

Abstract
The budding yeast, Saccharomyces cerevisiae, is a convenient system for coupling heterologous G protein-coupled receptors (GPCRs) to the pheromone response pathway to facilitate empirical ligand screening and/or GPCR mutagenesis studies. However, few studies have applied this system to define GPCR-G protein-coupling preferences and furnish information on ligand affinities, efficacies, and functional selectivity. We thus used different S. cerevisiae strains, each expressing a specific human Gα/yeast Gpa1 protein chimera, and determined the pharmacology of various ligands of the coexpressed human adenosine A1 receptor. These assays, in conjunction with the application of quantitative models of agonism and antagonism, revealed that (−)-N6-(2-phenylisopropyl)adenosine was a high-efficacy agonist that selectively coupled to Gpa/1Gαo, Gpa1/Gαi1/2, and Gpa1/Gαi3, whereas the novel compound, 5′-deoxy-N6-(endo-norborn-2-yl)-5′-(2-fluorophenylthio)adenosine (VCP-189), was a lower-efficacy agonist that selectively coupled to Gpa1/Gαi proteins; the latter finding suggested that VCP-189 might be functionally selective. The affinity of the antagonist, 8-cyclopentyl-1,3-dipropylxanthine, was also determined at the various strains. Subsequent experiments performed in mammalian Chinese hamster ovary cells monitoring cAMP formation/inhibition, intracellular calcium mobilization, phosphorylation of extracellular signal-regulated kinase 1 and 2 or 35S-labeled guanosine 5′-(γ-thio)triphosphate binding, were in general agreement with the yeast data regarding agonist efficacy estimation and antagonist affinity estimation, but revealed that the apparent functional selectivity of VCP-189 could be explained by differences in stimulus-response coupling between yeast and mammalian cells. Our results suggest that this yeast system is a useful tool for quantifying ligand affinity and relative efficacy, but it may lack the sensitivity required to detect functional selectivity of low-efficacy agonists.
Adenosine is a purine nucleoside that is vital in regulating numerous physiological processes, especially in the cardiovascular and central nervous systems (Haskó et al., 2008). In pathophysiological settings, adenosine is commonly regarded as a tissue-protective molecule whose actions are mostly mediated by the adenosine A1 receptor, one of four G protein-coupled receptor (GPCR) subtypes recognized by adenosine. The A1 receptor is nearly ubiquitously expressed throughout the body, with highest levels in brain, spinal cord, atria, and adipose tissue (Baraldi et al., 2000; Kourounakis et al., 2001). Activation of the adenosine A1 receptor remains an attractive therapeutic approach for treating conditions such as ischemia reperfusion injury, paroxysmal superventricular tachycardia, chronic pain, and non-insulin-dependent diabetes mellitus (Gao and Jacobson, 2007; Elzein and Zablocki, 2008). For this reason, A1 receptor-selective compounds have generated much interest as potential novel therapeutic agents.
Approaches for selectively targeting the A1 receptor have traditionally exploited differences in the binding properties of this receptor relative to other subtypes, either by focusing on the receptor's orthosteric site or, alternatively, on potential allosteric sites (Gao et al., 2005; Aurelio et al., 2009). More recently, there has been a growing appreciation that ligands may be designed that are not only subtype-selective, but also signal pathway-selective. This reflects the recognition that GPCRs adopt multiple active states that can be differentially stabilized such that only a subset of the entire signaling repertoire associated with a given receptor is activated in a ligand-specific manner, a phenomenon termed “stimulus-trafficking” or “functional selectivity” (Kenakin, 1995; Urban et al., 2007). As with most GPCRs, the A1 receptor is known to couple promiscuously to multiple signaling pathways (reviewed in Schulte and Fredholm, 2003), and it is thus conceivable that selectively targeting only some of these could potentially lead to a reduction in the side-effect profile associated with indiscriminate activation of the A1 receptor. However, in general, multiple assay types are required to study the promiscuous coupling of GPCRs, such that pharmacological parameters (e.g., ligand affinities and relative efficacies) can be obtained. Determination of such information is required to facilitate structure-activity relationships and to understand functional selectivity. Nonetheless, many assays can be influenced by intracellular signaling cross talk arising from the promiscuous nature of GPCR signaling. Therefore, it would be useful to use a single system that could generate the requisite information on selective signaling with minimal contribution from potential confounding influences.
The yeast, Saccharomyces cerevisiae, expresses a single type of GPCR that, upon activation, signals to the pheromone response pathway via coupling to a single heterotrimeric G protein (Dowell and Brown, 2002). It is noteworthy that yeast can be adapted to accommodate mammalian GPCR signaling via this one-GPCR–one-G protein pathway. Brown et al. (2000) have previously modified this system to allow expression of a human/yeast Gα protein chimera, consisting of five C-terminal amino acids of a given human Gα protein fused with the truncated yeast Gα protein, Gpa1(1–467), (Gpa1/Gα) (Brown et al., 2000). This modification allows specificity of binding to potentially any desired mammalian GPCR, while maintaining the capacity to couple to the endogenous yeast Gβγ subunits (Dowell and Brown, 2002), which then signal to a mitogen-activated protein kinase pathway to activate reporter gene expression. This yeast signaling assay is thus an attractive system for studying specific pairs of GPCRs and G proteins in the absence of other GPCRs and signal cross-talk.
Despite these properties, the yeast signaling system has not been widely explored to date for quantification of GPCR agonist and antagonist pharmacological properties and G protein-coupling profiles. Thus, the aim of the current study was to determine the pharmacological characteristics of A1 adenosine receptors expressed in various yeast strains together with individual G protein chimeras for each of the main mammalian Gα subunits. Our results suggest that the yeast signaling assay is a robust platform for determining relative efficacies of agonists and affinity values for both agonists and antagonists. The assay, in general, is predictive of coupling preferences that are also seen in mammalian cell assays. However, a potential for a lack of sensitivity also exists in the ability of the yeast system to detect functional selectivity between some Gα protein subtypes.
Materials and Methods
Materials.
The Surefire ERK1/2 phosphorylation kit was kindly donated by Dr. Michael Crouch (TGR Biosciences, Adelaide, Australia). All AlphaScreen beads and [35S]GTPγS were purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA). VCP-189 was synthesized (as compound 12c) as described previously by Ashton et al. (2008). Flp-In Chinese hamster ovary (CHO) cells, Gateway plasmids, BP clonase kit, LR clonase kit, hygromycin B, zeocin, Fluo-4-AM, S. cerevisiae EasyComp transformation kit, and fluorescein di(β-d-galactopyranoside) were obtained from Invitrogen (Carlsbad, CA). cDNA constructs of the human adenosine A1 receptor and pertussis toxin (PTX)-insensitive Gαi/o proteins were purchased from the Missouri University of Science and Technology cDNA Resource Center (http://cdna.org). Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were purchased from Invitrogen and JRH Biosciences (Lenexa, KS), respectively. Metafectene reagent was obtained from Biontex (Martinsried/Planegg, Germany). Ultima gold scintillation cocktail was purchased from Packard Bioscience (Meriden, CT). Bicinchoninic acid protein reagents were obtained from Pierce Biotechnology (Rockford, IL) and adenosine deaminase (ADA), derived from calf intestine, was purchased from Roche (Basel, Switzerland). All other reagents were purchased from Sigma-Aldrich (St Louis, MO).
Cell Culture and Transfections.
The sequence of the human adenosine A1 receptor was amplified by PCR and cloned into the Gateway entry vector, pDONR201, using the BP clonase kit according to manufacturer's instructions. The A1 receptor construct was subsequently transferred in the Gateway destination vector, pEF5/FRT/V5-dest, using the LR clonase kit in accordance with manufacturer's instructions. The construct was then transfected into Flp-In CHO cells by use of methods described previously (Nawaratne et al., 2008). Flp-In CHO cells stably expressing adenosine A1 receptors (CHO A1R) were cultured at 37°C in 5% CO2 in DMEM supplemented with 5% (v/v) FBS, 16 mM HEPES, and were selected by use of 400 μg ml−1 hygromycin B, but maintained by use of 200 μg ml−1 hygromycin B. For ERK1/2 phosphorylation experiments requiring transfection of PTX-insensitive G proteins, CHO cells expressing the A1 receptor were transfected with either pcDNA3.1+ (vector only) or pcDNA3.1+ encoding PTX-insensitive Gαi/o family proteins [described in Wise et al. (1997)], in 96-well plates with use of Metafectene reagent according to the manufacturer's instructions for CHO cells.
Yeast Transformations and Signaling Assay.
Production of S. cerevisiae strains expressing chimeras of five C-terminal amino acids of human Gα protein with the yeast Gpa1, 1–467, (Gpa1/Gα) has been described previously in Brown et al. (2000). The gene encoding the human adenosine A1 receptor was cloned into the p426GPD vector by use of traditional cloning methods. Yeast strains were then transformed with this construct with use of the S. cerevisiae EasyComp transformation kit in accordance with the manufacturer's instructions. The conditions for the signaling component of the assay were as described previously (Brown et al., 2000), except that fluorescein di(β-d-galactopyranoside) was used as the β-galactosidase substrate rather than chlorophenolred-β-d-galactopyranoside. After appropriate treatment with ligand, the cells were incubated at 30°C for 18 to 24 h. Fluorescence was read on an EnVision plate reader (PerkinElmer Life and Analytical Sciences) at 475-nm excitation and 520-nm emission.
Membrane Preparation.
CHO A1R cells were grown to 90% confluence, harvested, and centrifuged at 300g for 3 min. The intact cell pellet was suspended in homogenization buffer (20 mM HEPES; 10 mM EDTA; 0.1 mg ml−1 saponin, pH 7.7) and further centrifuged (300g, 3 min). Cells were then resuspended in homogenization buffer and homogenized by use of a Polytron PT1200 homogenizer (Kinematica, Littau-Lucerne, Switzerland) for two 10-s intervals at maximum setting (6), with 30-s cooling periods on ice between each burst. The homogenate was then treated for 30 min with 1U ml−1 ADA. The homogenate was then centrifuged (40,000g, 1 h, 4°C). The resulting pellet was resuspended in 5 ml of GTPγS buffer (100 mM NaCl, 20 mM HEPES, 10 mM MgCl2, pH 7.4), and the protein content was determined by use of a bicinchoninic acid assay kit (Pierce Biotechnology) according to the manufacturer's instructions. The homogenate was then divided into 1-ml aliquots and either used immediately or stored frozen at −80°C until required.
[35S]GTPγS Immunoprecipitation Assay.
CHO A1R membranes (20 μg/sample) were incubated in GTPγS buffer with ligand (or buffer) and 10 μM GDP for 30 min at 30°C, before addition of 0.1 nM [35S]GTPγS (final concentration) for a further 30 min at 30°C, in a final volume of 500 μl. The reaction was terminated by membrane solubilization in GTPγS buffer with 1.25% Igepal CA630 [(octylphenoxy)polyethoxyethanol, octylphenyl-polyethylene glycol] (Sigma-Aldrich). Samples were then placed on a rotor at 4°C for 30 min, before being incubated with 2 μg of appropriate anti-Gα subunit antibody for a further 90 min at 4°C. A slurry of protein A Sepharose in assay buffer was added to each sample to achieve a 3% (v/v) final concentration and incubated at 4°C for 1 h. Samples were centrifuged three times at 60g at 4°C, each time washed with 500 μl of ice-cold GTPγS buffer with 1.25% Igepal CA630. The final pellet was resuspended and added to 4 ml of scintillation cocktail (Ultima Gold, PerkinElmer Life and Analytical Sciences), and radioactivity was then determined by scintillation counting.
Extracellular Signal-Regulated Kinase 1/2 Phosphorylation Assays.
Initial ERK1/2 phosphorylation time course experiments were performed to determine the time at which ERK1/2 phosphorylation was maximal after stimulation by each agonist. Cells were seeded into transparent 96-well plates at 5 × 104 cells per well and grown overnight or until confluent. Cells were then washed twice with phosphate-buffered saline (PBS) and incubated in serum-free DMEM at 37°C for at least 4 h to allow FBS-stimulated phosphorylated ERK1/2 levels to subside. Before stimulation, cells were treated with 1 U ml−1 ADA for 30min. Cells were then stimulated with agonist for 5 min and incubated at 37°C in 5% CO2. For antagonist interaction studies, cells were incubated with 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) for 30 min at 37°C in 5% CO2, before agonist stimulation. Experiments using PTX-insensitive Gα protein subunits were transfected as described above. Twenty-four hours after transfection, cells were washed with PBS, and cultured overnight in serum-free media containing 100 ng ml−1 PTX. The assay was then performed as described above. For receptor alkylation experiments, CHO A1R cells were cultured overnight to approximately 90% confluence in 96-well plates, washed with PBS, and treated for 15 or 30 min (VCP-189 alkylation assays) with 10 μM8-cyclopentyl-3-(3-((4(fluorosulfonylbenzoyl)oxy)propyl)-1-propylxanthine (FSCPX), an irreversible A1 receptor antagonist, or 5 μM or 10 μM FSCPX for 30 min [(−)-N6-(2-phenylisopropyl)adenosine (R-PIA) alkylation assays] at 37°C. The cells were washed extensively with PBS, and bathed in serum-free media 4 h before experimentation. The assay was then performed as described above. For all experiments, 3% (v/v) FBS was used as a positive control, and vehicle controls were also performed. The reaction was terminated by removal of drugs and lysis of cells with 100 μl of SureFire lysis buffer (TGR Biosciences). The lysates were agitated for 1 to 2 min and were diluted at a ratio of 4:1 (v/v) lysate/Surefire activation buffer in a total volume of 50 μl. Under low-light conditions a 1:240 (v/v) dilution of AlphaScreen beads/Surefire reaction buffer was prepared and this was mixed with the activated lysate mixture in a ratio of 6:5 (v/v), respectively, in a 384-well opaque Optiplate. Plates were incubated in the dark at 37°C for 1 h before the fluorescence signal was measured by use of a Fusion-α plate reader (PerkinElmer Life and Analytical Sciences) with standard AlphaScreen settings. Data were normalized to the maximal response elicited by 3% (v/v) FBS at the same time point.
cAMP Accumulation Assay.
CHO A1R cells were plated into 96-well plates and cultured overnight at 37°C in 5% CO2. Cells were washed with PBS, and cultured overnight in serum-free media in the absence or presence of 100 ng ml−1 PTX. Thirty minutes before assaying, the culture medium was replaced with phenol red-free DMEM with 0.1% bovine serum albumin (BSA), 1 U ml−1 ADA, and 500 μM 3-isobutyl-1-methylxanthine, and incubated at 37°C in 5% CO2. Cells were treated with R-PIA and/or forskolin, and incubated for 30 min at 37°C in 5% CO2. Medium was aspirated and cells were lysed in lysis buffer (dH2O, 0.3% Tween 20, 5 mM HEPES, 0.1% BSA). Lysates were transferred to a 384-well plate and mixtures of lysis buffer/donor bead-conjugated anti-cAMP antibody and lysis buffer/biotinylated cAMP/acceptor bead-conjugated streptavidin were added to the lysates according to the PerkinElmer cAMP Alphascreen protocol. Plates were incubated in the dark at room temperature overnight before the fluorescence signal was measured by use of a Fusion-α plate reader (PerkinElmer Life and Analytical Sciences) by use of standard AlphaScreen settings. Data were normalized to the response elicited by 10 μM forskolin at the same time point.
Ca2+ Mobilization Assay.
CHO A1R cells were cultured overnight in 96-well plates at 37°C in 5% CO2. Cells were washed with PBS, and cultured overnight in serum-free media in the absence or presence of 100 ng ml−1 PTX. Cells were washed twice in Ca2+ assay buffer [150 mM NaCl, 2.6 mM KCl, 1.2 mM MgCl2, 10 mM dextrose, 10 mM HEPES, 2.2 mM CaCl2, 0.5% (w/v) BSA, and 4 mM probenecid]. The final wash replaced with Ca2+ assay buffer containing 1 μM Fluo-4-AM and incubated for 1 h at 37°C in 5% CO2. Cells were washed twice more and replaced with 37°C Ca2+ assay buffer containing 1 U ml−1 ADA. R-PIA was added and fluorescence was measured in a Flexstation (Molecular Devices, Sunnyvale, CA).
Data Analysis.
Agonist concentration-response curves, in the absence of antagonist, were fitted via nonlinear regression to the following three-parameter logistic function by use of Prism 5.01 (GraphPad Software, San Diego, CA):
where E is response, Emax and basal are the top and bottom asymptotes of the curve, respectively, log[A] is the logarithm of the agonist concentration, and pEC50 is the negative logarithm of the agonist concentration that gives a response halfway between Emax and basal.
Experiments measuring the interaction between R-PIA and DPCPX, or R-PIA and VCP-189 at the adenosine A1 receptor were globally fitted to the following logistic equation of agonist-antagonist interaction:
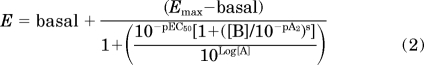 |
where basal, Emax, log[A], and 50are as described previously. B is the molar concentration of antagonist. pA2 is the negative logarithm of the concentration of antagonist that requires a 2-fold increase in the concentration of agonist to achieve an equal response to that in absence of antagonist. The parameter, s, is analogous to the Schild slope factor (Motulsky and Christopoulos, 2004).
To generate agonist affinity and efficacy estimates the following form of an operational model of agonism (Black and Leff, 1983) was applied to the relevant data (see Results):
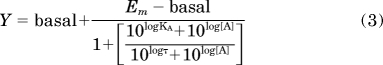 |
where log KA is the logarithm of the equilibrium dissociation constant of the agonist, Em is the maximal response of the system, and τ is an operational measure of efficacy, equal to the total receptor concentration divided by the concentration of agonist-receptor complexes required to achieve 50% of the maximal system response. All other parameters are as described previously. For application to receptor alkylation experiments, eq. 3 was globally fitted to the entire family of curves for a given agonist's responses determined in the absence or after treatment with FSCPX, with all parameters being shared except log τ. For experiments in yeast comparing the responses of VCP-189 to those of the full agonist, R-PIA, eq. 3 was applied to the VCP-189 responses whereas eq. 1 was applied to the R-PIA responses, with the basal parameters shared across datasets and the Emax parameter of eq. 1 constrained to equal the Em parameter of eq. 3; in this manner, estimates of log KA and log τ could be derived for VCP-189 (Leff et al., 1990; Motulsky and Christopoulos, 2004).
Statistical comparisons between parameters were performed by use of a one-way analysis of variance with Bonferroni's multiple comparisons or Dunnett's post test, as appropriate, and a probability (p) less than 0.05 was taken to indicate significance.
Results
Determination of Agonist and Antagonist Pharmacology Using S. cerevisiae.
Concentration-response curves were constructed to the agonists, R-PIA and VCP-189 in yeast strains expressing chimeras of Gpa1/Gαq, Gpa1/Gα12, Gpa1/Gαo, Gpa1/Gαi1/2, Gpa1/Gαi3, Gpa1/Gαs, or the full-length endogenous yeast Gα protein Gpa1, to test the ability of these compounds to elicit a response when coupled to an individual subtype of G protein. R-PIA produced detectable responses in strains expressing Gpa1/Gαo, Gpa1/Gαi1/2, and Gpa1/Gαi3. VCP-189 was a partial agonist compared with R-PIA, and only produced significant responses in yeast strains expressing Gpa1/Gαi subunits (Fig. 1); Table 1 lists the estimates of agonist potency derived from these experiments. Subsequent application of an operational model of agonism (eq. 3) to the data also allowed for the determination of VCP-189 affinity (log KA) and relative efficacy (log τ) at the Gpa1/Gαi1/2 and Gpa1/Gαi3 strains, yielding the following values: for Gpa1/Gαi1/2, log KA = −6.40 ± 0.16; log τ = −0.37 ± 0.07 (τ = 0.42); for Gpa1/Gαi3, log KA = −5.76 ± 0.22; log τ = −0.61 ± 0.17 (τ = 0.24), n = 3–5. Neither the affinity or relative efficacy values were significantly different from one another between strains (p > 0.05).
Fig. 1.

Influence of G protein subtype on R-PIA (●) and VCP-189 (▲) concentration-response curves in yeast strains expressing either: A, Gpa1/Gαo; B, Gpa1/Gαi1/2; or C, Gpa1/Gαi3. Each yeast strain was incubated with a range of ligand concentrations for 18 to 24 h at 30°C, before obtaining fluorescence measurements. Data points represent the mean ± S.E.M. obtained from three to five experiments conducted in duplicate and are normalized to the fluorescence measure in the absence of ligand. RFU, relative fluorescence unit.
TABLE 1.
Potency (pEC50) values generated from concentration-response curves from yeast signaling assays performed in strains expressing the adenosine A1 receptor with either Gpa1/Gα o, Gpa1/Gα i1/2, or Gpa1/Gαi3
Values represent the mean ± S.E. obtained from three to five experiments conducted in duplicate.
| Yeast Strain | R-PIA | VCP-189 |
|---|---|---|
| Gαo | 7.38 ± 0.18 | N.D. |
| Gαi1/2 | 8.41 ± 0.17 | 6.97 ± 0.01 |
| Gαi3 | 8.08 ± 0.07 | 6.29 ± 0.25 |
N.D., not determined.
The lack of response to VCP-189 in the Gpa1/Gαo strain may reflect either a lack of interaction of the agonist with the receptor in that yeast strain, or a lack of signaling efficacy via the Gpa1/Gαo protein. To differentiate between these two possibilities, interaction experiments were performed between R-PIA and VCP-189 at the Gpa1/Gαo strain. As shown in Fig. 2, increasing concentrations of VCP-189 led to a progressive rightward shift of the R-PIA concentration-response curve in a manner consistent with a competitive interaction, indicating that VCP-189 was indeed able to interact with the A1 receptor expressed in this yeast strain. Application of eq. 2 to the data yielded a −pA2 estimate of −5.98 ± 0.06 (n = 3) for VCP-189. Neither this value, nor the log KA values determined from the operational model analysis of the data from the other two yeast strains, were significantly different from one another (p > 0.05).
Fig. 2.

Effect of VCP-189 on R-PIA concentration-response curves in the absence or presence of the indicated concentrations of VCP-189 in yeast strains expressing Gpa1/Gαo. The yeast strain was incubated with a range of R-PIA concentrations in the absence or presence of VCP-189 for 18 to 24 h at 30°C, before fluorescence was measured. Data are expressed as a percentage of the maximal response attained by R-PIA in the absence of VCP-189. Data points represent the mean ± S.E.M. obtained from three experiments conducted in duplicate. RFU, relative fluorescence unit.
The interaction paradigm used above was next extended to investigate the properties of a prototypical A1 receptor orthosteric antagonist, DPCPX, at each of the Gpa/Gαo, Gpa/Gαi1/2, or Gpa/Gαi3 strains. Figure 3shows that, in each instance, the antagonist caused parallel rightward shifts of the R-PIA concentration-response curves in a concentration-dependent manner, characterized by the following pA2 estimates (n = 4): 8.51 ± 0.15, 9.15 ± 0.29, and 8.58 ± 0.32, for Gpa/Gαo, Gpa/Gαi1/2, and Gpa/Gαi3, respectively. Statistical analysis revealed no significant difference between these values (p > 0.05).
Fig. 3.

Effect of DPCPX on R-PIA concentration-response curves in the absence or presence of the indicated concentrations of DPCPX performed in yeast strains expressing the adenosine A1 receptor and either: A, Gpa1/Gαo; B, Gpa1/Gαi1/2; or C, Gpa1/Gαi3. A range of R-PIA concentrations in the absence or presence DPCPX was incubated with the aforementioned yeast strains for 18 to 24 h at 30°C, before obtaining fluorescence measurements. Data are represented as a percentage of the maximal response elicited by R-PIA in the absence of DPCPX, and each point is the mean ± S.E.M. collected from four experiments. RFU, relative fluorescence unit..
Validation of Gαi/o Coupling Preference in Chinese Hamster Ovary A1 Cells.
The yeast data indicated that the A1 receptor preferentially couples to members of the Gαi/o protein family with no appreciable interaction with Gαq and Gαs proteins. To ascertain whether this finding is relevant to a mammalian cell background, we used a CHO cell line stably expressing the human A1 receptor and monitored intracellular Ca2+ mobilization as a surrogate of either Gαq activation and/or Gβγ-mediated activation (Selbie and Hill, 1998; Migita et al., 2005); inhibition of forskolin-stimulated cAMP accumulation, which indicates Gαi/o and Gαs activation (Levitzki, 1988; Cordeaux et al., 2000); and ERK1/2 phosphorylation, which can result from activation of various G proteins and even non-G protein signaling (Schulte and Fredholm, 2003; Werry et al., 2006). Figure 4shows that the ability of R-PIA to promote ERK1/2 phosphorylation, intracellular Ca2+ mobilization or inhibition of forskolin-stimulated cAMP accumulation was completely abolished by pretreatment with PTX. Furthermore, in the absence of forskolin stimulation with or without PTX treatment, R-PIA was not able to elicit a cAMP accumulation response (not shown), indicating that the A1 receptor does not couple to Gαs proteins in this cell line. Taken together, these results suggest that A1 receptor-mediated responses in this mammalian cell line are wholly dependent on Gαi/o activation, as predicted by the findings in the yeast assays.
Fig. 4.

Impact of PTX pretreatment on R-PIA-induced responses in A1 receptor-mediated ERK1/2 phosphorylation (A), inhibition of forskolin-induced cAMP accumulation (B), and intracellular Ca2+ mobilization (C). CHO cells expressing the A1 receptor were serum-starved overnight in the absence (●) or presence (▲) of 100 ng ml−1 PTX. For B, ▾ refers to PTX treated with no forskolin stimulation. Data points represent the mean ± S.E.M. of three to four experiments performed in duplicate. RFU, relative fluorescence unit..
To confirm that the pharmacology of the antagonist, DPCPX, studied in the yeast cellular background is retained in a mammalian background, we also performed Schild analysis on the ability of DPCPX to inhibit R-PIA-mediated ERK1/2 phosphorylation in the CHO cells. As shown in Fig. 5, the antagonist produced a concentration-dependent, parallel, rightward shift of the agonist concentration-response curve, characterized by a pA2 of 9.11 ± 0.09 (n = 4), which was not significantly different from the values derived in the yeast assays.
Fig. 5.

Effect of DPCPX on R-PIA-induced ERK1/2 phosphorylation concentration-response curves in the absence or presence of the indicated concentrations of DPCPX performed in CHO cells expressing the adenosine A1 receptor. CHO cells expressing the adenosine A1 receptor were pretreated for 30 min with DPCPX before a 5-min stimulation of R-PIA at a range of concentrations. Data are expressed as percentage mean of ERK1/2 phosphorylation elicited by 3% FBS ± S.E.M. collected from four experiments performed in duplicate.
Assessment of Agonist Functional Selectivity.
The yeast data indicated that VCP-189 had a lower efficacy than R-PIA and a preferential coupling to Gαi proteins relative to Gαo proteins. However, a comparison of the ability of each agonist to mediate ERK1/2 phosphorylation in CHO cells (Fig. 6 ) indicated robust and equiefficacious agonism for both ligands. The differences between these observations in yeast and mammalian cells may reflect differences in stimulus-response coupling between the cellular backgrounds and/or true functional selectivity by VCP-189 for specific Gα subunits that is not detected because of the multipathway-convergent nature of ERK1/2 signaling. To first determine the influence of stimulus-response coupling on our observations, we used the irreversible antagonist, FSCPX, to occlude the A1 receptor's orthosteric site and thus effectively reduce the pool of accessible receptor-binding sites that can be activated by each agonist (Fig. 6). Application of the operational model of agonism to the datasets obtained before and after receptor alkylation yielded the following parameters for R-PIA: log KA = −6.96 ± 0.34, log τ = 1.71 ± 0.34 (τ = 51), whereas for VCP-189, the following values were determined: log KA = −5.85 ± 0.21, log τ = 0.61 ± 0.18 (τ = 4). The differences between the τ values under control conditions (in the absence of alkylation) for the two agonists clearly indicate that VCP-189 is a lower-efficacy agonist than R-PIA for ERK1/2 phosphorylation in CHO cells, and that the equivalent maximal agonist responses under the control conditions are thus most likely due to a high degree of stimulus-response coupling and/or receptor expression level in this cellular background compared with the yeast.
Fig. 6.

Impact of A1 receptor alkylation by FSCPX on: A, R-PIA-induced ERK1/2 phosphorylation concentration-response curves (5-min incubation) in the absence (●) or presence of 5 μM (■) or 10 μM (▴) FSCPX pretreatment (30 min) in CHO A1R cells. B, VCP-189-induced ERK1/2 phosphorylation concentration-response curves (5-min incubation) in the absence (○) or presence of 10 μM FSCPX pretreatment for 15 (□) or 30 (▵) min. Each data point is expressed as a percentage mean of ERK1/2 phosphorylation elicited by 3% FBS ± S.E.M. obtained from three to five experiments.
Finally, to investigate whether the preferential coupling of VCP-189 to Gαi over Gαo proteins identified in yeast was also indicative of functional selectivity for the former G proteins over the latter, we used two different experimental approaches in CHO cells. The first was to investigate agonist ERK1/2 phosphorylation responses in CHO A1 cells transfected with cDNA encoding specific PTX-insensitive Gαi/o subunits after PTX treatment. These experiments revealed that both R-PIA and VCP-189 were, in fact, able to promote ERK1/2 phosphorylation via either Gαo splice variants (GαoA and GαoB), and Gαi2 to varying extents (Fig. 7). Both ligands had comparable efficacies when coupled to either GαoA or Gαi2, but VCP-189 was a lower-efficacy agonist compared with R-PIA when coupled to GαoB. The second approach that we used was to generate a G protein-coupling profile for the A1 receptor by use of [35S]GTPγS immunoprecipitation, which determines activation of native G proteins. Figure 8 shows the results from these assays with use of CHO A1 cell membranes treated with either buffer (basal), 10 μM R-PIA, or 10 μM VCP-189. The results show that R-PIA and VCP-189 could only significantly alter [35S]GTPγS binding at Gαi3 G proteins compared with basal, in contrast to their effects on Gαo Gαi1, Gαi2, and Gαq proteins. However, there did seem to be a trend toward an increase in [35S]GTPγS binding to Gαo Gαi1, and Gαi2 proteins. Collectively, these results suggest that any preferential agonist coupling to Gαi proteins over Gαo proteins identified in yeast cells are most likely due to differences in strength of coupling rather than functional selectivity due to stimulus bias.
Fig. 7.

Effect of individual PTX-insensitive Gαi/o proteins on R-PIA (●)- and VCP-189 (○)-induced ERK1/2 phosphorylation in PTX-pretreated CHO A1R cells. The cDNA constructs used were: A, pcDNA3.1 (vector only); B, GαoAC351I; C, αoBC351I; D, Gαi1C351I; E, Gαi2C352I; and F. Gαi3C351I. cDNA constructs were transfected into CHO cells stably expressing the A1 receptor 48 h before experimentation. ERK1/2 phosphorylation was determined at the 5-min time point for each concentration of ligand. Each data point is expressed as a percentage mean of ERK1/2 phosphorylation elicited by 3% FBS ± S.E.M. obtained from three experiments.
Fig. 8.

Contribution of native mammalian Gαi/o proteins to A1 receptor-mediated [35S]GTPγS binding with use of immunoprecipitation. Basal (no ligand) (■), 10 μM R-PIA (□)- and 10 μM VCP-189 (▦)-induced [35S]GTPγS binding at specific Gαi/o proteins in CHO A1R membranes. Each data point is expressed as the mean DPM ± S.E.M. from three to six experiments. ∗∗, P < 0.01 determined by one-way analysis of variance with a Dunnett's post test for basal Gαi3 [35S]GTPγS binding compared with R-PIA- or VCP-189-stimulated [35S]GTPγS binding.
Discussion
This is the first study to use a yeast system to characterize adenosine A1 receptor ligand affinities, efficacies, and G protein-coupling profiles. Although there may be issues associated with a potential lack of sensitivity for agonists that are weakly coupled to a particular G protein, the yeast system is a valid platform for determining G protein coupling and generating affinity estimates for A1 receptor ligands in a manner that is consistent with coupling identified in mammalian cells. This suggests that yeast studies may have applicability to the determination of GPCR ligand-G protein functional selectivity profiles.
There are numerous approaches to estimating affinities of ligands, the most traditional of which are based on radioligand binding. However, this assay does not readily provide information about downstream events, it can be expensive, and, for many receptors, it is limited by a lack of appropriate radioligands. Another approach to estimate affinity values is to use functional assays that generate concentration-response data such that appropriate quantitative models can be applied to furnish agonist or antagonist affinity values (Kenakin, 2003). This approach may also be costly, time-consuming, and/or of insufficient throughput if multiple curves are required. In this regard, the use of a yeast-based assay for determining GPCR ligand pharmacology may prove useful, because it is inexpensive, lacks interacting mammalian proteins, and is able to reconstitute many GPCRs with high fidelity (Dowell and Brown, 2002). The majority of studies investigating GPCRs in yeast in the past have exploited the assays for facilitating random mutagenesis studies (Erlenbach et al., 2001; Schmidt et al., 2003; Armbruster et al., 2007), or as an empirical screening tool with a single-pathway endpoint (Campbell et al., 1999). However, to our knowledge, no study has investigated the ability of the system to ascertain quantitative, system-independent, pharmacological ligand properties.
Although an earlier study used S. cerevisiae to identify adenosine A1 receptor antagonists (Campbell et al., 1999), it was restricted to the empirical determination of antagonist potencies. We have now shown that it is possible to apply classic Schild analysis in yeast to yield antagonist affinity values that are similar to those obtained in CHO cells. Moreover, these affinity estimates are consistent with values derived for DPCPX in the past (e.g., Townsend-Nicholson and Shine, 1992; de Ligt et al., 2005), indicating that the G protein subtype present has little bearing on antagonist affinity. In addition to allowing appropriate quantification of antagonist pharmacology, the yeast system also profiled the G protein-coupling preferences for A1 receptor orthosteric agonists, indicating a role only for PTX-sensitive G proteins that was subsequently confirmed in our CHO cells. These results are consistent with previous (Akbar et al., 1994; Jockers et al., 1994), although there is some evidence to suggest that A1 receptors may couple to Gαs (Cordeaux et al., 2000) or Gαq proteins (Minelli et al., 2008); this is probably cell background-dependent.
Agonist potency estimates derived from functional studies traditionally cannot be related directly to agonist affinity estimates for a given GPCR because of the nonlinear nature of stimulus-response coupling in most systems (Kenakin, 2003). However, a recent study by Yu et al. (2008) suggested that a property of S. cerevisiae is an apparent lack of signal amplification. This relates to the cell biology of S. cerevisiae, which involves a negative-feedback loop in the pheromone response pathway contingent on the phosphorylated Fus3 protein inhibiting Sst2-mediated Ste5 recruitment, resulting in a pronounced decrease in amplification of signal. A consequence of this feedback loop is to “align” the yeast concentration-response and concentration-occupancy relationships. Yu et al. (2008) have also shown that deletion of the Sst2 protein provides equivalent results to that of a system where Fus3 is constitutively activated. Given that the yeast system used in our study is Sst2-negative, this suggests that the actual potency values obtained from agonist concentration-response curves should be approximations of agonist affinity for A1 receptor expressed in our strains. If this is the case, then a number of interesting observations arise from our results. Although the R-PIA potency estimates spanned an approximately 10-fold range across the various strains, they were generally comparable with the “high-affinity” binding constant we derived previously using membrane-based binding assays in CHO cells (May et al., 2005). However, these potency values are not consistent with the log KA estimates derived from the receptor alkylation assays of R-PIA-induced ERK1/2 phosphorylation in CHO cells (Fig. 6); the latter were, instead, consistent with the “low-affinity” binding constant we previously determined (May et al., 2005). If the hypothesis of a lack of signal amplification in yeast is valid, then it may be that the high potency of R-PIA as an agonist in yeast, compared with its estimated log KA for the A1 receptor from the operational model analysis in the CHO cells, reflects a preformed high-affinity state of the A1 receptor-G protein complex in the intact S. cerevisiae cells that may not be present, or only transiently so, in CHO cells. It is worth noting that a previous study in HEK-293 cells has suggested that the A1 receptor can form precoupled GPCR-G protein complexes (Nobles et al., 2005).
In addition to allowing for the quantification of parameters describing the drug-receptor interaction, another utility of the yeast assay system is to profile ligands acting unambiguously via a single type of G protein in a common cellular background. This offers the potential to detect G protein-mediated functional selectivity, which could be manifested as a change in ligand potency or efficacy orders across different yeast strains. However, the validation of the phenomenon in mammalian cells must consider the impact of differences in stimulus-response coupling and/or receptor expression levels between the cell types on the resultant pharmacology. Indeed, VCP-189 generated the same maximal response as R-PIA when studied as an agonist of ERK1/2 phosphorylation in our CHO cells, but was clearly of lower efficacy than R-PIA in each of the yeast strains tested. The most parsimonious explanation for this difference is that the CHO cells are characterized by a higher degree of signal amplification than yeast, such that low-efficacy agonists can still generate the maximal cellular response. The results of the receptor alkylation experiments and associated analysis are in agreement with this mechanism; even small degrees of alkylation resulted in a collapse of the VCP-189 concentration-response curve, whereas R-PIA displayed a rightward shift in potency with no collapse of the maximal agonist effect until the extent of receptor alkylation was more pronounced.
Despite the differences in the strength of signal between the two agonists studied, it is still possible that functional selectivity was operative in determining their coupling preferences to various G protein subtypes. Data acquired from the yeast studies indicated that VCP-189 could discriminate between Gpa1/Gαo and Gpa1/Gαi proteins, whereas R-PIA did not. However, the functional data obtained in CHO A1 cells either transfected with PTX-insensitive Gαi/o subunits or following [35S]GTPγS immunoprecipitation were not in agreement with data collected from the yeast assay, in that VCP-189 also activated Gαo coupling in CHO cells. There are a number of possible reasons for this discrepancy, but the most likely is that, because all Gpa1/Gα chimeras contain only five C-terminal amino acids of the mammalian Gα protein, the sequence in the Gpa1/Gαo chimera is suboptimal for ensuring sufficient affinity for the A1 receptor for low-efficacy agonists; if an agonist has very low efficacy, it may be undetectable in the yeast system. Thus, we conclude that there is no evidence from these studies of true functional selectivity of either agonist in their coupling preferences for Gαi/o protein subtypes.
The preceding discussion highlights important considerations when trying to relate agonist efficacy determinations made in recombinant systems to more physiologically relevant settings, such as isolated tissues or in vivo. Specifically, changes in receptor expression and/or stimulus-response coupling are likely to be the most obvious causes for dissimilitude between behavior determined in cell-based screening assays and behavior observed in a more native cellular environment. Mechanistically, this may also involve differences in G protein complement and stoichiometry, receptor-effector compartmentalization, or a role for accessory cellular proteins that are not present in recombinant systems. Nonetheless, the quantitative approaches used in the current study at least allow for the generation of hypotheses to model and predict the impact of changes in stimulus-response coupling on the appearance, or lack thereof, of selectivity in the actions of a given agonist between cell types. If subsequent experiments reveal that changes in receptor expression levels or coupling efficiency are insufficient to account for the observed pharmacology in native systems, then mechanisms such as stimulus-bias/functional selectivity can be invoked.
Taken together, our findings indicate that the yeast signaling system is a useful and convenient tool to add to the pharmacological armamentarium, especially in regard to the quantification of agonist and antagonist affinity and relative efficacy estimates. This is likely to apply not only to the A1 receptor, but also to any GPCR that can be expressed in yeast. In theory, the assay can also be used to routinely profile G protein-coupling selectivity, although potential issues of sensitivity for detecting low-efficacy agonists need to be considered.
Acknowledgments
We thank Elizabeth Guida for generating the CHO FlpIn A1 receptor cell line.
This work was supported by the National Health and Medical Research Council (NHMRC) [Program Grant 519461]. A.C. is an NHMRC Senior Research Fellow, and P.S. is an NHMRC Principal Research Fellow. G.S. received an Australian Postgraduate Award (Industry) from the Australian Research Council.
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
- GPCR
- G protein-coupled receptor
- ADA
- adenosine deaminase
- BSA
- bovine serum albumin
- CHO
- Chinese hamster ovary
- DMEM
- Dulbecco's modified Eagle's medium
- DPCPX
- 8-cyclopentyl-1,3-dipropylxanthine
- ERK1/2
- extracellular signal-regulated kinase 1 and 2
- FBS
- fetal bovine serum
- FSCPX
- 8-cyclopentyl-3-(3-((4-(fluorosulfonylbenzoyl)oxy)propyl)-1-propylxanthine
- GTPγS
- guanosine 5′(γ-thio)triphosphate
- PBS
- phosphate-buffered saline
- PTX
- pertussis toxin
- R-PIA
- (−)-N6-(2-phenylisopropyl)adenosine
- VCP-189
- 5′-deoxy-N6-(endo-norborn-2-yl)-5′-(2-fluorophenylthio)adenosine.
References
- Akbar M, Okajima F, Tomura H, Shimegi S, Kondo Y. ( 1994) A single species of A1 adenosine receptor expressed in Chinese hamster ovary cells not only inhibits cAMP accumulation but also stimulates phospholipase C and arachidonate release. Mol Pharmacol 45: 1036–1042 [PubMed] [Google Scholar]
- Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL. ( 2007) Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci U S A 104: 5163–5168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashton TD, Baker SP, Hutchinson SA, Scammells PJ. ( 2008) N6-substituted C5′-modified adenosines as A1 adenosine receptor agonists. Bioorg Med Chem 16: 1861–1873 [DOI] [PubMed] [Google Scholar]
- Aurelio L, Valant C, Flynn BL, Sexton PM, Christopoulos A, Scammells PJ. ( 2009) Allosteric modulators of the adenosine A(1) receptor: synthesis and pharmacological evaluation of 4-substituted 2-amino-3-benzoylthiophenes. J Med Chem 52: 4543–4547 [DOI] [PubMed] [Google Scholar]
- Baraldi PG, Zaid AN, Lampronti I, Fruttarolo F, Pavani MG, Tabrizi MA, Shryock JC, Leung E, Romagnoli R. ( 2000) Synthesis and biological effects of a new series of 2-amino-3-benzoylthiophenes as allosteric enhancers of A1-adenosine receptor. Bioorg Med Chem Lett 10: 1953–1957 [DOI] [PubMed] [Google Scholar]
- Black JW, Leff P. ( 1983) Operational models of pharmacological agonism. Proc R Soc Lond B Biol Sci 220: 141–162 [DOI] [PubMed] [Google Scholar]
- Brown AJ, Dyos SL, Whiteway MS, White JH, Watson MA, Marzioch M, Clare JJ, Cousens DJ, Paddon C, Plumpton C, et al. ( 2000) Functional coupling of mammalian receptors to the yeast mating pathway using novel yeast/mammalian G protein alpha-subunit chimeras. Yeast 16: 11–22 [DOI] [PubMed] [Google Scholar]
- Campbell RM, Cartwright C, Chen W, Chen Y, Duzic E, Fu JM, Loveland M, Manning R, McKibben B, Pleiman CM, et al. ( 1999) Selective A1-adenosine receptor antagonists identified using yeast Saccharomyces cerevisiae functional assays. Bioorg Med Chem Lett 9: 2413–2418 [DOI] [PubMed] [Google Scholar]
- Cordeaux Y, Briddon SJ, Megson AE, McDonnell J, Dickenson JM, Hill SJ. ( 2000) Influence of receptor number on functional responses elicited by agonists acting at the human adenosine A(1) receptor: evidence for signaling pathway-dependent changes in agonist potency and relative intrinsic activity. Mol Pharmacol 58: 1075–1084 [DOI] [PubMed] [Google Scholar]
- de Ligt RA, Rivkees SA, Lorenzen A, Leurs R, IJzerman AP. ( 2005) A “locked-on,” constitutively active mutant of the adenosine A1 receptor. Eur J Pharmacol 510: 1–8 [DOI] [PubMed] [Google Scholar]
- Dowell SJ, Brown AJ. ( 2002) Yeast assays for G-protein-coupled receptors. Receptors Channels 8: 343–352 [PubMed] [Google Scholar]
- Elzein E, Zablocki J. ( 2008) A1 adenosine receptor agonists and their potential therapeutic applications. Expert Opin Investig Drugs 17: 1901–1910 [DOI] [PubMed] [Google Scholar]
- Erlenbach I, Kostenis E, Schmidt C, Serradeil-Le Gal C, Raufaste D, Dumont ME, Pausch MH, Wess J. ( 2001) Single amino acid substitutions and deletions that alter the G protein coupling properties of the V2 vasopressin receptor identified in yeast by receptor random mutagenesis. J Biol Chem 276: 29382–29392 [DOI] [PubMed] [Google Scholar]
- Gao ZG, Jacobson KA. ( 2007) Emerging adenosine receptor agonists. Expert Opin Emerg Drugs 12: 479–492 [DOI] [PubMed] [Google Scholar]
- Gao ZG, Kim SK, Ijzerman AP, Jacobson KA. ( 2005) Allosteric modulation of the adenosine family of receptors. Mini Rev Med Chem 5: 545–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haskó G, Linden J, Cronstein B, Pacher P. ( 2008) Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov 7: 759–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jockers R, Linder ME, Hohenegger M, Nanoff C, Bertin B, Strosberg AD, Marullo S, Freissmuth M. ( 1994) Species difference in the G protein selectivity of the human and bovine A1-adenosine receptor. J Biol Chem 269: 32077–32084 [PubMed] [Google Scholar]
- Kenakin T. ( 1995) Agonist-receptor efficacy. II. Agonist trafficking of receptor signals. Trends Pharmacol Sci 16: 232–238 [DOI] [PubMed] [Google Scholar]
- Kenakin TP. ( 2003) A Pharmacology Primer: Theory, Application and Methods, Academic Press, San Diego [Google Scholar]
- Kourounakis A, Visser C, de Groote M, IJzerman AP. ( 2001) Differential effects of the allosteric enhancer (2-amino-4,5-dimethyl-trienyl)[3-trifluoromethyl) phenyl]methanone (PD81,723) on agonist and antagonist binding and function at the human wild-type and a mutant (T277A) adenosine A1 receptor. Biochem Pharmacol 61: 137–144 [DOI] [PubMed] [Google Scholar]
- Leff P, Prentice DJ, Giles H, Martin GR, Wood J. ( 1990) Estimation of agonist affinity and efficacy by direct, operational model-fitting. J Pharmacol Methods 23: 225–237 [DOI] [PubMed] [Google Scholar]
- Levitzki A. ( 1988) From epinephrine to cyclic AMP. Science 241: 800–806 [DOI] [PubMed] [Google Scholar]
- May LT, Sexton PM, Christopoulos A. ( 2005) Effects of urea pretreatment on the binding properties of adenosine A1 receptors. Br J Pharmacol 146: 1119–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migita K, Lu L, Zhao Y, Honda K, Iwamoto T, Kita S, Katsuragi T. ( 2005) Adenosine induces ATP release via an inositol 1,4,5-trisphosphate signaling pathway in MDCK cells. Biochem Biophys Res Commun 328: 1211–1215 [DOI] [PubMed] [Google Scholar]
- Minelli A, Bellezza I, Collodel G, Fredholm BB. ( 2008) Promiscuous coupling and involvement of protein kinase C and extracellular signal-regulated kinase 1/2 in the adenosine A1 receptor signalling in mammalian spermatozoa. Biochem Pharmacol 75: 931–941 [DOI] [PubMed] [Google Scholar]
- Motulsky H, Christopoulos A. ( 2004) Fitting Models to Biological Data Using Linear and Nonlinear Regression. A Practical Guide to Curve Fitting, Oxford University Press, New York [Google Scholar]
- Nawaratne V, Leach K, Suratman N, Loiacono RE, Felder CC, Armbruster BN, Roth BL, Sexton PM, Christopoulos A. ( 2008) New insights into the function of M4 muscarinic acetylcholine receptors gained using a novel allosteric modulator and a DREADD (designer receptor exclusively activated by a designer drug). Mol Pharmacol 74: 1119–1131 [DOI] [PubMed] [Google Scholar]
- Nobles M, Benians A, Tinker A. ( 2005) Heterotrimeric G proteins precouple with G protein-coupled receptors in living cells. Proc Natl Acad Sci U S A 102: 18706–18711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt C, Li B, Bloodworth L, Erlenbach I, Zeng FY, Wess J. ( 2003) Random mutagenesis of the M3 muscarinic acetylcholine receptor expressed in yeast. Identification of point mutations that “silence” a constitutively active mutant M3 receptor and greatly impair receptor/G protein coupling. J Biol Chem 278: 30248–30260 [DOI] [PubMed] [Google Scholar]
- Schulte G, Fredholm BB. ( 2003) Signalling from adenosine receptors to mitogen-activated protein kinases. Cell Signal 15: 813–827 [DOI] [PubMed] [Google Scholar]
- Selbie LA, Hill SJ. ( 1998) G protein-coupled-receptor cross-talk: the fine-tuning of multiple receptor-signalling pathways. Trends Pharmacol Sci 19: 87–93 [DOI] [PubMed] [Google Scholar]
- Townsend-Nicholson A, Shine J. ( 1992) Molecular cloning and characterisation of a human brain A1 adenosine receptor cDNA. Brain Res Mol Brain Res 16: 365–370 [DOI] [PubMed] [Google Scholar]
- Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, et al. ( 2007) Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther 320: 1–13 [DOI] [PubMed] [Google Scholar]
- Werry TD, Christopoulos A, Sexton PM. ( 2006) Mechanisms of ERK1/2 regulation by seven-transmembrane-domain receptors. Curr Pharm Des 12: 1683–1702 [DOI] [PubMed] [Google Scholar]
- Wise A, Watson-Koken MA, Rees S, Lee M, Milligan G. ( 1997) Interactions of the alpha2A-adrenoceptor with multiple Gi-family G-proteins: studies with pertussis toxin-resistant G-protein mutants. Biochem J 321: 721–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu RC, Pesce CG, Colman-Lerner A, Lok L, Pincus D, Serra E, Holl M, Benjamin K, Gordon A, Brent R. ( 2008) Negative feedback that improves information transmission in yeast signalling. Nature 456755–761 [DOI] [PMC free article] [PubMed] [Google Scholar]