
Figure 2.
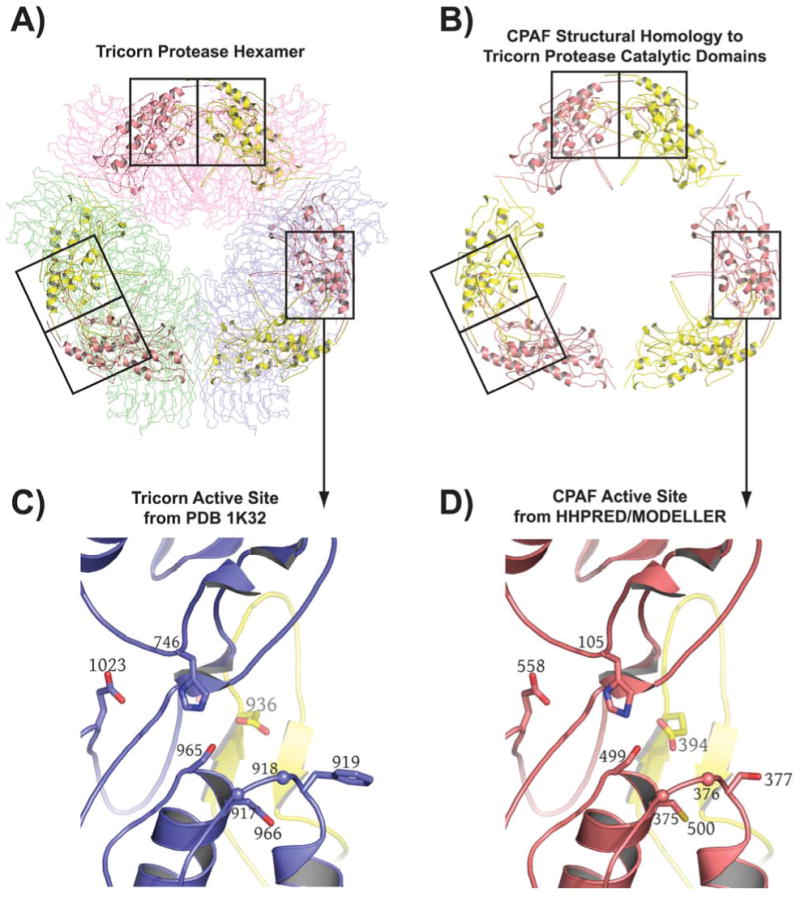
Computational prediction of CPAF fold and catalytic residues. A) The highest scoring hit returned for the CPAF amino acid sequence by HHPRED [26, 27] is the structure of the tricorn protease [25], a 720 kDa proteolytic system from Thermoplasma acidophilum that degrades cytosolic proteins analogously to the proteasome [43] (see text). The tricorn protease structure is a hexamer formed by a trimer of dimmers that has the shape of a distorted hexagon. The dimers forming the long sides of the hexagon are shown as Cα-traces and are colored in light pink, light green, and blue, respectively. The amino acid residues of CPAF that map onto the catalytic domain of the tricorn protease are shown in cartoon format. B) The amino acid residues of CPAF that map onto the catalytic domains of the tricorn protease taken out of the context of the tricorn protease. The functional active site requires contributions from both a yellow and a salmon subunit, corresponding to the catalytic domains of two-fold related tricorn protease subunits, suggesting that for CPAF, the minimal functional proteolytic unit is a CPAFn:CPAFc dimer (see text). C) The tricorn protease active site. Note that the specificity-determining residue, E936 shown in yellow, comes from a two-fold related catalytic domain subunit of the tricorn protease. D) CPAF active site identified by HHPRED [26, 27] and modeled by the program MODELLER [28]. The CPAF H105, S499, E558 catalytic triad and the E394 specificity-determining residue of CPAF corresponds to the H746, S945, D1023 catalytic triad and E936 specificity-determining residue of tricorn protease. This figure was prepared using the program PyMol (Delano Scientific, www.pymol.org).