

Abstract
Recent X-ray crystal structures and solution NMR spectroscopy data for calcium- and integrin-binding protein 1 (CIB1) have all revealed a common EF-hand domain structure for the protein. However, the orientation of the two protein domains, the oligomerization state, and the conformations of the N- and C-terminal extensions differ among the structures. In this study, we examine whether the binding of glutathione or auxiliary Ca2+ ions as observed in the crystal structures, occur in solution, and whether these interactions can influence the structure or dimerization of CIB1. In addition, we test the potential phosphatase activity of CIB1, which was hypothesized based on the glutathione binding site geometry observed in one of the crystal structures of the protein. Biophysical and biochemical experiments failed to detect glutathione binding, protein dimerization, or phosphatase activity for CIB1 under several solution conditions. However, our data identify low affinity (Kd, 10−2M) Ca2+ binding events that influence the structures of the N- and C-terminal extensions of CIB1 under high (300 mM) Ca2+ crystallization conditions. In addition to providing a rationale for differences amongst the various solution and crystal structures of CIB1, our results show that the impact of low affinity Ca2+ binding events should be considered when analyzing and interpreting protein crystallographic structures determined in the presence of very high Ca2+ concentrations.
Keywords: calcium- and integrin-binding protein, CIB1, calmyrin, calcium, NMR spectroscopy, X-ray crystallography, light scattering
Introduction
Calcium- and integrin-binding protein 1 (CIB1) is a small, ubiquitous, multifunctional protein with several hypothesized cell signaling roles through its interaction with integrin receptors, protein kinases, transcription factors, and other signaling proteins.1,2 The protein structure consists of four helix-loop-helix “EF-hand” Ca2+-binding motifs (EF-I to EF-IV),3 as well as a myristoylated N-terminal extension,4 and a short C-terminal extension which folds back on the protein and increases target-binding specificity.5 EF-III is a mixed Mg2+/Ca2+-binding site (Kd,Mg2+, ∼10−5M; Kd,Ca2+, ∼10−6M), EF-IV is a higher affinity Ca2+-specific site (Kd,Ca2+, ∼10−7M), and the N-terminal EF-I and EF-II domains deviate from the canonical EF-hand amino acid sequence and are incapable of high-affinity divalent metal ion-binding.6,7
A high-resolution X-ray crystal structure of Ca2+-CIB1 was first determined by Parise and coworkers (PDB: 1XO5), who reported a monomeric, bilobal conformation with the N- and C-domains positioned side-by-side similar to calcineurin B and many related EF-hand proteins [Fig. 1(A)].8 A second crystal structure was subsequently published by Bahnson and coworkers (PDB: 1Y1A), revealing a head-to-tail Ca2+-CIB1 dimer with a similar EF-hand structure to PDB: 1XO5, but with a large rotation of the N- and C-domains with respect to each other, and distinct N- and C-terminal extensions [Fig. 1(B)].9 Another unique characteristic of the PDB: 1Y1A structure was the presence of a glutathione (GSH) molecule noncovalently bound to the N-domain of Chain B but not Chain A. The orientation of the GSH, with the free thiol facing Cys35 in EF-I, implied that CIB1 might be redox regulated in vivo. In addition, the authors noted that the GSH binding site geometry resembled a cysteine-nucleophile phosphatase active site, suggesting that CIB1 might possess intrinsic protein phosphatase activity.9 This is a particularly intriguing possibility since CIB1 shares no sequence or structural homology with known phosphatases, and such an activity has not yet been demonstrated for any other member of the EF-hand protein family.
Figure 1.
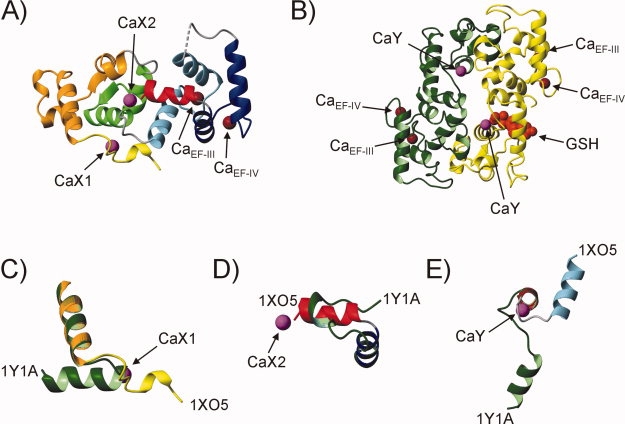
X-ray crystal structures of Ca2+-CIB1. A: PDB: 1XO5: Ca2+-CIB1 was crystallized as a monomer with two molecules in the asymmetric unit (only Chain A is shown).8 Color-coding is as follows: yellow (N-terminal extension), orange (EF-I), green (EF-II), blue (EF-III), navy (EF-IV), and red (C-terminal extension). Part of the loop connecting EF-III to EF-IV (G137-T142) is disordered in the crystal and is shown as a dotted line. B: PDB: 1Y1A: Ca2+-CIB1 was crystallized as a head-to-tail dimer consisting of Chain A (green) and Chain B (yellow), with a GSH molecule bound to Chain B shown in red space fill representation.9 C: Residues 9–36 of PDB: 1XO5 (Chain A) and PDB: 1Y1A (Chain A) are aligned with respect to residues 24–36 to show the distinct orientation of the N-terminal extension as a result of Ca2+ binding to the CaX1 site in PDB: 1XO5. Note that the N-terminal eight residues of CIB1 (MGGSGSRL) are highly flexible in solution,5 and the removal of these residues was necessary to successfully crystallize the protein.8,9 D: Residues 170–191 of PDB: 1XO5 (Chain A) and PDB: 1Y1A (Chain A) are aligned with respect to residues 170–178 to show how Ca2+ binding to the CaX2 site in PDB: 1XO5 influences the structure of the C-terminal extension. E: Residues 88–115 of PDB: 1XO5 (Chain A) and PDB: 1Y1A (Chain A) are aligned with respect to residues 88–98 to show how Ca2+ binding to site CaY influences the conformation of the central linker between the N- and C-domains of Ca2+-CIB1. In each panel, the Ca2+ ions bound to sites CaX1, CaX2, and CaY are labeled and shown in magenta, and the Ca2+ ions bound to EF-III and EF-IV are labeled (CaEF-III and CaEF-IV) and shown in brown. The color-coding of the secondary structure elements in panels C, D, and E is the same as in panels A and B. This figure was generated using the program MolMol 2K.2.10
Using triple resonance nuclear magnetic resonance (NMR) spectroscopy and highly sensitive residual dipolar coupling NMR experiments, we recently showed that Ca2+-CIB1 exists as a monomer near physiological solution conditions, with its N- and C-domains oriented side-by-side like PDB: 1XO5.5 However, the N-terminal extension adopts an α-helical structure oriented similarly to PDB: 1Y1A, and the C-terminal extension is somewhat flexible and only partially α-helical in solution.5 Interestingly, the regions of Ca2+-CIB1 which are structurally distinct under solution and crystallization conditions (the N- and C-terminal extensions and linker between the N- and C-domains) each contain extra Ca2+ ions bound in one of the crystal structures of the protein, in addition to the high-affinity Ca2+ ions bound to EF-III and EF-IV (Fig. 1). In the PDB: 1XO5 structure, one of these Ca2+ ions is coordinated in the second shell by the N-terminal extension (hereafter referred to as site CaX1), and another Ca2+ is bound between the C-terminal end of H5 and the acidic COO− C-terminus of the protein (hereafter referred to as site CaX2) [Fig. 1(C,D)]. Ca2+ binding to sites CaX1 or CaX2 was not observed in the PDB: 1Y1A structure, but an additional Ca2+ ion was found coordinated by the central linker between the N- and C-domains of each protein chain at the dimer interface (hereafter referred to as site CaY). The binding of Ca2+ to site CaY may contribute to the formation of the distinct turn in the central linker region which causes the different domain orientation in comparison to PDB: 1XO5 [Fig. 1(E)].
In this study, we have used NMR spectroscopy and dynamic light scattering (DLS) experiments to examine whether Ca2+ can bind to sites CaX1, CaX2, or CaY, or if GSH can bind to CIB1 in solution, and whether these interactions influence the structure or dimerization of CIB1. In addition, we have investigated the potential phosphatase activity of CIB1 by testing the protein's ability to hydrolyze the substrates p-nitrophenyl phosphate (pNPP) and 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP). Our data provide evidence that Ca2+ can bind to sites CaX1 and CaX2 of CIB1 with low affinity (Kd, 10−2M) in solution, these interactions induce chemical exchange effects in NMR spectra of the protein, and likely contribute to the formation of the non-native N- and C-terminal structures in the PDB: 1XO5 crystal.
Results
Characterization of auxiliary Ca2+-binding to CIB1
High-affinity Ca2+ binding to the metal-binding loops of EF-III and EF-IV induces a large conformational change from the molten globule apo-CIB1 structure to the well-defined structure of Ca2+2-CIB1, and this transition is in slow chemical exchange on the NMR timescale as is typical for high-affinity interactions.6 To detect low-affinity auxiliary Ca2+ binding sites on the protein, uniformly 15N-labeled Ca2+2-CIB1 (with EF-III/EF-IV Ca2+-bound, see Materials and Methods) was titrated with excess Ca2+ and backbone amide chemical shift changes were monitored by NMR spectroscopy. The excess Ca2+ induced small but significant chemical shift changes in several HSQC peaks, which could be monitored throughout the titration due to fast chemical exchange behavior [Fig. 2(A,B)]. The largest residue-specific chemical shift perturbations (1H, 15N CSP >0.1) were for D18 at the C-terminal end of α-helix H0, L144 in the loop connecting EF-III to EF-IV, and for F187, K188, V190, and L191 at the extreme C-terminus of CIB1 [Fig. 2(C)]. Peaks corresponding to residues in the ancestral Ca2+-binding loops of EF-I (L38-Q57) and EF-II (S78-D90) experienced smaller than average chemical shift changes, confirming that these sites have completely lost the ability to bind Ca2+. Since Mg2+ can compete for Ca2+ binding sites on many proteins in vivo, we also examined the effect of Mg2+ on the NMR spectra of CIB1. However, high Mg2+ concentrations were shown to have a much smaller effect on the HSQC spectrum than high Ca2+ concentrations, with 1H, 15N CSP values less than 0.07 for all backbone amides even in the presence of 50 mM MgCl2, suggesting that the auxiliary metal binding sites on CIB1 bind Ca2+ more strongly than Mg2+ (data not shown).
Figure 2.
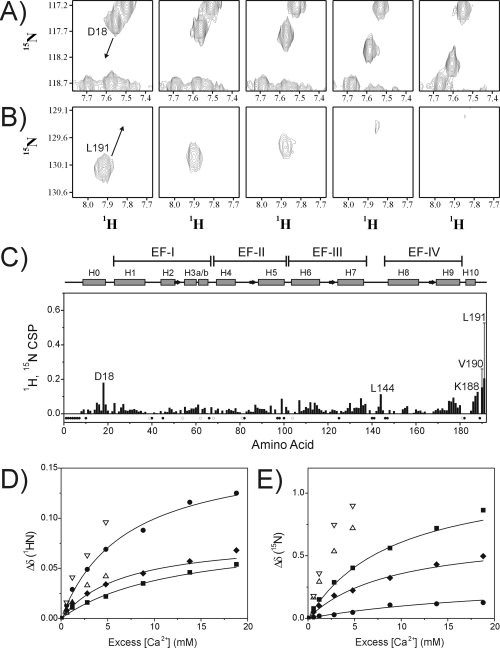
NMR characterization of auxiliary Ca2+ binding to CIB1. A: Selected region of the 1H, 15N HSQC NMR spectrum of 0.6 mM15N-CIB1 showing the Ca2+-dependent chemical shift changes for D18: spectra were recorded in the presence of (from left to right) 1.2, 2.4, 4.0, 10, and 20 mM CaCl2. B: Ca2+-dependent chemical shift changes for L191: spectra were recorded in the presence of (from left to right) 1.2, 1.8, 2.4, 4.0, and 6.0 mM CaCl2. The arrows in panels A and B indicate the direction of peak movement. C: 1H, 15N CSP analysis of the backbone amide chemical shift differences for 0.6 mM15N-CIB1 in the presence of 1.2 mM CaCl2 in comparison to 20 mM CaCl2. The solid bars for V190 and L191 represent the 1H, 15N CSP between 1.2 and 6 mM CaCl2, with the vertical lines indicating the extrapolated 1H, 15N CSP value at 20 mM CaCl2. Solid circles in the lower part of panel C indicate amino acid residues that could not be assigned in the triple resonance NMR data for Ca2+-CIB1,5 and open circles indicate proline residues. Chemical shift changes Δδ are plotted as a function of excess Ca2+ concentration (see Materials and Methods) for the backbone (D) 1HN and (E) 15N nuclei of D18 (▪), L144 (♦), K188 (•), V190 (▵), and L191 (▿). Solid lines represent the best fit as determined using the program Caligator, with all Kd values determined to be in the range of 11 ± 6 mM. The Ca2+-binding curves for V190 and L191 could not be completely defined because of resonance line broadening at high Ca2+ concentrations.
With the exception of the C-terminus of CIB1 (see below), there were no significant Ca2+ or Mg2+-dependent increases in resonance linewidth in the HSQC spectra of CIB1 (see Supporting Information Fig. S1). This indicates that high Ca2+ or Mg2+ concentrations do not induce the dimerization of CIB1, since dimerization would lengthen the rotational correlation time for CIB1 resulting in shorter T2 relaxation rates, which would broaden all of the protein's resonance signals. This lack of resonance line broadening holds true for the D18 and L144 signals which could be monitored throughout the entire titration [Fig. 2(A)]. In contrast, signals for the C-terminal F187, K188, V190, and L191 residues became considerably broadened, in some cases beyond detection (V190 and L191), suggesting that excess Ca2+ influences conformational exchange in the C-terminal extension of CIB1 [Fig. 2(B)]. Dissociation constant (Kd) values calculated from the Ca2+-dependence of the chemical shift changes for D18, L144, and K188 were all in the range of Kd = 11 ± 6 mM [Fig. 2(D,E)].
Although there was no NMR evidence for soluble CIB1 dimers, we observed a time-dependent accumulation of an insoluble protein precipitate in NMR samples of Ca2+-CIB1 or Mg2+-CIB1, which increased at higher metal concentrations and was not observed in NMR samples of apo-CIB1. This aggregation of CIB1 was accompanied by a moderate decrease in the integrated signal intensity in the HSQC spectra, for example, a ∼20% reduction in intensity over 24 h in 20 mM CaCl2. This suggests that the major molar fraction of CIB1 remains monomeric under these conditions, whereas a minor molar fraction of CIB1 forms large insoluble aggregates in a Ca2+- or Mg2+-dependent manner. This aggregation behavior was also observed in DLS experiments as described later.
CIB1 is unable to bind to GSH in solution
The binding of GSH to Ca2+-CIB1 in the PDB: 1Y1A crystal displaces amino acids 39–51 in EF-I by as much as 16 Å, with V45–S48 (α-helix H2) unfolding from the native α-helical structure of Chain A into a loop structure in the GSH-bound Chain B.9 This GSH-induced conformational change should be readily detectable using 1H, 15N HSQC NMR experiments, which are a highly sensitive measure of backbone conformation. However, the addition of GSH had no effect on the backbone amide chemical shift values or resonance linewidths even at GSH:CIB1 ratios of 100:1 [Supporting Information Fig. S2(A)]. Isothermal titration calorimetry experiments also failed to detect any interaction between CIB1 and GSH under a variety of experimental conditions, including conditions where CIB1 adopts a native structure and can bind other biological targets [Supporting Information Fig. S2(B,C)].
CIB1 is monomeric in solution
To directly examine whether Ca2+, Mg2+, or GSH can induce dimerization of CIB1 as observed in PDB: 1Y1A, DLS experiments were performed. At high protein concentrations similar to those used in NMR and X-ray studies (0.3–0.5 mM CIB1), the DLS data for apo-CIB1 revealed a monomodal size distribution with ∼99% of the protein (by mass) having an average hydrodynamic radius (Rh) near 2.4–2.5 nm (Table 1). This is slightly larger than the predicted Rh of a spherical CIB1 monomer (2.3 nm) because of the somewhat elongated structure of the protein. However, the Rh is much smaller than the theoretical size of a spherical CIB1 dimer (3.1 nm), indicating that apo-CIB1 exists as a monomeric protein in solution. Similar Rh values were observed in the presence of up to 6 mM Ca2+ or Mg2+, or in the presence of up to 30 mM GSH, suggesting that CIB1 remains monomeric under each of these conditions (Fig. 3, Table 1). However, at very high Ca2+ or Mg2+ concentrations (10–100 mM), the light scattering intensity exceeded the detector limit of the DLS instrument, indicating that there was an increase in the size of the scattering molecules. Decreasing the protein concentration below 0.1 mM combined with a very low laser power (11%) reduced the scattering intensity enough to record DLS data at high Ca2+ or Mg2+ concentrations. Under these conditions the autocorrelation function decayed much more slowly than at lower metal concentrations, and revealed a time-dependent increase in molecular size with high polydispersity and Rh values of several hundred nanometers or more [Fig. 3(B)]. Monomeric CIB1 could not be detected in these samples, presumably because the weak signal from the small monomeric CIB1 molecules was obscured by the much more intense scattering from the very large protein aggregates.
Table I.
Hydrodynamic Radii (Rh) for CIB1 in Solution Determined Using DLS
| Conditions | [CIB] (mM) | Rh (nm)a |
|---|---|---|
| 2.5 mM EDTA + 2.5 mM EGTA | 0.3 | 2.4 ± 0.1b |
| 2.5 mM EDTA + 2.5 mM EGTA | 0.5 | 2.5 ± 0.1b |
| 1.5 mM Ca2+ | 0.3 | 2.3 ± 0.2b |
| 3 mM Ca2+ | 0.3 | 2.4 ± 0.1b |
| 3 mM Ca2+ | 0.5 | 2.4 ± 0.2b |
| 6 mM Ca2+ | 0.3 | 2.3 ± 0.1b |
| 10 mM Ca2+ | 0.07–0.5 | Large aggregatesc |
| 20 mM Ca2+ | 0.07–0.5 | Large aggregatesc |
| 100 mM Ca2+ | 0.07–0.5 | Large aggregatesc |
| 1.5 mM Mg2+ | 0.3 | 2.5 ± 0.3b |
| 3 mM Mg2+ | 0.3 | 2.5 ± 0.0b |
| 6 mM Mg2+ | 0.3 | 2.3 ± 0.1b |
| 6 mM Mg2+ | 0.5 | 2.2 ± 0.1b |
| 10 mM Mg2+ | 0.07–0.5 | Large aggregatesc |
| 20 mM Mg2+ | 0.07–0.5 | Large aggregatesc |
| 100 mM Mg2+ | 0.07–0.5 | Large aggregatesc |
| 3 mM Ca2+ 0.3 mM GSH | 0.3 | 2.3 ± 0.1b |
| 3 mM Ca2+ 3 mM GSH | 0.3 | 2.3 ± 0.1b |
| 3 mM Ca2+ 30 mM GSH | 0.3 | 2.4 ± 0.1b |
All measurements represent that average and standard deviation of at least three independent DLS measurements of at least 40 acquisitions each.
Numbers represent ˜99% of the CIB1 protein (by mass) with less than 15% polydispersity.
Large and variably sized aggregates with Rh values of 100 nm to greater than 1000 nm were observed.
Figure 3.
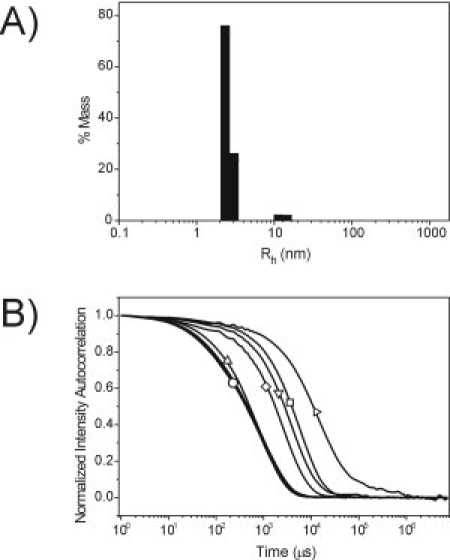
Dynamic light scattering data for CIB1. A: Size distribution histogram for CIB1 in the presence of 3 mM CaCl2. 99% of the protein (by mass) is represented by an average Rh of 2.4 nm (9.9% polydispersity), with a minor species (1% mass) at 12.4 nm. B: Normalized autocorrelation curves for CIB1 in the presence of 2.5 mM EDTA + 2.5 mM EGTA, 1.5 mM CaCl2, or 3.0 mM CaCl2 (overlapped curves collectively labeled with ○), 6 mM CaCl2 (▵), 10 mM CaCl2 (⋄), 20 mM CaCl2 (▿), 100 mM CaCl2 at time zero (□), and 100 mM CaCl2 at time 35 min (▵).The increased time for autocorrelation curve decay at greater than 6 mM CaCl2 indicates that there is an increase in size of the scattering molecules in these samples. Note the log scale on the x-axis.
CIB1 does not possess phosphatase activity against pNPP or DiFMUP
The chromogenic protein phosphatase substrate pNPP and the fluorogenic substrate DiFMUP were used to examine the potential phosphatase activity of CIB1 in solution. In comparison to the calf intestinal alkaline phosphatase positive control, we found that neither apo-CIB1, Mg2+-CIB1, nor Ca2+-CIB1 were capable of hydrolyzing the pNPP or DiFMUP substrates, even when high CIB1 concentrations were assayed (see Supporting Information Figure S3).
Discussion
The highly solvated Ca2+ coordination spheres at sites CaX1 and CaX2 in PDB: 1XO5 suggest that these Ca2+ ions bind with very low affinity to CIB1.8,11 The first Ca2+ coordination sphere at site CaX1 is entirely composed of water molecules, with second shell coordination by the D18 side chain representing the only connection that is observed in both Chains A and B of the crystal (note that the entire Chain B protein in the PDB: 1XO5 structure has weaker electron density and higher temperature factors than Chain A, presumably due to fewer intermolecular contacts in the crystal8). Ca2+ coordination at site CaX2 is better defined than at site CaX1, with first shell coordination by the backbone carboxyl of L191 (bidentate) and the F98 backbone carbonyl, and second shell coordination by the D100 side chain and the K188 backbone carbonyl in both Chains A and B.
The Ca2+-dependent chemical shift changes for D18 and the C-terminal F187-L191 residues, and the apparent Kd values of 10−2M, are consistent with the weak binding of Ca2+ at sites CaX1 and CaX2 in solution. These Kd values are well outside the normal physiological Ca2+ concentration range (10−8 to 10−5M) indicating that these two sites would never be occupied in vivo, and should not contribute to the structure or biological function of CIB1. However, these sites should be occupied under the crystallization conditions (300 mM CaCl2) utilized by Parise and coworkers, explaining the presence of Ca2+ bound to sites CaX1 and CaX2 in PDB: 1XO5. Consequently, we suggest that Ca2+ binding at site CaX1 influences the non-native extended conformation of the N-terminal extension in the PDB: 1XO5 crystal, which differs from the native α-helical structure that extends away from the C-domain under lower Ca2+ level solution conditions and in PDB: 1Y1A [Fig. 1(C)].5 The N-terminal extensions of many CIB1 homologs also adopt α-helical structures oriented away from the C-domain, the placement of which is necessary to properly orient these proteins with respect to the membrane-anchoring N-terminal myristoyl group, see for example Ref.12.
The binding of Ca2+ at site CaX2 might also stabilize the continuous α-helical conformation from D182-V190 in the PDB: 1XO5 crystal, since this C-terminal extension is only partially α-helical (D182-S185) followed by a more flexible extended conformation (S186-L191) in solution5 and in PDB: 1Y1A [Fig. 1(D)].9 Indeed the chemical exchange broadening of the C-terminal residues suggests that Ca2+ binding at site CaX2 influences the conformation of this C-terminal extension [Fig. 2(B)]. In addition, the weak binding of Ca2+ at site CaX2 may contribute to chemical exchange-broadening that leads to the absence of some HSQC signals for residues in α-helix H5 including V97 and F98. Interestingly, the binding of the αIIb cytoplasmic domain target to Ca2+-CIB1 eliminates the chemical exchange broadening of the V97-F98 and the I189-V190-L191 HSQC signals5 presumably because displacement of the C-terminal extension away from the protein dismantles the CaX2 binding site.
The NMR data also showed a distinct Ca2+-dependent chemical shift change for the L144 HSQC peak, albeit a smaller shift than the D18 or C-terminal signals [Fig. 2(C), Supporting Information Fig. S1]. This suggests that the flexible loop connecting EF-III to EF-IV of CIB1 might also bind Ca2+ with low affinity in solution. This weak interaction could lead to structural heterogeneity in this region and contribute to the poorly defined electron density for G137-T142 in PDB: 1XO5.8
The absence of significant chemical shift changes for CIB1 residues proximal to the CaY Ca2+ binding site (see Fig. 2) or the GSH binding site (Supporting Information Fig. S2), the monomeric state of CIB1 in solution (Table 1, Fig. 3) and the lack of phosphatase activity for CIB1 (Supporting Information Fig. S3), suggest that the dimerization and unique interactions with Ca2+ and GSH observed in PDB: 1Y1A are likely artifacts of crystal packing. In contrast, our solution experiments clearly show that the low-affinity auxiliary Ca2+ binding sites CaX1 and CaX2 on CIB1 can become occupied at very high Ca2+ concentrations, and likely influence parts of the protein structure in conjunction with crystal packing forces. This finding emphasizes the importance of considering the impact of low-affinity Ca2+ binding events when analyzing and interpreting crystallographic structures of acidic proteins determined in the presence of very high Ca2+ (and perhaps other cation) concentrations.
Materials and Methods
Materials
Unlabeled and uniformly 15N-labeled CIB1 (15N-CIB1) were expressed and purified as previously described.6 Reduced l-GSH and all other buffer reagents were purchased from Sigma.
NMR spectroscopy
NMR spectra were recorded at 37°C on Bruker AVANCE 500 MHz or Bruker AVANCE 700 MHz NMR spectrometers each equipped with a triple resonance inverse cryoprobe with single axis z-gradient. Ca2+ or Mg2+ titration experiments were performed by titrating samples of 0.6 mM15N-CIB1 in 20 mM HEPES, 100 mM KCl, 0.1 mM 2,2-dimethyl-2-silapentane-5-sulfonic acid, 10 mM DTT, 10% D2O, pH 7.5 with concentrated stock solution of CaCl2 or MgCl2, and 1H, 15N HSQC NMR spectra were recorded at each titration point. The pH was continuously monitored and found to remain constant throughout the Ca2+ and Mg2+ titrations. Experimental conditions for GSH titrations of CIB1 as monitored by NMR are described in the Supporting Information.
Backbone amide chemical shift perturbations (1H, 15N CSP) were calculated as [(ΔHN)2 + (ΔN/5)2]1/213 using previously determined resonance assignments for Ca2+-CIB15 and Mg2+-CIB1.14 The Kd values for the low-affinity Ca2+ binding sites were determined by plotting the 1H or 15N amide chemical shift changes as a function of “excess” Ca2+ concentration, assuming that 0.6 mM CIB1 in 1.2 mM CaCl2 contained no free Ca2+. This assumption is valid since 99% of the Ca2+ in this sample would be bound to the high-affinity sites EF-III (Kd, 10−6M) and EF-IV (Kd, 10−7M) under these conditions.6,7 Ca2+ binding curves were fitted using the program Caligator15 to obtain log(Ka) values, which were converted into Kd values using the relationship Kd = 1/Ka. All spectra were processed and analyzed using NMRPipe/NMRDraw16 and NMRView17 software.
Dynamic light scattering
DLS experiments were performed on a DynaPro Titan instrument equipped with an Ambient MicroSampler (Wyatt Technologies, Santa Barbara). Lyophilized CIB1 was first dissolved to concentrations of 0.4–0.6 mM in 20 mM HEPES, 100 mM KCl, 10 mM βME, pH 7.5. These samples were diluted to the appropriate concentration in the same buffer and supplemented with CaCl2, MgCl2, EDTA, EGTA, or GSH from 0.5 to 1M stock solutions immediately before DLS analysis to give the final sample compositions reported in Table 1. Each DLS experiment consisted of at least three individual measurements of at least 40 acquisitions with 10 readings each. Intensity autocorrelation functions were fitted using the “Regularization” algorithm in the Dynamics software version 6.7.3 (Wyatt Technologies).
Acknowledgments
Hans J. Vogel holds a Scientist award from the Alberta Heritage Foundation for Medical Research. The authors thank George Templeton and Dr. Greg Moorhead for advice on performing the phosphatase assays and for providing the pNPP reagent.
Glossary
Abbreviations:
- CIB1
calcium- and integrin-binding protein 1
- DiFMUP
6,8-difluoro-4-methylumbelliferyl phosphate
- DLS
dynamic light scattering
- GSH
glutathione
- NMR
nuclear magnetic resonance
- pNPP
p-nitrophenyl phosphate.
References
- 1.Yamniuk AP, Vogel HJ. Insights into the structure and function of calcium- and integrin-binding proteins. Calcium Bind Prot. 2006;1:150–155. [Google Scholar]
- 2.Leisner TM, Yuan W, DeNofrio JC, Liu J, Parise LV. Tickling the tails: cytoplasmic domain proteins that regulate integrin alphaIIbbeta3 activation. Curr Opin Hematol. 2007;14:255–261. doi: 10.1097/MOH.0b013e3280dce543. [DOI] [PubMed] [Google Scholar]
- 3.Naik UP, Patel PM, Parise LV. Identification of a novel calcium-binding protein that interacts with the integrin alphaIIb cytoplasmic domain. J Biol Chem. 1997;272:4651–4654. doi: 10.1074/jbc.272.8.4651. [DOI] [PubMed] [Google Scholar]
- 4.Stabler SM, Ostrowski LL, Janicki SM, Monteiro MJ. A myristoylated calcium-binding protein that preferentially interacts with the Alzheimer's disease presenilin 2 protein. J Cell Biol. 1999;145:1277–1292. doi: 10.1083/jcb.145.6.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamniuk AP, Ishida H, Vogel HJ. The interaction between calcium- and integrin-binding protein 1 and the alphaIIb integrin cytoplasmic domain involves a novel C-terminal displacement mechanism. J Biol Chem. 2006;281:26455–26464. doi: 10.1074/jbc.M603963200. [DOI] [PubMed] [Google Scholar]
- 6.Yamniuk AP, Nguyen LT, Hoang TT, Vogel HJ. Metal ion binding properties and conformational states of calcium- and integrin-binding protein. Biochemistry. 2004;43:2558–2568. doi: 10.1021/bi035432b. [DOI] [PubMed] [Google Scholar]
- 7.Yamniuk AP, Gifford JL, Linse S, Vogel HJ. Effects of metal-binding loop mutations on ligand binding to calcium- and integrin-binding protein 1 Evolution of the EF-hand? Biochemistry. 2008;47:1696–1707. doi: 10.1021/bi701494m. [DOI] [PubMed] [Google Scholar]
- 8.Gentry HR, Singer AU, Betts L, Yang C, Ferrara JD, Sondek J, Parise LV. Structural and biochemical characterization of CIB1 delineates a new family of EF-hand-containing proteins. J Biol Chem. 2005;280:8407–8415. doi: 10.1074/jbc.M411515200. [DOI] [PubMed] [Google Scholar]
- 9.Blamey CJ, Ceccarelli C, Naik UP, Bahnson BJ. The crystal structure of calcium- and integrin-binding protein 1: insights into redox regulated functions. Protein Sci. 2005;14:1214–1221. doi: 10.1110/ps.041270805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 11.Gifford JL, Walsh MP, Vogel HJ. Structures and metal-ion-binding properties of the Ca2+-binding helix-loop-helix EF-hand motifs. Biochem J. 2007;405:199–221. doi: 10.1042/BJ20070255. [DOI] [PubMed] [Google Scholar]
- 12.Stephen R, Palczewski K, Sousa MC. The crystal structure of GCAP3 suggests molecular mechanism of GCAP-linked cone dystrophies. J Mol Biol. 2006;359:266–275. doi: 10.1016/j.jmb.2006.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grzesiek S, Bax A, Clore GM, Gronenborn AM, Hu JS, Kaufman J, Palmer I, Stahl SJ, Wingfield PT. The solution structure of HIV-1 Nef reveals an unexpected fold and permits delineation of the binding surface for the SH3 domain of Hck tyrosine protein kinase. Nat Struct Biol. 1996;3:340–345. doi: 10.1038/nsb0496-340. [DOI] [PubMed] [Google Scholar]
- 14.Yamniuk AP, Silver DM, Anderson KL, Martin SR, Vogel HJ. Domain stability and metal-induced folding of calcium- and integrin-binding protein 1. Biochemistry. 2007;46:7088–7098. doi: 10.1021/bi700200z. [DOI] [PubMed] [Google Scholar]
- 15.Andre I, Linse S. Measurement of Ca2+-binding constants of proteins and presentation of the CaLigator software. Anal Biochem. 2002;305:195–205. doi: 10.1006/abio.2002.5661. [DOI] [PubMed] [Google Scholar]
- 16.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 17.Johnson BA, Blevins RA. NMRView: a computer program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]