemical purity of diverse starting materials, and relatively well-precedented etherification reactions as compared with carbon–carbon bond forming reactions.
Gratifyingly, biological evaluation of glucoside 6 against the SRIF receptors on membranes from both the cerebral cortex and the pituitary, as well as receptors on the surface of AtT-20 cells, demonstrated that 6 indeed binds the SRIF receptors. 3-Deoxy analog 7 (Figure 3) also possessed comparable affinity. Importantly, a functional assay performed at the Salk Institute revealed that 6 acted as an SRIF partial agonist.14b The success in the design of the peptidomimetic provided crucial experimental evidence that β-D-glucose does indeed represent a β-turn mimetic. The result confirmed our design premises, notably that only three of the four β-turn side chains of SRIF are necessary for binding, and hydrogen bonding with the backbone of the receptor is not mandatory for activity. An overlay of glucoside 6 with SRIF is shown in Figure 6.30
Figure 6.
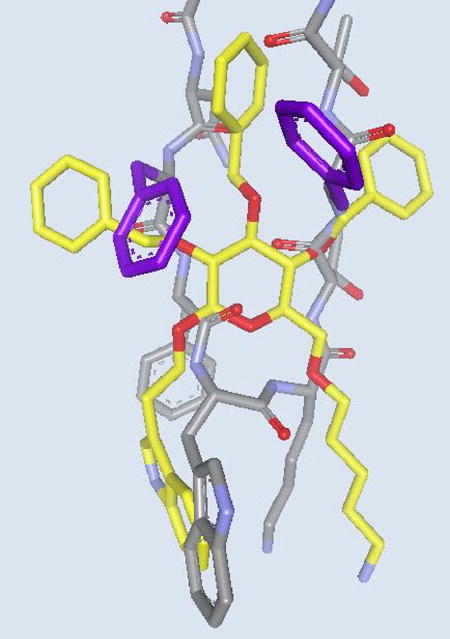
Comparison of SRIF-14 and glucoside 6 (yellow carbons). Note the overlap of the two essential side chains (Trp8 and Lys9 and their mimics in 6. Phe6 and Phe11 of SRIF-14, which are thought to stabilize its bioactive conformation, are shown in purple.
The expected β-turn mimicry of the design was validated by correlating the effects of side chain modifications on the potency of the glucoside 7 with the changes to binding affinities induced by the corresponding changes to L-363,301 (see Figure 7). As with the cyclic hexapeptides, the relative importance of the Lys9 mimicking side chain for binding is readily apparent. An additional pleasing result was the affinity enhancement induced both by the replacement of Phe 7 in L-363,301 with His7 (i.e. 10, Figure 8) and the analogous replacement of the C2 benzyl group of 7 by a methylimidazole side chain (i.e. 8).14c Taken together, the SARs of the cyclic hexapeptide and the corresponding pyranoside comprise a proof of concept for a designed mimetic whereby the side chains of Phe7-D-Trp8-Lys9 correspond to those attached to the sugar at C2, C1 and C6, respectively. Furthermore, there is substantial evidence suggesting that the benzyl group at C4 mimics Phea.
Figure 7.

Comparison of the SAR profiles of peptide analogs and β-D-glucose-based peptidomimetics.
Figure 8.

Incorporation of a methylimidazole group and the resulting enhancement in activity and affinity.
Pseudosymmetry: an Element of Privilege
Early on, we made two unexpected observations which we now understand to have a common chemical origin. First, we discovered that 11 (Figure 9), which lacks a side chain mimicking Trp8 of SRIF (a residue thought to be an absolute requirement for binding SRIF receptors), has higher affinity than 7 for SRIF receptors on AtT-20 cells.14b,31 At the time, we explained this result by proposing an “alternative binding mode” involving a reorientation of 11 to give 11′, which places the C4 benzyl group in a position so as to mimic the spatial relationship between the Trp 8 and Lys9 side chains of SRIF and D-Trp8-SRIF.
Figure 9.

des-Indole compound 11 shown (a) in a binding mode which is unable to present an aromatic, tryptophan-mimicking side chain to the receptor and (b) in an alternative binding mode (11′).
More recently, we have recognized that this alternative binding mode of 11 (i.e. 11′) places the C4-benzyl group into the Trp binding pocket (see Figure 9), a manifestation of what we have termed the pseudosymmetry of the glucose scaffold.32 We were able to confirm this rationalization experimentally by preparing the C3-O-methylimidazole congener (structure not shown), which we were pleased to find possessed increased affinity for the SRIF receptor.31 Failure of the 4-des-benzyl analog of 11 to bind the SRIF receptor is also consistent with the alternate binding mode hypothesis.14c,d
Equally unexpected was the discovery that 6 also binds the human neurokinin 1 (hNK1) receptor of substance P (SP) as an antagonist and with a higher affinity (IC50 150 nM) than for the SRIF receptors.14a This was surprising since the peptides SRIF and SP are chemically unrelated and do not bind each other’s receptors. Although hNK1 and hSST1–5 are both GPCRs, they share little sequence homology in the ligand binding domain.33 Nonetheless, there are likely to be similarities between GPCRs, as evidenced by the existence of “privileged” structures and the proposed common small molecule binding domain of GPCRs.34 We have recently proposed that evolutionarily conserved ligand binding sites provide regions that accommodate the projections of side chains from the exposed surface of β- and γ-turns and their components or mimics, and helices may be similarly available for binding interactions.32 We further suggested that the ability of GPCRs to recognize privileged ligand scaffolds makes them complementary to privileged platforms. We initially interpreted the ability of 6 to bind the hNK1 receptor to reflect only such similarities between the SRIF and hNK1 receptors. Additionally, we now propose that the ability of 6 to bind the SRIF, SP, and β2-adrenergic receptors14c reflects not only a similarity between the three corresponding GPCRs, but also the high degree of pseudosymmetry in the sugar scaffold.
The term “privileged structure” was introduced by Evans et al. to describe the fact that the benzodiazepine scaffold can be made to bind diverse receptors, especially (but by no means exclusively) GPCRs, by modulating the precise structure of the scaffold and substituents.35 We use the word “polyvalent” (an extension of bivalent/divalent)36 to describe compounds which bind more than one receptor or enzyme. Other structures such as the steroid scaffold, cyclic hexapeptides, and diketopiperazines37 have long been recognized as being privileged; other structures have recently been added to the list.38 The benzopyran core was identified by Murcko, who used shape description methods to identify molecular frameworks that are common among commercially available drugs,39 and Nicolaou and collaborators later described natural product-like combinatorial libraries incorporating benzopyrans.40 To our knowledge, there has been no report of a unifying chemical basis common to all of these scaffolds. We believe, however, that it may be possible to identify structural elements which, if one or more are present in a scaffold, confer polyvalency.
Believing that the ability of glucosides 6 and 7 to bind both the SRIF and hNK1 receptors reflects a hitherto unrecognized similarity between these receptors, we sought to convert the cyclic hexapeptide L-363,301, which binds only SRIF receptors, into a selective ligand for the hNK1 receptor. This was readily accomplished by replacing Lys 9 with Phe9 (12, Figure 10) or p-F-Phe9 (13).14d These results, though pleasing, raised new questions. Why does glucoside 6 bind both receptors, whereas an amino acid substitution is required in the i+2 position of L-363,301 (Lys9 → Phe9) to generate a ligand for the hNK1 receptor? Moreover, why were we unable to use the discovery that 13 is a more potent peptidal hNK1 receptor antagonist than 12 to design a superior glucoside?
Figure 10.
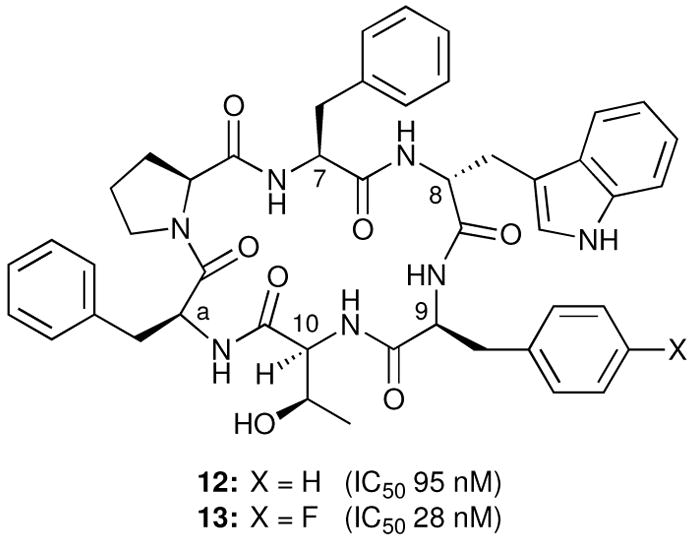
L-363,301-based hNK1 receptor ligands that do not bind to SRIF receptors.
We attribute this difference between the peptides and the glucosides to the fact that the sugar scaffold, but not constrained cyclic hexapeptides such as L-363,301, presents a multitude of binding modes (i.e. high pseudosymmetry), as shown in Figure 11.32 It is important to note that the cyclic hexapeptides L-363,301, 12 and 13 all bind their respective receptors through the same key interactions, namely those of the i+1 and i+2 residues of their β-turns. In contrast, glucoside 6 binds the SRIF receptors via the C1 and C6 side chains, but binds the hNK1 receptor via the C2 and C1 side chains.
Figure 11.

The advantage of pseudosymmetry in lead discovery. Shown are (a) the ten possible ways one pyranoside can present i+1 and i+2 mimicking motifs (marked in bold) to a GPCR and (b) the four possible ways one cyclic hexapeptide can display the i+1 and i+2 residues of a β-turn.
The existence of multiple binding modes (i.e. pseudosymmetry) within the glucoside scaffold arises from the presence of nearest neighbor diols which present appended functionality in a β-turn-like orientation in a similar, but non-identical, manner. This property of glucoside 6 explains its ability to bind the SRIF and NK1 receptors via different side chains. Thus, the pseudosymmetry of a monosaccharide scaffold allows a single functionalized sugar to present a multitude of turn-mimicking side chains to a receptor, enhancing the probability of finding a favorable interaction. Therefore, for a compound library generated for lead discovery in diverse screens, incorporation of highly pseudosymmetric scaffolds which display several functionalities in a β-turn-like manner should increase the likelihood of obtaining one or more leads.
Combinatorial chemistry/parallel synthesis, high throughput screening and database mining41 have emerged as the principal underpinnings for the discovery of new leads.38 A scaffold exhibiting multiple potential binding motifs should confer an enhanced opportunity to bind one or more receptors. We see such polyvalency as an advantage, not a liability, because we have demonstrated the ability to subsequently incorporate specificities into 6. For example, 14 (Figure 12), lacking a C4-benzyl substituent, binds the hNK1 receptor (IC 50 22 nM) but does not bind the SRIF receptors.14d Conversely, the C3 benzyl substituent is important for hNK1 receptor binding but not SRIF receptor binding, and incorporation of a methyl imidazole at C2 enhances SRIF affinity while eliminating hNK1 receptor binding.
Figure 12.

Development of a receptor-specific peptidomimetic (14).
Summary and Prospects
In 1990, we reported the design and synthesis of glucose derivative 6, a ligand that binds SRIF receptors, albeit weakly. It represented the first use of a sugar scaffold to mimic a β-turn. We found glucosides, like cyclic hexapeptides, to be a privileged class. Furthermore, we demonstrate herein that two surprising biological results, namely the unexpected finding that a glucoside lacking a Trp-mimicking side chain (11) is a better ligand than 6 for hSSTR4 and the serendipitous discovery that 6 also binds the hNK1 receptor, have a common chemical basis. These results are explained by pseudosymmetry, which is present to a larger extent in suitably substituted glucosides than in cyclic hexapeptides. This element of privilege allows a single functionalized sugar to offer many different combinations of β-turn mimicking side chains to a given receptor and appears to be a significant advantage for lead discovery.
Acknowledgments
We thank our many coworkers who have contributed to this project, and whose names appear in the cited references. Financial support for this work was provided by the National Institutes of Health through Grant GM-41821. R.F.H. also thanks the Merck Research Laboratories for an unrestricted grant. We are indebted to Benjamin Dyme for his assistance in the preparation of this manuscript and Madeleine Joullié for her invaluable support and constant encouragement.
Biographies
Ralph F. Hirschmann was born in Bavaria, Germany, and came to the U.S. in his teens. He graduated from Oberlin College and then served in the U.S. Army in the Pacific Theater during World War II. He resumed his education at the University of Wisconsin, Madison, where he was the Sterling Winthrop Fellow. He completed his Ph.D. studies in 1950 under the guidance of W. S. Johnson and joined Merck & Co., Inc. In 1987, at age 65, he retired from Merck, where he was Senior Vice President for Basic Research, and joined the faculty at the University of Pennsylvania as the Makineni Professor. At Merck, his team discovered Mevacor®, Vasotec®, Prinivil®, Primaxin®, Proscar®, and Ivermectin®. In 1969, Robert G. Denkewalter, Hirschmann, and their collaborators reported the first total synthesis of an enzyme in solution.
K. C. Nicolaou was born in Cyprus in 1946. He studied chemistry at the University of London (B.Sc., 1969, Bedford College; Ph.D., 1972, University College, Professors F. Sondheimer and P. J. Garratt). After postdoctoral appointments at Columbia University (1972–1973, Professor T. J. Katz) and Harvard University (1973–1976, Professor E. J. Corey), he joined the faculty at the University of Pennsylvania, where he became the Rhodes–Thompson Professor of Chemistry. In 1989, he accepted joint appointments at the University of California, San Diego, where he is Distinguished Professor of Chemistry, and The Scripps Research Institute, where he is Chairman of the Department of Chemistry and holds the Skaggs Professorship of Chemical Biology and the Darlene Shiley Chair in Chemistry. His dedication to chemical education is reflected in his training of hundreds of graduate students and postdoctoral fellows.
Angie R. Angeles was born in the Philippines in 1975. She received her B.S. degree (with distinction) in biochemistry/chemistry from the University of California, San Diego in 1999, where she conducted research as a Howard Hughes fellow under the direction of Professor Charles L. Perrin. After stints at Memorial Sloan–Kettering Cancer Center (Avon Foundation fellow, Professor Jon Thorson) and The Scripps Research Institute (Professor Jeffery W. Kelly), she pursued graduate studies at the University of Pennsylvania, where she obtained her Ph.D. degree in organic chemistry in 2005 under the mentorship of Professors Ralph F. Hirschmann and Amos B. Smith, III. After a NIH postdoctoral fellowship with Professor Samuel J. Danishefsky at Memorial Sloan–Kettering Cancer Center, she joined Schering–Plough in Cambridge, MA, where she is a Senior Scientist.
Jason S. Chen was born in Taipei, Taiwan in 1979. He received his A.B. and A.M. degrees in 2001 from Harvard University, where he performed research under the supervision of Professor Matthew D. Shair. He then joined Enanta Pharmaceuticals (Watertown, MA) as a medicinal chemist studying novel cyclosporine A analogs. He joined Professor K. C. Nicolaou’s group at The Scripps Research Institute in 2003, where he was a National Defense Science and Engineering Graduate (NDSEG) Fellow. In 2008, he completed his Ph.D. studies on the total synthesis and biological evaluation of uncialamycin. He is currently a research associate in Professor Nicolaou’s laboratory.
Amos B. Smith, III was born in Lewisburg, PA in 1944 and completed Bucknell University’s inaugural B.S.–M.S. degree in chemistry in 1966. After a year in medical school at the University of Pennsylvania, he entered The Rockefeller University, completing his Ph.D. degree in 1972. After a year as a Research Associate at Rockefeller, he joined the Department of Chemistry and the Monell Chemical Senses Center at the University of Pennsylvania, where he is currently the Rhodes–Thompson Professor of Chemistry and a Full Member of the Monell Center. From 1988 to 1996, he served as Chair of the Department of Chemistry. In addition, he is Associate Director of the Penn Center for Molecular Discovery, holds Honorary Membership in the Kitasato Institute and the Pharmaceutical Society of Japan, and serves as the inaugural Editor-in-Chief of Organic Letters.
References
- 1.(a) Veber DF, Saperstein R, Nutt RF, Freidinger RM, Brady SF, Curley P, Perlow DS, Paleveda WJ, Colton CD, Zacchei AG, Tocco DJ, Hoff DR, Vandlen RL, Gerich JE, Hall L, Mandarino L, Cordes EH, Anderson PS, Hirschmann R. A Super Active Cyclic Hexapeptide Analog of Somatostatin. Life Sciences. 1984;34:1371–1378. doi: 10.1016/0024-3205(84)90009-2. [DOI] [PubMed] [Google Scholar]; (b) Veber DF, Freidinger RM. The Design of Metabolically-Stable Peptide Analogs. TINS. 1985:392–396. [Google Scholar]; (c) Stein WD. The Movement of Molecules Across Cell Membranes; Eds. Academic; New York: 1967. pp. 65–125. [Google Scholar]; (d) Diamond JM, Wright EM. Molecular Forces Governing Non-Electrolyte Permeation Through Cell Membranes. Proc R Soc B. 1969;172:273–316. doi: 10.1098/rspb.1969.0022. [DOI] [PubMed] [Google Scholar]
- 2.Shemyakin MM, Ovchinnikov YA, Ivanov VJ. Topochemical Investigations on Peptide Systems. Angew Chem Int Ed Engl. 1969;8:492–499. doi: 10.1002/anie.196904921. [DOI] [PubMed] [Google Scholar]
- 3.(a) Freidinger RM, Perlow DS, Veber DF. Protected Lactam-Bridged Dipeptides for Use as Conformational Constraints in Peptides. J Org Chem. 1982;47:104–109. [Google Scholar]; (b) Pelton JT, Gulya K, Hruby V, Duckles SP, Yamamura HI. Conformationally Restricted Analogs of Somatostatin with High μ-Opiate Receptor Specificity. Proc Natl Acad Sci USA. 1985;82:236–239. doi: 10.1073/pnas.82.1.236. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Li W, Hanau CE, d’Avignon A, Moeller KD. Conformationally Restricted Peptide Mimetics: The Incorporation of 6,5-Bicyclic Lactam Ring Skeletons into Peptides. J Org Chem. 1995;60:8155–8170. [Google Scholar]
- 4.Smith AB, III, Keenan TP, Holcomb RC, Sprengeler PA, Guzman MC, Wood JL, Carrol PJ, Hirschmann R. Design, Synthesis, and Crystal Structure of a Pyrrolinone-Based Peptidomimetic Possessing the Conformation of a β-Strand: Potential Application to the Design of Novel Inhibitors of Proteolytic Enzymes. J Am Chem Soc. 1992;114:10672–10674. [Google Scholar]
- 5.Hirschmann R. Medicinal Chemistry in the Golden Age of Biology. Lessons from Steroid and Peptide Research. Angew Chem Int Ed Engl. 1991;30:1278–1301. [Google Scholar]
- 6.Sawyer TK. In: Structure-Based Drug Design: Diseases, Targets, Techniques and Developments. Veerapandian P, editor. Marcel Dekkar; New York: 1997. pp. 559–634. [Google Scholar]
- 7.Farmer PS. Bridging the Gap Between Bioactive Peptides and Nonpeptides: Some Perspectives in Design. In: Ariens EJ, editor. Drug Design. Academic; New York: 1980. pp. 119–143. [Google Scholar]
- 8.Belanger PC, Dufresne C. Preparation of Exo-6-benzyl-Exo-2-(m-Hydroxyphenyl)-1-Dimethylaminomethylbicyclo[2.2.2]octane. A Non-Peptide Mimic of Enkephalins. Can J Chem. 1986;64:1514–1520. [Google Scholar]
- 9.Olson GL, Cheung HC, Chiang E, Madison VS, Sepinwall J, Vincent GP, Winkokur A, Gary KA. Peptide Mimetics of Thyrotropin-Releasing Hormone Based on a Cyclohexane Framework: Design, Synthesis, and Cognition-Enhancing Properties. J Med Chem. 1995;38:2866–2879. doi: 10.1021/jm00015a009. [DOI] [PubMed] [Google Scholar]
- 10.(a) Rudinger J. Design of Peptide Hormone Analogs. In: Ariens EJ, editor. Drug Design, Vol 2. Vol. 2. Academic Press; New York: 1971. pp. 319–419. [Google Scholar]; (b) Urry DW, Walter R. Proposed Conformation of Oxytocin in Solution. Proc Natl Acad Sci USA. 1971;68:956–958. doi: 10.1073/pnas.68.5.956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hughes J, Smith TW, Kosterlitz HW, Fothergill LA, Morgan BA, Morris HR. Identification of Two Related Pentapeptides from the Brain with Potent Opiate Agonist Activity. Nature. 1975;258:577–579. doi: 10.1038/258577a0. [DOI] [PubMed] [Google Scholar]
- 12.Eisenmesser EZ, Bosco DA, Akke M, Kern D. Enzyme Dynamics During Catalysis. Science. 2002;295:1520–1523. doi: 10.1126/science.1066176. [DOI] [PubMed] [Google Scholar]
- 13.(a) Bikker JA, Trumpp-Kallmeyer S, Humblet C. G-Protein Coupled Receptors: Models, Mutagenesis, and Drug Design. J Med Chem. 1998;41:2911–2927. doi: 10.1021/jm970767a. [DOI] [PubMed] [Google Scholar]; (b) Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal Structure of Rhodopsin A: A G Protein-Coupled Receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 14.(a) Nicolaou KC, Salvino JM, Raynor K, Pietranico S, Reisine T, Freidinger RM, Hirschmann R. In: Design and Synthesis of a Peptidomimetic Employing β-D-Glucose for Scaffolding. Rivier JE, Marshall GR, editors. ESCOM; Leiden: 1990. pp. 881–884. [Google Scholar]; (b) Hirschmann R, Nicolaou KC, Pietranico S, Salvino J, Leahy EM, Sprengeler PA, Furst G, Smith AB, III, Strader CD, Cascieri MA, Candelore MR, Donaldson C, Vale W, Maechler L. Nonpeptidal Peptidomimetics with β-D-Glucose Scaffolding. A Partial Somatostatin Agonist Bearing a Close Structural Relationship to a Potent, Selective Substance P Antagonist. J Am Chem Soc. 1992;114:9217–9218. [Google Scholar]; (c) Hirschmann R, Nicolaou KC, Pietranico S, Leahy EM, Salvino J, Arison B, Cichy MA, Spoors PG, Shakespeare WC, Sprengeler PA, Hamley P, Smith AB, III, Reisine T, Raynor K, Maechler L, Donaldson C, Vale W, Freidinger RM, Cascieri MA, Strader CD. De Novo Design and Synthesis of Somatostatin Non-Peptide Peptidomimetics Utilizing β-D-glucose as a Novel Scaffolding. J Am Chem Soc. 1993;115:12550–12568. [Google Scholar]; (d) Hirschmann R, Yao WQ, Cascieri MA, Strader CD, Maechler L, Cichy-Knight MA, Hynes J, Jr, van Rijn RD, Sprengeler PA, Smith AB., III Synthesis of Potent Cyclic Hexapeptide NK-1 Antagonists. Use of a Minilibrary in Transforming a Peptidal Somatostatin Receptor Ligand into an NK-1 Receptor Ligand via a Polyvalent Peptidomimetic. J Med Chem. 1996;39:2441–2448. doi: 10.1021/jm960281e. [DOI] [PubMed] [Google Scholar]
- 15.Brazeau P, Vale W, Burgus R, Guillemin R. Isolation of Somatostatin (a Somatotropin Release Inhibiting Factor) of Ovine Hypothalamic Origin. Can J Biochem. 1974;52:1067–1072. doi: 10.1139/o74-148. [DOI] [PubMed] [Google Scholar]
- 16.Patel YC. Molecular Pharmacology of Somatostatin Receptor Subtypes. J Endocrinol Invest. 1997;20:348–367. doi: 10.1007/BF03350317. [DOI] [PubMed] [Google Scholar]
- 17.(a) Csaba Z, Dournaud P. Cellular Biology of Somatostatin Receptors. Neuropeptides. 2001;35:1–23. doi: 10.1054/npep.2001.0848. [DOI] [PubMed] [Google Scholar]; (b) Raynor K, Reisine T. Somatostatin Receptors. Crit Rev Neurobiol. 1992;6:273–289. [PubMed] [Google Scholar]
- 18.Reisine T, Bell GI. Molecular Biology of Somatostatin Receptors. Endocr Rev. 1995;16:427–442. doi: 10.1210/edrv-16-4-427. [DOI] [PubMed] [Google Scholar]
- 19.Yamada Y, Post SR, Wang K, Tager HS, Bell GI, Seino S. Cloning and Functional Characterization of a Family of Human and Mouse Somatostatin Receptors Expressed in Brain, Gastrointestinal Tract, and Kidney. Proc Natl Acad Sci USA. 1992;89:251–255. doi: 10.1073/pnas.89.1.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rivier J, Brown M, Vale W. D-Trp8-Somatostatin, Analog of Somatostatin More Potent than the Native Molecule. Biochim Biophys Res Commun. 1975;65:746–751. doi: 10.1016/s0006-291x(75)80208-7. [DOI] [PubMed] [Google Scholar]
- 21.Veber DF, Holly FW, Paleveda WJ, Nutt RF, Bergstrand SJ, Torchiana M, Glitzer MS, Saperstein R, Hirschmann R. Conformationally Restricted Bicyclic Analogs of Somatostatin. Proc Natl Acad Sci USA. 1978:2636–2640. doi: 10.1073/pnas.75.6.2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chandrasekaran R, Lakshminarayanan AV, Pandya UV, Ramachandran GN. Conformation of the LL and LD Hairpin Bends with Internal Hydrogen Bonds in Proteins and Peptides. Biochim Biophys Acta. 1973;303:14–27. doi: 10.1016/0005-2795(73)90143-8. [DOI] [PubMed] [Google Scholar]
- 23.(a) Vale W, Rivier J, Ling N, Brown M. Biologic and Immunologic Activities and Applications of Somatostatin Analogs. Metabolism. 1978;27:1391–1401. doi: 10.1016/0026-0495(78)90081-1. [DOI] [PubMed] [Google Scholar]; (b) Bauer W, Briner U, Doepfiner W, Haller R, Huguenin R, Marbach P, Petcher TJ, Pless J. SMS 201–995: A Very Potent and Selective Octapeptide Analog of Somatostatin with Prolonged Action. Life Sci. 1982;31:1133–1140. doi: 10.1016/0024-3205(82)90087-x. [DOI] [PubMed] [Google Scholar]
- 24.(a) Veber DF, Freidinger RM, Perlow DS, Paleveda WJJ, Holly FW, Strachan RG, Nutt RF, Arison BH, Homnick C, Randall WC, Glitzer MS, Saperstein R, Hirschmann R. A Potent Cyclic Hexapeptide Analog of Somatostatin. Nature. 1981;292:55–58. doi: 10.1038/292055a0. [DOI] [PubMed] [Google Scholar]; (b) Veber DF, Holly FW, Nutt RF, Bergstrand SJ, Brady SF, Hirschmann R, Glitzer MS, Saperstein R. Highly Active Cyclic and Bicyclic Somatostatin Analogs of Reduced Ring Size. Nature. 1979;280:512–514. doi: 10.1038/280512a0. [DOI] [PubMed] [Google Scholar]
- 25.Arison BH, Hirschmann R, Paleveda WJ, Brady SF, Veber DF. On the Low Energy Solution Conformation of Somatostatin. Biochem Biophys Res Commun. 1981;100:1148–1153. doi: 10.1016/0006-291x(81)91944-6. [DOI] [PubMed] [Google Scholar]
- 26.Hirschmann R. Some Recent Developments in the Chemistry and Biology of Somatostatin-Related Peptides. In: Yu W, editor. Chemistry of Natural Products: Proceedings of the Sino-American Symposium on the Chemistry of Natural Products. Gordon and Breach, Science Publishers; New York: 1982. pp. 44–54. [Google Scholar]
- 27.Veber DF. Design of a Highly Active Cyclic Hexapeptide Analog of Somatostatin. In: Rich DH, Gross VJ, editors. Peptides, Synthesis, Structure and Function; Proceedings of the 7th American Peptide Symposium; 1981. pp. 685–694. [Google Scholar]
- 28.Lamberts SW, van der Lely A-J, de Herder WW, Hofland LJ. Octreotide. New Engl J Med. 1996;334:246–254. doi: 10.1056/NEJM199601253340408. [DOI] [PubMed] [Google Scholar]
- 29.Freidinger RM, Colton CD, Perlow DS, Whitter WL, Paleveda WJ, Veber DF. Modified Retro Enantiomers are Potent Somatostatin Analogs. In: Hruby VJ, Rich D, editors. Peptides, Structure and Function; Proceedings of the 8th American Peptide Symposium; 1983. pp. 349–352. [Google Scholar]
- 30.Prasad V, Birzin ET, McVaugh CT, van Rijn RD, Rohrer SP, Chicchi G, Underwood DJ, Thornton ER, Smith AB, III, Hirschmann R. Effects of Heterocyclic Aromatic Substituents on Binding Affinities at Two Distinct Sites of Somatostatin Receptors. J Med Chem. 2003;46:1858–1869. doi: 10.1021/jm0205088. [DOI] [PubMed] [Google Scholar]
- 31.Hirschmann R, Hynes J, Jr, Cichy-Knight MA, van Rijn RD, Sprengeler PA, Spoors PG, Shakespeare WC, Pietranico-Cole S, Barbosa J, Liu J, Yao WQ, Rohrer S, Smith AB., III Modulation of Receptor and Receptor Subtype Affinities Using Diasteromeric and Enantiomeric Monosaccharide Scaffolds as a Means to Structural and Biological Diversity. A New Route to Ether Synthesis. J Med Chem. 1998;41:1382–1391. doi: 10.1021/jm9800346. [DOI] [PubMed] [Google Scholar]
- 32.Liu J, Underwood DJ, Cascieri MA, Rohrer SP, Cantin L-D, Chicchi G, Smith AB, III, Hirschmann R. Synthesis of a Substance P Antagonist with a Somatostatin Scaffold: Factors Affecting Agonism/Antagonism at GPCRs and the Role of Pseudosymmetry. J Med Chem. 2000;43:3827–3831. doi: 10.1021/jm000316h. [DOI] [PubMed] [Google Scholar]
- 33.Sequence alignment was performed using CLUSTAL W. Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position-Specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673.
- 34.Cascieri MA, Fong TM, Strader CD. Identification of Critical Functional Groups within Binding Pockets of G-Protein-Coupled Receptors. Drugs Future. 1996;21:521–527. [Google Scholar]
- 35.Evans BE, Rittle KE, Bock MG, DiPardo RM, Freidinger RM, Whitter WL, Lundell GF, Veber DF, Anderson PS, Chang RSL, Lotti VJ, Cerino DJ, Springer JP, Hirshfield J. Methods for Drug Discovery: Development of Potent, Selective, Orally Effective Cholecystokinin Antagonists. J Med Chem. 1988;31:2235–2246. doi: 10.1021/jm00120a002. [DOI] [PubMed] [Google Scholar]
- 36.(a) Woltersdorf OW, DeSolms SJ, Schultz EM, Cragoe EJ., Jr (Acylaryloxy)Acetic Acid Diuretics. 1. (2-Alkyl-and 2,2-Dialkyl-1-Oxo-5-Indanyloxy)Acetic Acids. J Med Chem. 1977;20:1400–1408. doi: 10.1021/jm00221a010. [DOI] [PubMed] [Google Scholar]; (b) Baldwin JJ, Hirschmann R, Engelhardt EL, Ponticello GS, Sweet CS, Scriabine A. Approaches to Vasodilating/β-Adrenergic Blocking Agents: Examples of the Dihydrolutidine Type. J Med Chem. 1981;24:628–631. doi: 10.1021/jm00137a028. and references cited. [DOI] [PubMed] [Google Scholar]
- 37.Szardenings AK, Harris D, Lam S, Shi L, Tien D, Wang Y, Patel DV, Navre M, Campbell DA. Rational Design and Combinatorial Evaluation of Enzyme Inhibitor Scaffolds: Identification of Novel Inhibitors of Matrix Metalloproteinases. J Med Chem. 1998;41:2194–2200. doi: 10.1021/jm980133j. [DOI] [PubMed] [Google Scholar]
- 38.(a) Mason JS, Morize I, Menard PR, Cheney DL, Hulme C, Labaudiniere RF. New 4-Point Pharmacophore Method for Molecular Similarity and Diversity Applications: Overview of the Method and Applications, Including a Novel Approach to the Design of Combinatorial Libraries Containing Privileged Substructures. J Med Chem. 1999;42:3251–3264. doi: 10.1021/jm9806998. [DOI] [PubMed] [Google Scholar]; (b) Dolle RE, Nelson KH., Jr Comprehensive Survey of Combinatorial Library Synthesis: 1998. J Comb Chem. 1999;1:235–282. doi: 10.1021/cc9900192. [DOI] [PubMed] [Google Scholar]; (c) Lewell XQ, Judd DB, Watson SP, Hann MM. RECAP-Retrosynthetic Combinatorial Analysis Procedure: A Powerful New Technique for Identifying Privileged Molecular Fragments with Useful Applications in Combinatorial Chemistry. J Chem Inf Comput Sci. 1998;38:511–522. doi: 10.1021/ci970429i. [DOI] [PubMed] [Google Scholar]
- 39.Bemis GW, Murcko MA. The Properties of Known Drugs. 1. Molecular Frameworks. J Med Chem. 1996;39:2887–2893. doi: 10.1021/jm9602928. [DOI] [PubMed] [Google Scholar]
- 40.Nicolaou KC, Pfefferkorn JA, Roecker AJ, Cao GQ, Barluenga S, Mitchell HJ. Natural Product-Like Combinatorial Libraries Based on Privileged Structures. 1. General Principles and Solid-Phase Synthesis of Benzopyrans. J Am Chem Soc. 2000;122:9939–9953. [Google Scholar]
- 41.Mosley RT, Miller MD, Kearsley SK, Prendergast K, Underwood DJ. New Lead Discovery in Drug Development. In: Witten M, editor. Computational Medicine, Public Health and Biotechnology. World Scientific; Singapore: 1995. pp. 101–125. [Google Scholar]