

Abstract
Gastrointestinal stromal tumours (GISTs) are the most common mesenchymal tumours of the gastrointestinal tract. Formerly GISTs were commonly classified histologically as leiomyosarcomas; however, they are now known to arise from the interstitial cells of Cajal. Majority of GISTs overexpress KIT and have characteristic mutations within the gene, which are the targets of drug treatment with tyrosine kinase inhibitors. Leiomyosarcoma is a malignant tumour of smooth muscle differentiation and falls into a group of sarcomas that show complex karyotypic changes with no consistent recurrent genetic abnormality. We have used comparative genomic hybridization in combination with fluorescence in situ hybridization to determine genetic differences between the tumour types. We found leiomyosarcomas and GISTs share common regions of chromosomal 13q and 11q imbalance, in addition to more specific 1p and 8p losses in leiomyosarcoma and 15q and 22q losses in GISTs. More importantly, we have shown for the first time a deletion in the ataxia telangiectasia mutated (ATM) gene locus with decreased/absent expression of ATM protein, and amplification in the region 13q21–q32 in both tumour types, suggesting both regions may play a role in leiomyosarcoma and GIST biology.
Keywords: 11q, 13q, comparative genomic hybridization, fluorescence in situ hybridization, gastrointestinal stromal tumour, leiomyosarcoma
Leiomyosarcoma and gastrointestinal stromal tumour (GIST) are malignant tumours of mesenchymal origin. Until relatively recently, the distinction between leiomyosarcoma and GIST was not clear and GISTs were commonly classified as smooth muscle tumours based on histological criteria. However, GISTs are now thought to arise from the interstitial cells of Cajal, the pacemaker cells of the gastrointestinal tract (Kindblom et al. 1998; Robinson et al. 2000), whereas leiomyosarcomas show features of smooth muscle differentiation.
Leiomyosarcomas occur in a wide variety of anatomic locations, most commonly in the retroperitoneum, uterus, skin and superficial soft tissues, and in the deep compartments of the extremities, whereas GISTs are the most common mesenchymal tumours of the gastrointestinal tract and may occur anywhere along it, most commonly in the stomach and the small intestine.
The majority of GISTs (95%) are characterized by gain of function mutations in the proto-oncogene KIT (CD117) which encodes a receptor tyrosine kinase, KIT, resulting in its constitutive activation and expression (Hirota et al. 1998; Sarlomo-Rikala et al. 1998). Mutations within exon 11 of KIT are the most common, and are the target of drug treatment with the tyrosine kinase inhibitors imatinib mesylate and more recently sunitinib malate. A subset of GISTs show activating mutations in the platelet-derived growth factor receptor α gene, a member of the same receptor tyrosine kinase family as KIT. Leiomyosarcoma, on the contrary, falls into a group of sarcomas that show complex genetic and karyotypic changes. To date leiomyosarcomas lack a recurrent genetic alteration that may be used as a marker or a target for diagnostic and therapeutic intervention (Yang et al. 2009).
Ataxia telangiectasia mutated (ATM), located in the chromosome region 11q22, is the gene mutated in the autosomal recessive disorder ataxia telangiectasia (AT; Savitsky et al. 1995). Ataxia telangiectasia has characteristic features of increased radiosensitivity, high genomic instability and an increased risk of cancer. Ataxia telangiectasia mutated is a 350-kDa protein with a major role in cell cycle regulation following DNA damage, and in particular DNA double-strand breaks, by the activation of a number downstream signal transduction pathways including p53 and breast cancer associated 1 (BRCA1; Kastan & Lim 2000).
In this study, we used comparative genomic hybridization (CGH) to screen for genetic abnormalities in both leiomyosarcoma and GIST. Deletion in the ATM region was confirmed with fluorescence in situ hybridization (FISH). Using immunohistochemistry we showed ATM to be down-regulated in both tumours, suggesting ATM may have a role in the pathogenesis of leiomyosarcomas and GISTs.
Materials and methods
Patients and tumour samples
Formalin fixed paraffin embedded tissue blocks were obtained from the Royal Hallamshire Hospital, Sheffield, having been collected during the period 1994–2006. Ethical approval for the study was obtained and all tissue was collected and stored according to the principles of the Declaration of Helsinki. Comparative genomic hybridization analysis consisted of 22 leiomyosarcoma samples from 20 patients (13 female, 7 male), including two primary tumours and corresponding recurrences, and one recurrent case without a primary tumour. Thirteen cases of GISTs were investigated (eight female, five male), which included a primary tumour with corresponding recurrence. Immunohistochemical analysis involved additional cases of leiomyosarcoma and GIST, and assigned as LMS/GIST B1, B2, etc.
DNA preparation
DNA was extracted from six 5 μm sections mounted onto 3′-aminopropyltriethoxysilane (APES)-coated glass slides. The sections were de-waxed in two washes of xylene for 5 min, followed by hydration through an alcohol series (two 5 min washes in 100% ethanol, one 5 min wash in 85% ethanol, one 5 min wash in 70% ethanol). Sections were washed and kept hydrated in deionized water, whereas sections of interest (of at least 50% tumour cells) were dissected manually, confirmed by examination of H&E stained sections. Genomic DNA was isolated with proteinase K digestion using Qiagen Tissue Kit (Qiagen, West Sussex, UK).
Comparative genomic hybridization analysis
Comparative genomic hybridization was performed as previously described (Al-Assar et al. 2005). Briefly, tumour and normal reference DNA (Promega, Southampton, UK) were labelled by nick translation with Spectrum Green dUTP (Vysis/Abbott, Wessex, UK) and SpectrumRed dUTP (Vysis/Abbott), respectively. Digestion time and enzyme concentration were adjusted to obtain an average fragment size of 200–3000 bp. Fluorescently labelled tumour and matched normal reference DNA were co-precipitated with COT-1 DNA (Roche Diagnostics Ltd, East Sussex, UK), redissolved in hybridization mixture and hybridized to normal human metaphase chromosomes (Vysis) at 37 °C for 4 days. Following posthybridization washes, the slides were counterstained with 4′6-diamidino-2-phenylindole (DAPI; Vysis).
Digital images of the target metaphases were captured using charged-coupled device (CCD) camera attached to an epifluorescence microscope (Zeiss Axioskop; Germany). Dedicated software QUIPS CGH analysis software (Vysis) and Cytovision (Applied Imaging, Newcastle upon Tyne, UK) were used for analysis to calculate the ratio of green (tumour DNA) to red (normal DNA) fluorescence along the length of each chromosome. Mean ratio profiles were calculated using 10–15 metaphases for each sample. Based on control experiments using pooled normal DNA, green-to-red ratios of 0.8 and 1.2 were used as threshold values for determining regions of loss and gain, respectively. A ratio of 1.0 represents the balanced state of chromosome copy number. Centromeric and heterochromatic regions were excluded from analysis, as highly repetitive DNA sequences present in these areas can give false-positive results.
Fluorescence in situ hybridization analysis
Following CGH, seven leiomyosarcoma and two GIST cases with 11q deletion and four cases each of leiomyosarcoma and GIST with 13q gain were available for confirmation of the CGH findings by FISH. Interphase FISH was carried out on 4-μm paraffin-embedded tumour sections prepared onto glass slides. Chromosome 11 centromeric [CEP 11 (D11Z1); Vysis] and LSI ATM (11q22.3; Vysis) were used to confirm chromosome 11 copy number and deletion of 11q22, respectively. LSI 13 (13q14; Vysis) probe was used to confirm amplification of 13q. Chromosome 13 centromeric probe was not used because of complication with cross-reactivity.
The sections were de-waxed twice in xylene for 5 min, followed by dehydration in 100% ethanol for 5 min. The sections were washed in 0.2 N HCl at room temperature for 23 min, and in deionized water for 2 min. Pretreatment solution (40 ml; Zymed ‘Spotlight’ Paraffin Section Pre-treatment Kit; Invitrogen Ltd, Paisley, UK) at 95 °C was added to each section for 3 h. Following two 3 min washes in deionized water, the sections were treated with Enzyme Reagent (Zymed ‘Spotlight’ Paraffin Section Pre-treatment Kit) for 2.5 h. Sections were washed and dehydrated through an alcohol series (two 5 min washes in 70% ethanol, 95% ethanol, 100% ethanol). Probes were hybridized onto the sections at 37 °C overnight according to the manufacturer’s guidelines. Following posthybridization washes (2 min in 0.4XSSC/0.3% Tween 20 at 73 °C and 1 min 2XSSC/0.1% Tween 20 at room temperature), the slides were counterstained with DAPI (Vysis). The FISH signals were scored from 200 to 300 non-overlapping intact nuclei, in each case by Zeiss fluorescence microscope and Cytovision (Applied Imaging) software.
Immunohistochemistry
Paraffin embedded sections (4 μm) were used for immunostaining. The sections were de-paraffinized in two washes of xylene for 5 min, followed by hydration through an alcohol series (one 5 min wash in 100% ethanol, one 5 min wash in 95% ethanol, one 5 min wash in 70% ethanol). Endogenous peroxidase activity was blocked using 0.3% hydrogen peroxide. Antigen retrieval was carried out by heating the slides in a solution of 10 mM/l tri-sodium citrate using a microwave oven (950 W). The sections were then incubated in normal blocking serum, followed by incubation in the primary antibody solution overnight at 4 °C. Ataxia telangiectasia mutated antibody was obtained from Abcam (Cambridge, UK) and breast tissue was used as positive controls. For the analysis of KIT, the sections were incubated with the primary antibody obtained from Dako (Cambridgeshire, UK) for 30 min at room temperature. Gastrointestinal stromal tumours were used as positive controls. For all negative controls, a solution containing IgG from the same primary antibody host species was used. Immunoreactivity of the primary antibody was detected following the manufacturer’s instructions using the Vectastain Elite ABC and 3,3′-diaminobenzidine Kits from Vector Laboratories (Peterborough, UK). The immunostained images were captured using a Nikon Cool Pix Digital Camera mounted on Nikon Eclipse E600 Microscope (Nikon UK Ltd, Surrey, UK). Sections were reviewed by two independent observers (David Hughes and Aliya Ul-Hassan) and assigned as negative (no detectable staining), weak (minority of cells showing detectable staining) or moderately positive (minority of cells showing strong staining or majority showing detectable staining). The staining evaluation of the sections considered both the staining intensity and percentage of positive tumour cells to take into consideration any heterogeneity of staining.
Results
Clinicopathologic data
The clinical details of the cases are presented Table 1. The average age of leiomyosarcoma patients was 64 years (range 29–85). Number of deep tumours (including extremity and central, i.e. abdomen, retroperitoneum, pelvic) was 14 including 7 uterine leiomyosarcoma, and 8 tumours were superficial. According to the French grading system (Trojani et al. 1984), 11 tumours were of grade III, 2 were grade II and 2 tumours were of grade I. Uterine leiomyosarcoma were graded as low or high grade, depending on the degree of cellular atypia, amount of tumour necrosis and the number of mitotic figures. All uterine leiomyosarcomas were of high grade in this study. Twelve tumours were 5 cm or above and 10 tumours were less than 5 cm. The average age of GIST patients was 63 years (range 37–82). According to the risk stratification system (Hornick & Fletcher 2007), nine cases were of high risk, three intermediate and one low risk. Eleven tumours were 5 cm or above and two tumours were less than 5 cm.
Table 1.
A summary of the clinical details, CGH, FISH and immunohistochemical data for leiomyosarcoma and GIST
| Tumour | Age | Sex | Tumour site | Tumour grade | CGH status of 11q | FISH status of 11q | ATM expression | cKit expression |
|---|---|---|---|---|---|---|---|---|
| Leiomyosarcoma | ||||||||
| LMS 1 | 79 | M | Abdominal | 3 | Del | Del | M | – |
| LMS 2 | 74 | F | Uterine | High | Del | Del | NA | – |
| LMS 4 | 65 | F | Abdominal | 3 | * | Del | NA | – |
| LMS 5 | 74 | F | Recurrent pelvic | 3 | * | N | N | |
| LMS 8 | 29 | F | Knee | 2 | Del | NS | M | – |
| LMS 10 | 85 | F | Uterine | 2 | Del | NS | N | N |
| LMS 11 | 51 | M | Small bowel | 3 | Del | Del | N | – |
| LMS 13 | 37 | M | Bladder | 3 | * | W | N | |
| LMS 15 | 64 | M | Abdominal | 3 | * | W | N | |
| LMS 16 | 48 | M | Shoulder | 3 | * | M | – | |
| LMS 17 | 82 | F | Calf | 3 | Del | M | N | |
| LMS 18 | 51 | M | Recurrent peritoneal | 3 | Del | Del | N | N |
| LMS 20 | 55 | F | Uterine | High | Del | Del | W | – |
| LMS 21 | 66 | F | Uterine | High | Del | Del | M | N |
| LMS 22 | 75 | M | Thigh | 1 | Del | N | – | |
| LMS B2 | 77 | F | Uterine | High | – | – | N | N |
| LMS B3 | 46 | M | Recurrent knee | 1 | – | – | N | N |
| LMS B4 | 37 | F | Uterine | High | – | – | W | – |
| LMS B6 | 71 | F | Bladder | 3 | – | – | N | N |
| LMS B7 | 76 | M | Thigh | 1 | – | – | N | N |
| LMS B8 | 92 | F | Breast | 2 | – | – | W | N |
| LMS B9 | 69 | F | – | – | W | N | ||
| LMS B10 | 62 | F | Uterine | – | – | W | N | |
| LMS B11 | 87 | M | Buttock | 3 | – | – | N | N |
| LMS B12 | 49 | F | Retroperitoneal | 3 | – | – | N | N |
| LMS B13 | 77 | F | Uterine | – | – | N | N | |
| LMS B14 | 58 | F | Uterine | – | – | N | N | |
| LMS B15 | 80 | F | Retroperitoneal | 2 | – | – | N | N |
| LMS B16 | 57 | F | – | – | N | N | ||
| LMS B17 | 54 | F | Recurrent pelvic | – | – | W | N | |
| LMS B18 | 55 | M | Buttock | 3 | – | – | N | – |
| Gastrointestinal stromal tumour | ||||||||
| GIST 1 | 49 | M | Stomach | High risk | Normal | M | ||
| GIST 2 | 64 | F | Small bowel | High risk | * | W | ||
| GIST 4 | 60 | M | Abdominal | High risk | * | N | ||
| GIST 5 | 59 | F | Small bowel | High risk | * | N | ||
| GIST 6 | 73 | F | Small bowel | High risk | Del | Del | M | |
| GIST 7 | 82 | F | Colon | High risk | Del | NS | W | |
| GIST 9 | 44 | M | Small bowel | Intermediate risk | Del | NS | N | |
| GIST 11 | 73 | F | Recurrent small bowel | High risk | Del | Del | M | |
| GIST 12 | 73 | M | Retroperitoneal | Intermediate risk | * | N | ||
| GIST 13 | 37 | F | Stomach | High risk | * | W | S | |
| GIST B1 | 66 | F | Gastric | Low risk | – | – | M | S |
| GIST B3 | 81 | F | Small bowel | Low risk | – | – | W | M |
| GIST B4 | 60 | M | Stomach | Low risk | – | – | N | M |
| GIST B5 | 76 | F | Stomach | Low risk | – | – | N | S |
| GIST B7 | 26 | M | Small bowel | Intermediate risk | – | – | M | S |
| GIST B8 | 80 | F | Stomach | Low risk | – | – | M | S |
| GIST B9 | 71 | F | Stomach | Low risk | – | – | W | S |
| GIST B10 | 69 | F | Stomach | High risk | – | – | M | W |
| GIST B11 | 57 | M | Liver metastasis | – | – | N | W | |
Del, deletion; S, strongly positive; M, moderately positive; W, weakly positive; N, negative; NA, tissue not available; NS, tissue not suitable.
CGH profile was just below the 0.8 threshold value for a deletion.
Comparative genomic hybridization
Chromosomal gains were found to be more frequent than losses for both leiomyosarcoma and GIST (Figure 1). The most common gains in leiomyosarcoma involved chromosome regions on 4q (59%), 6q (45%), 8q (45%), 5q (36%), 13q (32%), 1q (27%) and chromosome X. With GISTs, the common gains were within the regions 4q (100%), 6q (54%), 13q (46%), 12q (38%), 8q (38%) and chromosome X. The most common losses in leiomyosarcoma involved regions on 1p (55%), 11q (45%) and 8p (41%), and losses in GISTs included 22q (77%), 15q (62%), 1p (46%), 11q (31%) and 14q (31%). Chromosome 16 was excluded from analysis, as it contains a large heterochromatic region often resulting in false-positive results.
Figure 1.

A summary of the combined chromosomal imbalances from CGH analysis of LMS (blue) and GISTs (purple). Lines to the left of the chromosomes represent loss and lines to the right represent gain.
The pattern of chromosomal imbalance for both leiomyosarcoma and GISTs was comparable, with the most specific regions affected being losses of 1p, 8p, 11q and 22q with gains of 6q, 12q and 13q. Of these, the deletion of 11q and amplification of 13q have not been previously investigated in association with leiomyosarcoma and GISTs. In this study of the 17 high-grade leiomyosarcoma cases (including uterine), 35% (6/17) had 13q gain and 47% (8/17) had loss of 11q; however, as there were only two grade I and three grade II leiomyosarcoma tumours, no correlation between 11q deletion and 13q gain with tumour grade can be made. Twenty per cent (2/10) of leiomyosarcoma less than 5 cm showed gain of 13q, whereas 47% (5/12) of leiomyosarcoma 5 cm or above showed gain of 13q. Thirty per cent (3/10) of leiomyosarcoma less than 5 cm showed deletion of 11q, whereas 58% (7/12) of leiomyosarcoma 5 cm or above showed a deletion of 11q. For GISTs, a similar relationship with larger, more advanced tumours was seen for amplification of 13q, with 56% (5/9) of 13q gains in high-risk patients. The deletion of 11q was however less tightly linked with only 33% (3/9) of 11q losses in high-risk tumours. The number of GIST cases was however low and a clinical correlation between 11q loss and gain of 13q with tumour size, grade and location could not be made.
Fluorescence in situ hybridization analysis of 11q deletion and 13q amplification
Cut-off values for the probes were calculated from five normal controls (three tonsils and two placentas) as the mean ± 3SD for both probe sets. The cut-off value for chromosome 11 deletion was 15% and for chromosome 13 gain was 2%. Results above the cut-off values were positive. Fifteen to twenty per cent of cells in all cases studied for 13q gains (four leiomyosarcoma and four GISTs) showed extra copies of the 13q probe (Figure 2b). All leiomyosarcoma and GIST cases studied for 11q losses (seven leiomyosarcoma and two GISTs) showed loss of one ATM signal in 40–75% of the cells (Figure 2d) and loss of both signals in 5–20% of the cells.
Figure 2.
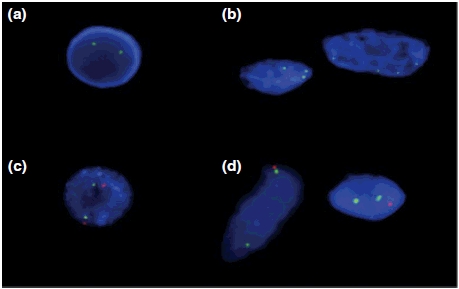
Interphase FISH results confirming gain of a region in 13q and deletion in 11q in LMS and GIST. (a) Normal lymphocyte with two copies of LSI 13 (13q14) (green). (b) Tumour cells with extra copies of LSI 13 (13q14) (green). (c) Normal lymphocyte with two copies each of CEP 11 (D1121) (green) and LSI ATM (11q22.3) (red). (d) Tumour cells with two copies of CEP 11 (D1121) (green) and loss of one copy of LSI ATM (11q22.3) (red).
Ataxia telangiectasia mutated protein expression
Sixty-three per cent (12/19) of GISTs showed either negative (37%, 7/19) or weak (26%, 5/19), and 37% (7/19) showed moderate staining for ATM expression. Eighty-three per cent (24/29) of leiomyosarcoma showed either negative (55%, 16/29) or weak (28%, 8/29) ATM expression, and 17% (5/29) showed moderate staining for ATM (Figure 3 and Table 1). Fifty per cent of the cases with an 11q deletion detected by CGH and FISH had decreased/absent expression of ATM (Table 1).
Figure 3.
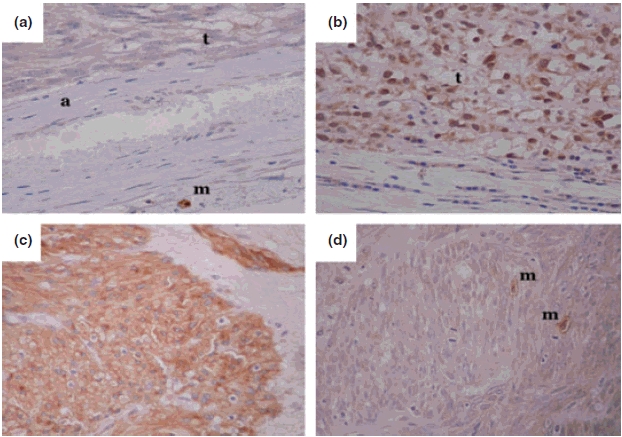
Immunostaining for ATM and KIT proteins in leiomyosarcomas and GISTs. (a) Tumour negative for ATM showing a positive mast cell as an internal control. (b) Tumour showing positive staining for ATM with blue lymphocytes/fibroblasts as internal negative controls. (a) A KIT positive GIST showing strong cytoplasmic staining. (d) A KIT negative leiomyosarcoma with positive mast cells as internal controls. (t, Area of tumour; a, arteriole, m, mast cell). Photographs were taken at 400 × original magnification.
Discussion
Soft tissue sarcomas are a heterogeneous group of tumours often difficult to classify, and while some can be classified by characteristic changes at the cytogenetic level, leiomyosarcoma often have a complex karyotype with no consistent abnormality. Most GISTs, on the contrary, have a mutation in the KIT gene, which has led to the use of targeted drug treatment and aided in classification of the malignancy as a separate tumour category. KIT expression has been reported in other sarcomas; however, KIT expression in leiomyosarcoma remains controversial (Hornick & Fletcher 2002). Here, we found no detectable expression of KIT in the leiomyosarcoma cases (Figure 3 and Table 1), despite both leiomyosarcoma and GIST sharing similar chromosomal changes.
The CGH aspect of the investigation found that chromosomal imbalance was widespread with gains more frequent than losses in both tumour types (Figure 1). Deletion in 13q14, the region containing the tumour suppressor retinoblastoma (RB1) gene, has been found to be a common occurrence in leiomyosarcomas; however in this study, 13q14 deletion was only present in two leiomyosarcoma cases and one case of GIST. As extensive karyotypic variation exists in leiomyosarcoma and the series of leiomyosarcomas investigated here is different, these differences may be attributable to chance occurrence. More importantly, we have shown the presence of amplification in the region 13q21–q32 for both leiomyosarcoma and GIST, confirming the findings with FISH by using a probe for RB1 mapping to 13q14. Although this region is just above the gain observed by CGH, it was found that approximately 15–20% of the cells were aneuploid and showed at least three signals for the probe. Although polyploidy cannot be entirely excluded, CGH findings did not indicate complete 13q gains. It is likely that amplification of the region extending above 13q21 may have been missed by CGH, as CGH is determined by the copy number and the size of the region involved reflected as an average. 13q amplification has been reported in other malignancies. In particular, a novel 13q31 gain has been reported in alveolar rhabdomyosarcoma detected by CGH (Gordon et al. 2000) that may involve the amplification of Glypican-5 (Williamson et al. 2007), and recently gains of 13q21, 13q22, 13q31 and 13q32 were found to be significantly associated with poor overall survival in liposarcoma (Schmidt et al. 2005). Chromosome 13 is a particular gene poor area having one of the lowest gene densities (Dunham et al. 2004); however, it is a site of some critical genes involved in cancer including breast cancer associated 2 (BRCA2) and RB1. The findings in this investigation of amplification for 13q in both leiomyosarcoma and GIST suggests that gene(s) present within this region may be involved in leiomyosarcoma and GIST biology that have not been reported previously. Although, as mentioned previously, the number of low-grade tumours was limited, it was seen here that the amplification of 13q was associated with larger leiomyosarcoma and high-risk GISTs, and therefore possibly indicative of later stages of tumour development and represent a potential new marker of poor prognosis. Additional cases will be required to refine the region of amplification and to correlate with prognosis.
Both leiomyosarcoma and GIST were found in this study to have a specific targeted deletion in 11q22–q24. Initial studies were undertaken using FISH with the ATM probe which maps within the target region in 11q. Deletion of 11q region was confirmed, as all cases studied by FISH had at least 50% of cells with deletion of one or both copy numbers of ATM but not the centromeric probe. Moreover, this frequency was higher than found with CGH for either tumour type and may suggest ATM is targeted specifically. The potential relevance of ATM to the tumours was further confirmed with immunohistochemical data where the protein expression was either absent or decreased in both leiomyosarcoma (83%) and GISTs (63%). Ataxia telangiectasia mutated has been found to be involved in rhabdomyosarcoma (Zhang et al. 2003), and mice lacking the ATM phosphorylation site on p53 have shown to develop sarcomas including leiomyosarcomas (Armata et al. 2007), suggesting loss of wild-type ATM function has a role in sarcoma pathogenesis. This is the first time that ATM downregulation has been implicated in leiomyosarcoma and GIST, suggesting it may have a role in the biology of both tumours. As the target deleted region on 11q detected by CGH is quite large, ATM although found here to be deleted may not be the exclusive target and other genes mapping to 11q, e.g. CHEK1 (11q22–q23), CASP 1, 4 and 5 (11q22), ACAT1 (11q22.3–q23.1) and TSG 11 (11q23) may be involved. Comparative genomic hybridization detection threshold is determined by the copy number and the size of the region involved. Comparative genomic hybridization for some of the cases identified a large area of deletion, but not all regions (including ATM) may have been detected. It is generally accepted that CGH detects genetic abnormalities when the size of chromosomal region affected is 10–12 Mb (Kallioniemi et al. 1994). Single copy deletions smaller than 2 Mb are unlikely to be detected by CGH (Bentz et al. 1998). Furthermore, ATM expression was found in tumours with deletion of 11q, which may suggest that regulatory changes are also of importance. In normal tissues and under normal conditions, ATM expression is ubiquitous (Human Protein Atlas; http://www.proteinatlas.org). The heterogeneity of ATM expression in both tumours also raises the possibility that different levels of expression may be of significance to the behaviour of these tumours. The significance of 11q deletion to ATM expression in leiomyosarcomas and GISTs and the significance of ATM expression to the biological behaviour of these tumours remain to be determined. A more extensive study using array-CGH to follow-up the findings will be required.
Other regions of imbalance identified by this study in both leiomyosarcomas and GISTs include losses of 1p, 8p and 22q and gain of 4q, 6q and 12q and X. These regions tended to be larger and less specific (4q, 6q, 12q, 22q and X), or to partially include the telomeric regions (1p and 8p), which are traditionally known to have differential hybridization (Kallioniemi et al. 1994). The amplification of 12q12–q21 found here in both tumours contains genes frequently detected as amplified in sarcomas, namely CDK4, SAS and oncogenes GLI and MDM2 (Berner et al. 1996; Taubert et al. 2003; Ragazzini et al. 2004). In addition, MDM2 has also been found to be amplified in GIST (Tornillo et al. 2005), and it is likely that the amplification detected in this study reflects involvement of MDM2 in the development of both leiomyosarcoma and GIST. The amplification of the X chromosome in males in this study is an interesting finding, and may imply, as has been found in other tumours (Koivisto et al. 1999; Richardson et al. 2006), that hormonal influences may be a factor in the development of both leiomyosarcomas and GISTs. Finally, gains of 4q occur frequently for both leiomyosarcomas (59%) and more commonly in GISTs (100%). One of the genes present within this region is KIT (4q12). However, the region involved in 4q is extensive suggesting a number of genes may be contributing.
Regions found differentially involved in leiomyosarcoma and GIST are also of interest. Deletion of 14q was more common in GISTs (31%) than leiomyosarcomas (14%), and is a common region of deletion reported in GISTs. Leiomyosarcomas had a higher frequency of 1p (55%) and 8p (41%) losses than GISTs (46% 1p and 23% 8p), and it has been suggested that loss of 1p and 8p relate to increased metastasis for leiomyosarcoma (Mandahl et al. 2000). Gastrointestinal stromal tumours have also been shown to have loss of 1p, but they often co-existed with losses of 15q and were more frequent in high-risk tumours. Loss of 15q was however very specific to GISTs (62% GIST, 9% leiomyosarcoma), as was loss of 22q (77% GIST, 27% leiomyosarcoma), and because these deletions are common in GISTs, it may be that in combination loss of 1p, 15q and 22q define GISTs, whereas losses of 1p and 8p associate with leiomyosarcoma.
In summary, leiomyosarcomas and GISTs share common regions of chromosomal imbalance, most importantly the loss of 11q and gain of 13q. This is the first study to report deletion of ATM, and a common region of amplification at 13q21–q32 in both tumour types, suggesting that they are important in the development of both leiomyosarcoma and GIST. Furthermore, although leiomyosarcomas and GISTs share similar genetic profiles, there are some distinct differences, particularly in the frequency of involvement for 15q and 22q abnormalities. These chromosomal imbalances may represent defining abnormalities that can be used to further categorize and distinguish between leiomyosarcomas and GISTs. A larger study is required to confirm the findings and correlate with clinical significance, and the regions involved require further investigation to elucidate the genes involved in the pathogenesis of both types of tumours.
Acknowledgments
This work was supported by a grant from Weston Park Cancer Appeal (RB106379) and equipment grant from Yorkshire Cancer Research.
References
- Al-Assar O, Ul-Hassan A, Brown R, Wilson GA, Hammond DW, Reilly JT. Gains on 9p are common genomic aberrations in idiopathic myelofibrosis: a comparative genomic hybridization study. Br. J. Haematol. 2005;129:66–71. doi: 10.1111/j.1365-2141.2005.05413.x. [DOI] [PubMed] [Google Scholar]
- Armata HL, Garlick DS, Sluss HK. The ataxia telangiectasia-mutated target site Ser18 is required for p53-mediated tumor suppression. Cancer Res. 2007;67:11696–11703. doi: 10.1158/0008-5472.CAN-07-1610. [DOI] [PubMed] [Google Scholar]
- Bentz M, Plesch A, Stilgenbauer S, Dohner H, Lichter P. Minimal sizes of deletions detected by comparative genomic hybridization. Genes Chromosomes Cancer. 1998;21:172–175. [PubMed] [Google Scholar]
- Berner JM, Forus A, Elkahloun A, Meltzer PS, Fodstad O, Myklebost O. Separate amplified regions encompassing CDK4 and MDM2 in human sarcomas. Genes Chromosomes Cancer. 1996;17:254–259. doi: 10.1002/(SICI)1098-2264(199612)17:4<254::AID-GCC7>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Dunham A, Matthews LH, Burton J, et al. The DNA sequence and analysis of human chromosome 13. Nature. 2004;428:522–528. doi: 10.1038/nature02379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon AT, Brinkschmidt C, Anderson J, et al. A novel and consistent amplicon at 13q31 associated with alveolar rhabdomyosarcoma. Genes Chromosomes Cancer. 2000;28:220–226. [PubMed] [Google Scholar]
- Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577–580. doi: 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- Hornick JL, Fletcher CD. Immunohistochemical staining for KIT (CD117) in soft tissue sarcomas is very limited in distribution. Am. J. Clin. Pathol. 2002;117:188–193. doi: 10.1309/LX9U-F7P0-UWDH-8Y6R. [DOI] [PubMed] [Google Scholar]
- Hornick JL, Fletcher CD. The role of KIT in the management of patients with gastrointestinal stromal tumors. Hum. Pathol. 2007;38:679–687. doi: 10.1016/j.humpath.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Kallioniemi OP, Kallioniemi A, Piper J, et al. Optimizing comparative genomic hybridization for analysis of DNA sequence copy number changes in solid tumors. Genes Chromosomes Cancer. 1994;10:231–243. doi: 10.1002/gcc.2870100403. [DOI] [PubMed] [Google Scholar]
- Kastan MB, Lim DS. The many substrates and functions of ATM. Nat. Rev. Mol. Cell Biol. 2000;1:179–186. doi: 10.1038/35043058. [DOI] [PubMed] [Google Scholar]
- Kindblom LG, Remotti HE, Aldenborg F, Meis-Kindblom JM. Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am. J. Pathol. 1998;152:1259–1269. [PMC free article] [PubMed] [Google Scholar]
- Koivisto PA, Schleutker J, Helin H, Ehren-van Eekelen C, Kallioniemi OP, Trapman J. Androgen receptor gene alterations and chromosomal gains and losses in prostate carcinomas appearing during finasteride treatment for benign prostatic hyperplasia. Clin. Cancer Res. 1999;5:3578–3582. [PubMed] [Google Scholar]
- Mandahl N, Fletcher CD, Dal Cin P, et al. Comparative cytogenetic study of spindle cell and pleomorphic leiomyosarcomas of soft tissues: a report from the CHAMP Study Group. Cancer Genet. Cytogenet. 2000;116:66–73. doi: 10.1016/s0165-4608(99)00114-4. [DOI] [PubMed] [Google Scholar]
- Ragazzini P, Gamberi G, Pazzaglia L, et al. Amplification of CDK4, MDM2, SAS and GLI genes in leiomyosarcoma, alveolar and embryonal rhabdomyosarcoma. Histol. Histopathol. 2004;19:401–411. doi: 10.14670/HH-19.401. [DOI] [PubMed] [Google Scholar]
- Richardson AL, Wang ZC, De Nicolo A, et al. X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell. 2006;9:121–132. doi: 10.1016/j.ccr.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Robinson TL, Sircar K, Hewlett BR, Chorneyko K, Riddell RH, Huizinga JD. Gastrointestinal stromal tumors may originate from a subset of CD34-positive interstitial cells of Cajal. Am. J. Pathol. 2000;156:1157–1163. doi: 10.1016/S0002-9440(10)64984-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarlomo-Rikala M, Kovatich AJ, Barusevicius A, Miettinen M. CD117: a sensitive marker for gastrointestinal stromal tumors that is more specific than CD34. Mod. Pathol. 1998;11:728–734. [PubMed] [Google Scholar]
- Savitsky K, Bar-Shira A, Gilad S, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- Schmidt H, Bartel F, Kappler M, et al. Gains of 13q are correlated with a poor prognosis in liposarcoma. Mod. Pathol. 2005;18:638–644. doi: 10.1038/modpathol.3800326. [DOI] [PubMed] [Google Scholar]
- Taubert H, Schuster K, Brinck U, et al. Loss of heterozygosity at 12q14-15 often occurs in stage I soft tissue sarcomas and is associated with MDM2 amplification in tumors at various stages. Mod. Pathol. 2003;16:1109–1116. doi: 10.1097/01.MP.0000096045.51700.66. [DOI] [PubMed] [Google Scholar]
- Tornillo L, Duchini G, Carafa V, et al. Patterns of gene amplification in gastrointestinal stromal tumors (GIST) Lab. Invest. 2005;85:921–931. doi: 10.1038/labinvest.3700284. [DOI] [PubMed] [Google Scholar]
- Trojani M, Contesso G, Coindre JM, et al. Soft-tissue sarcomas of adults: study of pathological prognostic variables and definition of a histopathological grading system. Int. J. Cancer. 1984;33:37–42. doi: 10.1002/ijc.2910330108. [DOI] [PubMed] [Google Scholar]
- Williamson D, Selfe J, Gordon T, et al. Role for amplification and expression of glypican-5 in rhabdomyosarcoma. Cancer Res. 2007;67:57–65. doi: 10.1158/0008-5472.CAN-06-1650. [DOI] [PubMed] [Google Scholar]
- Yang J, Du X, Chen K, et al. Genetic aberrations in soft tissue leiomyosarcoma. Cancer Lett. 2009;275:1–8. doi: 10.1016/j.canlet.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Bhakta KS, Puri PL, Newbury RO, Feramisco JR, Wang JY. Association of ataxia telangiectasia mutated (ATM) gene mutation/deletion with rhabdomyosarcoma. Cancer Biol. Ther. 2003;2:87–91. doi: 10.4161/cbt.231. [DOI] [PubMed] [Google Scholar]