

Abstract
Histone deacetylases (HDACs) play a key role in homeostasis of protein acetylation in histones and other proteins and in regulating fundamental cellular activities such as transcription. Imbalances in protein acetylation levels and dysfunctions in transcription are associated with a wide variety of brain disorders. Treatment with various HDAC inhibitors corrects these deficiencies and has emerged as a promising new strategy for therapeutic intervention in neurodegenerative diseases. Here, we review and discuss intriguing recent developments in the use of HDAC inhibitors to combat neurodegenerative conditions in cellular and disease models. HDAC inhibitors have neuroprotective, neurotrophic and anti-inflammatory properties, and improvements in neurological performance, learning/memory and other disease phenotypes are frequently seen in these models. We discuss the targets and mechanisms underlying these effects of HDAC inhibition and comment on the potential for some HDAC inhibitors to prove clinically effective in treating neurodegenerative disorders.
Introduction
Acetylation and deacetylation of histone proteins associated with chromatin plays a pivotal role in the epigenetic regulation of transcription and other functions in cells, including neurons [1–4]. Histone acetyltransferases (HATs) and histone deacetylases (HDACs) catalyze the acetylation and deacetylation, respectively, of histone proteins at Lys (K) residues. The interplay between HATs and HDACs alters the net balance of histone acetylation levels, thereby remodeling chromatin structure (Figure 1). In general, an increase in protein acetylation at the histone tails results in a more open and relaxed chromatin conformation, thus facilitating transcription factor interaction with specific gene promoters and activating gene expression. HDACs often function as a component of the transcriptional repressor complex to silence gene expression and induce chromatin compaction through histone protein deacetylation. Accordingly, HDAC inhibition shifts the balance towards enhanced histone acetylation, chromatin relaxation and gene expression. Imbalance between the activities of HATs and HDACs could lead to disease states. For example, mutation and loss of activity of the HAT, cyclic AMP response element binding protein (CREB)-binding protein (CBP), is causative for Rubinstein-Taybi syndrome, a developmental disorder characterized by mental retardation [5]. In addition to histones, HATs and HDACs also use a number of non-histone proteins as their substrates, notably tubulin and transcription factors such as the tumor suppressor p53, Sp1, Smad7, CREB, the pleiotropic transcription factor NF-κB, and signal transducers and activators of transcription-1 (STAT-1) [reviewed in 4, 6]. In this article, we first briefly describe the classification and isoforms of HDACs and properties of a number of isoform-nonselective and more selective HDAC inhibitors. We then review the current research using various HDAC inhibitors in cellular and animal models of neurodegenerative diseases. The beneficial effects and potential caveats of these studies are discussed. Finally, proposed future directions are addressed.
Figure 1. The effects of HDAC inhibitors on chromatin remodeling.

Levels of histone acetylation at Lys residues on histone tails are determined by interplays of acetylation and deacetylation catalyzed by histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively. Inhibition of HDACs by HDAC inhibitors results in a net increase in histone acetylation levels and a more open, relaxed chromatin conformation which favors transcriptional activation. By contrast, chromatin with a compact conformation is transcriptionally inactive.  : acetylated Lys residues of histone-tail proteins.
: acetylated Lys residues of histone-tail proteins.
HDACs and HDAC inhibitors
HDAC enzymes are evolutionarily conserved among many species. In humans, HDAC enzymes can be divided into four major classes based on their homology to yeast HDACs [reviewed in 7, 8]. Class I HDACs include HDAC1, 2, 3 and 8, which are related to the yeast enzyme Rpd3. Class II HDACs include HDAC4, 5, 6, 7, 9 and 10, and are related to the yeast protein HDA1; Class II HDACs are further divided into two subclasses – IIa (HDAC4, 5, 7 and 9) and IIb (HDAC6 and 10) – according to their structural similarities. Class I and II HDACs have been most extensively investigated for their roles in the central nervous system (CNS). Class III HDACs show dependence of nicotinamide adenine dinucleotide (NAD+) and are referred to as sirtuins owing to their homology to the yeast HDAC Sir2. This class includes SIRT1–SIRT7 [9]. HDAC11, the most recently identified isoform, is a Class IV HDAC due to its distinct structure [10]. Class I, II, and IV are zinc-dependent enzymes.
A variety of isoform-nonselective and selective HDAC inhibitors have been developed, both from natural sources and synthetically derived [reviewed in 6]. Among the relatively nonselective HDAC inhibitors, trichostatin A (TSA) and suberoylanilide hydroxamic acid (vorinostat, also known as SAHA) inhibit most zinc-dependent HDACs including HDAC6, and are permeable to the blood-brain barrier (BBB). The hydroxamate moiety of these compounds appears to bind the zinc ion at the HDAC active site to inactivate the enzyme. Sodium butyrate and 4-phenylbutyrate are fatty acid derivatives that inhibit most Class I and II HDACs. However, butyrate does not appear to inhibit HDAC6 because acetylation levels of α-tubulin, a substrate of HDAC6 [11], are unaffected by sodium butyrate treatment. Valproic acid, a fatty acid derivative with mood stabilizing and anticonvulsant properties, is another HDAC inhibitor that binds to the active site of the enzymes [12, 13]; valproic acid inhibits Class I and Class IIa HDACs, but not Class IIb [14]. Butyrates and valproic acid are also known to readily cross the BBB [reviewed in 15].
Advances have been made to design more selective HDAC inhibitors. MS-275, a synthetic benzamide derivative, preferentially inhibits HDAC1, compared with HDAC2, 3, and 9, and has little or no activity against HDAC4, 6, 7, and 8 [16]. This drug also passes the BBB easily, can be administered orally, and appears to produce no severe side effects. Apicidin, a cyclic tetrapeptide, inhibits HDAC2 and 3 in the low nanomolar range and HDAC8 in the high nanomolar range, but does not affect HDAC1 or Class II HDACs [16]. Romidepsin (FK-228), another cyclic tetrapeptide, also potently inhibits HDAC1 and 2 [6]. Tubacin is a catalytic domain-targeting small molecule inhibitor showing high selectivity for HDAC6 and deacetylation of α-tubulin, a microtubule component [17]. However, tubacin only has about 4-fold selectivity over HDAC1 and HDAC4 [18]. Suramin, a symmetric polyanionic naphthylurea, and its structural analogues inhibit human NAD+-dependent Class III SIRT1 and SIRT2 activity [19]. Nicotinamide, also known as niacinamide, is a precursor of NAD+ and a competitive Class III HDAC inhibitor that can be given orally [20]. It should be noted that the IC50 values of a given HDAC inhibitor for HDAC isoforms varied considerably among different reports. It is recommended that the Ki measurements of HDAC inhibitors be performed in future investigations to minimize the variations between studies. Table 1 lists the isoforms of these four classes of HDACs and their sensitivities to key HDAC inhibitors discussed in this review.
Table 1.
HDAC isoforms and isoform-specific and nonspecific HDAC inhibitorsa
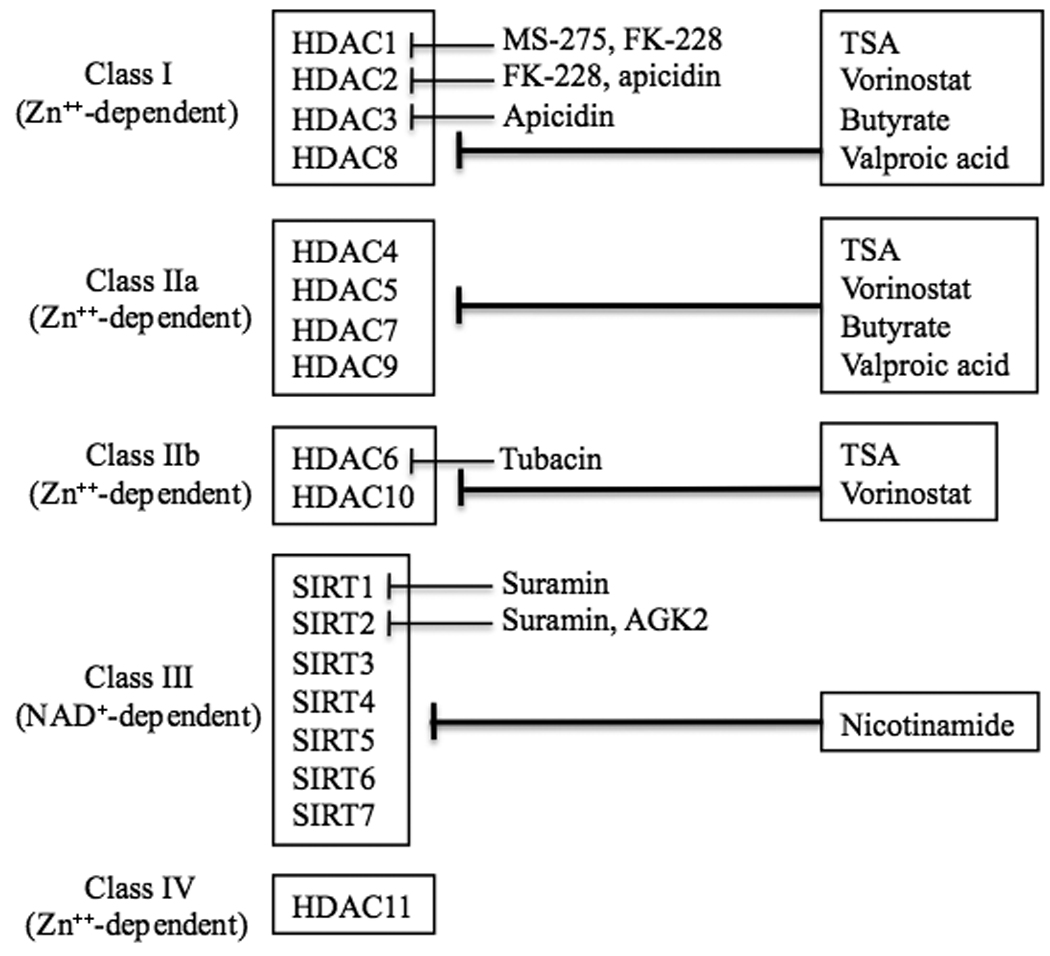 |
Detailed information and reference citations are described in the text.
Neuroprotection by HDAC inhibition in cellular models
HDAC inhibition has neuroprotective effects in both in vivo and in vitro models of brain disorders. One pioneering study noted that levels of the HATs CBP/p300 and histone protein acetylation were decreased during apoptosis induced by potassium deprivation in cultured primary cerebellar granule cells, and during signal activation of β-amyloid precursor protein (APP) in cultured primary cerebral cortical neurons from rodents [21]. Moreover, overexpression of CBP/p300 protected these neurons from apoptotic insults. In cultured cortical neurons, Ryu and colleagues showed that treatment with TSA, sodium butyrate, or vorinostat protected against glutathione depletion-induced oxidative stress, and that this neuroprotection involved acetylation and activation of the DNA binding activity of Sp1 [22]. However, it is well known that some HDAC inhibitors, such as TSA, have basal toxicity and their prolonged treatment at high doses often induces neuronal death, thus compromising their neuroprotective effects [23]. HDAC inhibitor-induced neurotoxicity could be partly due to “derepression” of genes involved in apoptosis including Bim and B-myb [24]. In an effort to side-step this issue, Langley and colleagues found that a two-hour pulse treatment with TSA sufficed to rescue cortical neurons from oxidative stress without obvious toxicity, and that this protection was associated with transcriptional activation of the cell-cycle inhibitor p21waf1/cipl [25]. Notably, p21waf1/cipl is sufficient, but not necessary, for protection by HDAC inhibition, and the action appears to be independent of its ability to inhibit cell-cycle progression. A very recent study showed that Class I/II HDAC inhibitors blocked Bax-dependent apoptosis of mouse cortical neurons by p53-dependent and -independent mechanisms [26]. This study identified Bax as a convergent target for these two distinct pathways in neuroprotection.
Glutamate-induced excitotoxicity has been implicated in the pathophysiology of many neurodegenerative and neuropsychiatric diseases, including stroke, Huntington’s disease, amyotrophic lateral sclerosis, spinal cord and traumatic brain injury, cerebellar degeneration and possibly Alzheimer’s disease, Parkinson’s disease and mood disorders [27]. Notably, Chuang and colleagues showed that the HDAC inhibitor valproic acid protected against excitotoxicity in cultured primary neurons induced by exposure to glutamate [28] or SYM 2081 [29], a blocker of excitatory amino acid transporters and an agonist of low-affinity kainate receptors. In the latter study, the protective effects of valproic acid were mimicked by treatment with structurally similar and dissimilar HDAC inhibitors and associated with decreased levels of excitotoxicity-induced accumulation of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in neuronal nuclei, an effect apparently due to the weakening of histone interactions after their hyperacetylation. Numerous studies established that overexpression and nuclear translocation of GAPDH, a glycolytic enzyme traditionally thought to be a house-keeping gene, have proapoptotic roles in cellular and animal models of neurodegenerative conditions, and the apoptotic effects were independent of the glycolytic activity of GAPDH [reviewed in 30]. Detailed nuclear mechanisms underlying GAPDH-induced neuronal apoptosis remain obscure. Of interest, Snyder, Sawa and colleagues recently reported that GAPDH is acetylated at K160 by interaction and activation of the HATs p300/CBP in the nucleus, resulting in acetylation and stabilization of the proapoptotic proteins such as p53 [31]. Although the exact role of GAPDH acetylation is undefined, their findings suggest that drugs that disrupt the nuclear GAPDH-p300/CBP complex may be developed as anti-apoptotic agents.
Leng and Chuang demonstrated that valproic acid, 4-phenylbutyrate, or TSA treatment protected against glutamate-induced, NMDA receptor-mediated excitotoxicity in cerebellar granule cells, with a concomitant transcriptional activation and induction of α-synuclein, a presynaptic protein of unknown function [28]. Knockdown with α-synuclein siRNA or antisense oligonucleotides confirmed that overexpression of endogenous α-synuclein plays a neuroprotective role which appears to involve upregulation of cytoprotective protein B-cell lymphoma 2 (Bcl-2), but downregulation of Ube2n, a proapoptotic, ubiquitin-conjugating enzyme. A subsequent study showed that α-synuclein protects cerebellar granule cells from 6-hydroxydopamine-induced death [32]. It appears that α-synuclein is neuroprotective in cytoplasm, but neurotoxic once translocated to the nucleus, where it inhibits HAT activity [28, 32, 33]. A more recent study by Leng et al. found that, under certain experimental conditions, valproic acid and other Class I and II HDAC inhibitors (e.g., sodium butyrate, 4-phenylbutyrate, and TSA) potentiated these neuroprotective effects against excitotoxicity when used in conjunction with lithium, another mood stabilizer with a robust neuroprotective profile [34]. This neuroprotective synergy was mediated, at least in part, by enhanced inhibition of glycogen synthase kinase-3 (GSK-3) to potentiate β-catenin-Lef-Tcf-dependent transcriptional activity, which is part of the Wnt signaling pathway.
Taken together, the evidence reviewed above suggests that HDAC inhibitors induce expression of multiple downstream targets that might work collectively to elicit neuroprotective effects. Lending further support to this view, HDAC inhibitors increase the expression of neurotrophins, which play prominent roles in neuronal development, synaptic plasticity, and neuronal survival. For instance, Yasuda et al. found that brain-derived neurotrophic factor (BDNF) was induced in rat cortical neurons by treatment with valproic acid, sodium butyrate or TSA, an induction involving activation of BDNF promoter IV [35]. Transfection of siRNA specific for HDAC1 also activated BDNF promoter IV, suggesting a regulatory role of this HDAC isoform in BDNF expression. Hong and colleagues found that both BDNF and glial cell line-derived neurotrophic factor (GDNF) were induced by Class I and II inhibitors in primary cultures of astrocytes from rat midbrain [36, 37]. GDNF induction by HDAC inhibition in astrocytes is associated with histone H3 hyperacetylation in the promoter of the gene encoding GDNF, and contributed to the trophic effects on midbrain dopaminergic neurons. In addition, emerging evidence supports the notion that HDAC inhibition plays a highly significant role in mediating anti-inflammatory effects by acting on microglia. Thus, HDAC inhibitors robustly protected against dopaminergic neuronal death and neuroinflammation induced by exposure to lipopolysaccharide (LPS) [36, 38]. The anti-inflammatory effects were characterized by inhibition of LPS-induced microglial activation, TNF-α release, and nitric oxide production, and were at least partially mediated by triggering apoptosis of overactivated microglia through disrupting mitochrondrial membrane potential [39]. Likewise, sodium butyrate was shown to be anti-inflammatory in LPS-treated brain-derived primary microglia [40]. The anti-inflammatory effects of HDAC inhibitors were also found in an animal model of cerebral ischemia (see below). Taken together, the in vitro studies demonstrate that HDAC inhibitors exert their neuroprotective effects through multiple mechanisms and, in addition to neurons, glia are also targets of HDAC inhibition and neuroprotection.
HDAC inhibition in animal models of neurodegenerative disorders
Stroke
Stroke, an acute neurological/neurodegenerative disease is the third leading cause of death in the USA, and most stroke cases are caused by cerebral ischemia. In a middle cerebral artery occlusion (MCAO) stroke model, reduced bulk histone acetylation was found at Lys residues in the ischemic brain of rats or mice, and these changes were restored by treatment with HDAC inhibitors, with a concomitant decrease in infarct volume [41–43]. In a rat MCAO model, Chuang and colleagues showed that post-insult treatment with valproic acid, sodium butyrate or TSA also improved behaviors [41, 42]. The long-term behavioral benefits in sodium butyrate-treated MCAO rats were associated with enhanced neurogenesis in the ischemic brain, which was abolished by blocking the BDNF-TrkB pathways [44]. In addition, administration of 4-phenylbutyrate in mice subjected to hypoxia-ischemia protected against endoplasmic reticulum (ER) stress [45], evidenced by decreased eIF2α phosphorylation and expression of the eIF2α-regulated proapoptotic protein CHOP.
It is increasingly recognized that neuroinflammation plays a causative role in neurodegeneration following ischemic injury. Kim et al. demonstrated that post-insult treatment with valproic acid or sodium butyrate suppressed permanent MCAO-induced activation of microglia and monocytes/macrophages and proinflammatory iNOS and COX-2 overexpression [42]. Treatment with HDAC inhibitors also markedly inhibited ischemia-induced p53 overexpression and superinduced heat shock protein 70 (HSP70) in the ischemic brain [41–43]. It is likely that superinduction of endogenous HSP70 by HDAC inhibition contributes to these anti-inflammatory effects. In support, one recent study noted that HSP70 overexpression inactivated NF-κB by stabilizing a complex of HSP70-IκBα-NF-κB in a mouse MCAO model [46].
The expression of cytoskeletal proteins has also been implicated in neuroprotection by HDAC inhibition under ischemic conditions. For instance, HDAC inhibition upregulated gelsolin, a protein involved in actin filament organization, which contributed to neuroprotection from ischemic brain injury [47, 48]. In addition, valproic acid was neuroprotective in an intracerebral hemorrhagic model of stroke by HDAC inhibition and transcriptional activation, and displayed anti-inflammatory actions by down-regulating proinflammatory factors, including Fas-L, IL-6, and MMP-9 [49]. The identity of the HDAC isoform(s) involved in HDAC inhibitor-mediated neuroprotection remains unclear. However, from the related cardiac field, it is noteworthy that knockdown of HDAC4 reduced infarct size following myocardial ischemia-induced reperfusion injury [50].
Huntington’s disease (HD)
HD is an inherited, autosomal-dominant fatal neurodegenerative disease characterized anatomically by a predominant loss of striatal medium-sized spiny neurons and cortical neurons, and clinically by hyperkinetic involuntary movement, cognitive impairment and memory loss, as well as psychosis and emotional deterioration. It is well known that the genetic mutation responsible for HD is an expansion of a CAG trinucleotide repeat encoding polyglutamine (polyQ) in the first exon of the huntingtin (HTT) gene. Transcriptional dysregulation plays a central role in the pathogenesis/pathophysiology of HD [51, 52]. For instance, HTT with an expanded polyQ repeat has been shown to interact with and impair neuroprotective transcription factors such as Sp1 and its co-activator TAFII130, as well as HATs such as CBP and p300/CBP associated factor CP/CAF [reviewed in 4, 53].
Treatment with vorinostat or TSA suppressed ongoing neuronal photoreceptor degeneration and reduced lethality in transgenic Drosophila expressing mutant HTT [54]. Complicating the picture, a subsequent study employing a Caenorhabditis elegans HD model demonstrated that knockdown of hda-3 suppressed neurodegeneration in response to HTT-Q150 [55]. Conversely, deletion of one copy of hda-1 enhanced polyQ neurotoxicity, which was unaffected by hda-3 loss of function. These results suggest that these two HDAC isoforms act on different targets to induce opposite effects on polyQ toxicity. Another report showed that neurodegeneration was sensitive to the zinc-dependent fly HDAC Rpd3, whereas genetic or pharmacological blockade by nicotinamide of NAD+-dependent Class III HDAC Sir2 or Sirt2 was neuroprotective in a Drosophila model [56]. Additional neuroprotection was achieved when Rpd3 and Sir2 were simultaneously inhibited. These results suggest that, in addition to Class I and II HDACs, Class III HDACs are also potential targets in HD and other diseases where polyQ is central to pathogenesis. Importantly, a very recent report by Jeong and colleagues showed that HTT was acetylated by the HAT, CBP, at K444 [57]. Enhanced K444 acetylation facilitated the trafficking of mutant HTT into autophagosomes for degradation and reduced the neurotoxicity of mutant HTT in primary neuronal cultures and a C. elegans HD model. These findings identify acetylation of HTT as a new mechanism for clearing accumulated HTT protein and suggest that increased HTT acetylation is a potential target for HDAC inhibition to elicit neuroprotective effects in HD.
The R6/2 mice express an N-terminal portion of human HTT with 150Q or more repeats and display early onset of the disease phenotype. Using R6/2 mice, Hockly and colleagues showed that vorinostat improved motor performance in a rotarod test but did not affect polyQ aggregation or downregulate the expanded HTT transgene expressed in the R6/2 mice [58]. Using the same model, Ferrante and colleagues reported that sodium butyrate treatment decreased the neurodegenerative phenotype and improved survival [59]. In R6/2 mice, the hypoacetylation associated with downregulated genes and mRNA aberrations in affected brain regions were corrected by treatment with 4-phenylbutyrate [60] or with a novel pimelic diphenylamide HDAC inhibitor, HDACi 4b [61]. Improvement of motor dysfunction, normalization of striatal atrophy, and prevention of brain weight loss were also observed using HDACi 4b, which is a synthetic benzamide derivative with relatively low toxicity. Furthermore, post-symptomatic (at 11 weeks) chemotherapy with 4-phenylbutyrate prolonged lifespan and ameliorated brain anatomical deficits, but failed to improve rotarod performance in N171-82Q HD transgenic mice [62].
It has been suggested that the pathophysiology of HD is intimately coupled to BDNF and HSP70 deficiency in affected brain regions [63–65]. Since both BDNF and HSP70 expression is regulated by Class I and II HDAC inhibitors, it is conceivable that restoring BDNF and HSP70 to their normal levels is part of the molecular mechanism underlying the beneficial effects elicited by HDAC inhibition in various HD models. In this context, it is notable that vorinostat and TSA, but not 4-phenylbutyrate or MS-275, increase vesicular transport of BDNF by inhibiting HDAC6 in particular, thereby increasing tubulin acetylation and compensating for the transport deficit in HD [66]. However, there is a flip side to this, and the role of HDAC6 in HD pathology is clearly complex – for example, HDAC6-dependent retrograde transport on microtubules is crucial for autophagic degradation of aggregated HTT [67] (hence is neuroprotective). Further, expression of HDAC6 rescues polyQ-induced neurodegeneration associated with dysfunction of the ubiquitin-proteasome system in a fly model of spinobulbar muscular atrophy [68]. The dual roles of HDAC6 in neurodegeneration and neuroprotection complicate the application of this subtype-specific inhibition in treating polyQ-induced neurodegenerative diseases.
Amyotrophic lateral sclerosis (ALS)
ALS is an adult-onset neurodegenerative disease characterized by progressive loss of motor neurons in the brain, brain stem, and spinal cord, resulting in generalized weakness, muscle atrophy, paralysis, and eventual mortality within five years of disease onset. Most ALS cases occur sporadically, with only about 10% of the patients categorized as having a familial form. Among them, approximately 20% are attributed to gain-of-function mutations in the gene encoding Cu/Zn superoxide dismutase 1 (SOD1), which is a critical antioxidant enzyme.
Mice expressing mutant Cu/Zn SOD1 exhibit ALS-like phenotypes, including the formation of intracellular aggregates of SOD1 in the brain and spinal cord, behavioral abnormalities, and premature death. Because transcriptional dysregulation may play a role in the pathophysiology of ALS, effects of HDAC inhibitors have been examined in transgenic ALS mouse models. Using SOD1/G93A transgenic mice, Ryu and colleagues injected 4-phenylbutyrate starting before or shortly after symptom onset, which resulted in extended survival and improved pathological phenotypes [69]. This study also found that 4-phenylbutyrate treatment ameliorated hypoacetylation, upregulated Bcl-2, NF-κB, p50 and phospho-IκB, and downregulated cytochrome c caspases in the spinal tissues of G93A mice. Using the same ALS transgenic mice, Ferrante and colleagues showed that combined treatment with phenylbutyrate and riluzole, the only FDA-approved drug for treating ALS, was more effective than either drug alone in increasing survival and improving pathological phenotypes [70]. Moreover, Petri and colleagues reported that co-treatment with 4-phenylbutyrate and AEOL 10150, a catalytic antioxidant, had a cumulative effect on survival time and reduced markers of oxidative damage in the lumbar spinal cord [71] of the ALS mice, suggesting the involvement of multiple molecular mechanisms in ALS pathophysiology. In support of this notion, a microarray analysis noted changes in a large number of genes involved in the progression of motor neuron injury including those involved in transcriptional and translational functions in SOD1/G93A mice, compared with controls [72].
Valproic acid treatment has variable effects on disease symptom onset and duration as well as survival in SOD1 mutant mice. For instance, pre- or post-symptomatic valproic acid treatment in drinking water increased lifespan, but pre-symptomatic treatment had no effect on the onset of motor symptoms in G93A mice [73]. Rouaux and colleagues found that, in G86R SOD1 mutant mice, valproic acid injections maintained normal levels of histone acetylation, restored the loss of CBP and significantly suppressed the death of motor neurons, although it did not prolong survival [74]. More recently, Feng et al. reported that valproic acid treatment in G93A mice had small, but significant, beneficial effects on motor dysfunction onset, motor deficits, and survival time [75]. These inconsistent results could arise from differences in valproic acid dosing, treatment method and duration, copies of the mutant SOD1 gene, and the strain of transgenic ALS mice used. In the latest study, combined treatment with valproic acid and lithium produced greater and more consistent benefits in delaying the onset of disease symptoms, prolonging lifespan, and decreasing neurological deficits than valproic acid alone [75]. Valproic acid and lithium co-treatment was also more effective than either drug alone in enhancing Ser9 phosphorylation of GSK-3β in the brain and lumbar spinal cord, suggesting enhanced inhibition of GSK-3 activity. These observations are reminiscent of the synergistic neuroprotective effects of valproic acid-lithium combinatorial treatment in cultured neurons (see above). This and other studies highlight the potential effectiveness of using combination treatments for ALS patients. A phase 2 study using sodium phenylbutyrate for ALS patients has been reported [76]. Following treatment for 20 weeks in 26 participants, phenylbutyrate was found to be safe and well tolerated in a dose-range of 9 to 21 g/day and increased blood histone acetylation levels.
Spinal muscular atrophy (SMA)
SMA is an autosomal-recessive inherited motor neuron disease caused by degeneration of α-motor neurons in the anterior horn of the spinal cord and characterized by weakness and atrophy of voluntary muscles. The genetic basis of SMA, a leading hereditary cause of infant mortality, is homozygous deletion of the SMN1 gene, encoding the full-length survival motor neuron protein, located on chromosome 5q13. Within the same chromosomal site, SMN2 is ubiquitously expressed and encodes an unstable SMN protein lacking C-terminal residues. Although SMA patients lacking SMN1 carry at least one copy of SMN2, the amount of functional SMN protein produced by SMN2 is insufficient to combat progressive motor neuron degeneration. Disease severity appears to be inversely correlated with SMN2 copy number and SMN expression, suggesting that SMN2 the potential to be a therapeutic target. A number of HDAC inhibitors increase SMN2 mRNA and protein levels in vitro, including sodium butyrate [77], 4-phenylbutyrate [78], valproic acid [79–81], M344 [81–83], vorinostat [81, 83], TSA [84] and romidepsin (FK-228) [83]. Interestingly, the SMN2 promoter is associated with HDAC1 and HDAC2, but not HDAC3-5 [85], indicating that HDAC isoforms may play a selective role in regulating SMN gene expression.
Chang and colleagues provided the first evidence that administering sodium butyrate to transgenic SMA-like mice (Smn1−/− SMN2) increased SMN1 protein expression, combated clinical symptoms and increased their lifespan [77]. Using similar SMA-like mice, Tsai and colleagues showed that oral administration of valproic acid in drinking water improved motor function, promoted motor-evoked potential and neuromuscular junction formation, as well as suppressed spinal motor neuronal degeneration and muscular atrophy [86, 87]. The beneficial effects of valproic acid were associated with elevated levels of SMN mRNA and protein and of the anti-apoptotic proteins Bcl-2 and Bcl-XL in spinal tissues. The upregulation of Bcl-XL probably contributes to valproic acid-induced neuroprotection, since transgenic overexpression of Bcl-XL also improved pathological features and lifespan in the mouse SMA model [88]. Avila and colleagues used repeated daily injections of TSA commencing after disease onset to show that HDAC inhibition activated spinal SMN2 gene expression and improved motor pathology and survival in SMA-like mice [84]. BDNF mRNA levels were also markedly increased after TSA treatment in the spinal cord and muscle of these mice.
A pilot trial using 4-phenylbutyrate increased SMN mRNA in the leukocytes of SMA patients and improved their functional performance scale [89, 90]. Similarly, valproic acid treatment increased SMN mRNA and protein in leukocytes of SMA patients [91] and improved the patients’ muscle power [92, 93]. It should be noted that the beneficial effects of valproic acid and butyrate in both preclinical and clinical studies of SMA are limited. Future SMA studies using other HDAC inhibitors, notably Class III inhibitors, seem warranted. Combinatorial treatment with an HDAC inhibitor in conjunction with another drug exhibiting different actions is also a rational approach.
Parkinson’s disease (PD)
PD is a prevalent neurodegenerative disease characterized by a relatively selective loss of dopaminergic neurons, mainly in the substantia nigra. Most cases of PD occur sporadically. Pioneering work by Beal and colleagues showed that administration of phenylbutyrate significantly attenuated the depletion of dopamine and loss of the dopamine biosynthetic enzyme tyrosine hydroxylase-positive neurons in the substantia nigra of mice treated with the classical dopaminergic toxin, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), which has been used as a model of PD [94]. Using a related dopaminergic toxin, 1-methyl-4-phenylpyridinium (MPP+), in an in vitro model of midbrain neuron–glia cocultures, Hong and colleagues found that MPP+-induced death of dopaminergic neurons was rescued by treatment with valproic acid, sodium butyrate or TSA, demonstrated by marked increases in dopamine uptake and the number of neurons staining positive for tyrosine hydroxylase [36, 37]. As discussed above, these treatments also induced GDNF in midbrain astrocytes through HDAC inhibition. Notably, the use of GDNF gene delivery has been considered as a potential therapy for neurodegenerative diseases, including PD [95]. Therefore, screening HDAC inhibitors that can more robustly upregulate endogenous GDNF in the brain seems warranted for drug development in PD and other brain disorders.
Mutations of a number of targets, including the presynaptic protein α-synuclein, have been linked to the familial forms of PD. One study reported that nuclear α-synuclein binds histones to inactivate HATs including CBP, p300 and P/CAF, causing histone hypoacetylation and apoptosis in human neuroblastoma cells, and these toxic effects are potentiated by α-synuclein mutations [33]. By contrast, cytoplasmic accumulation of α-synuclein is neuroprotective in both cultured cells and a transgenic Drosophila of PD. α-synuclein mutation linked to PD pathogenesis promotes nuclear targeting and neurotoxicity in cultured cells. Treatment with sodium butyrate or vorinostat reduced neuronal death in response to α-synuclein overexpression in vivo in vitro [33]. More recently, Outeiro and colleagues blocked SIRT2 with a specific inhibitor, AGK2, to increase α-tubulin acetylation and formation of large α-synuclein inclusions and rescue dopaminergic neurons both in vitro and in a Drosophila PD model [96]. Similarly, SIRT2 knockdown with its siRNA or HSP70 overexpression reduced α-synuclein-induced neurotoxic effects. Together, theses findings s suggest that both zinc-dependent HDACs as well as SIRT2 are potential targets for therapeutic intervention in PD.
Alzheimer’s disease (AD)
AD is the fourth most common cause of death in the USA and affects about 50% of the population over 85 years of age in industrialized countries. Clinically, it is characterized by progressive memory loss and personality changes, ultimately leading to dementia. Its neuropathological hallmarks include accumulation of extracellular β-amyloid (Aβ) and neurofibrillary tangles resulting from hyperphosphorylation of Tau protein. A number of transgenic mouse models of AD have been generated, which have dramatically advanced our understanding of the pathophysiological mechanisms of AD and of potential strategies to combat the progression of this disease.
Valproic acid, like lithium, appears to decrease Aβ production in HEK293 cells expressing Swedish APP751 and in the brains of PDAPP (APPV717F) AD transgenic mice [97]. More recently, Tsai and colleagues studied the role of chromatin remodeling in learning and memory in CK-p25 (CDK5 activator) transgenic mice in which the expression of p25, a protein implicated in various neurodegenerative conditions, can be switched on and off conditionally [98]. In this elegant study, environmental enrichment caused chromatin modification through increased histone-tail acetylation and reinstated learning and memory after significant neurodegeneration occurred in the CK-p25 mice. Moreover, treatment with sodium butyrate markedly improved associative and spatial learning. A recent study from the same group demonstrated that HDAC1 inactivation by p25 is part of the mechanism underlying the ability of p25 to elicit double-strand DNA breaks that precede neurotoxicity [99]. However, the mechanisms underlying p25-mediated HDAC1 inactivation are presently undefined and deserve further investigation. A follow-up study by Guan et al. convincingly demonstrated that mice overexpressing HDAC2, but not HDAC1, exhibit decreased dendritic spine density, synaptic number and synaptic plasticity, and show impaired memory formation [100]. Conversely, Hdac2 knockout mice show memory improvement. Further, HDAC2 regulates synaptic formation and plasticity in the mouse hippocampus and binds to promoters of a spectrum of genes involved in neuronal activity, synaptic formation and plasticity. The memory impairment in HDAC2 overexpressing mice was ameliorated by vorinostat through targeting HDAC2. These findings underscore the involvement of chromatin modification by HDAC2 in regulating synaptic plasticity and memory formation. The role of histone-tail acetylation in these processes is also supported by several independent studies showing that the HAT activity of CBP is essential for long-term potentiation (LTP) and long-term memory [101].
In Tg2576 AD mice, daily injections of 4-phenylbutyrate reversed spatial memory deficits by normalizing Tau hyperphosphorylation in the hippocampus without affecting Aβ levels [102]. 4-phenylbutyrate treatment also ameliorated the dramatic loss of histone H4 acetylation in the cortex and promoted GluR1, PSD95 and MAP2 expression, suggesting that the underlying neuroprotective mechanisms involve normalization of transcriptional dysfunction. In APP23 transgenic AD mice, daily injections with a relatively low dose of valproic acid (30 mg/kg, i.p.) robustly reduced Aβ plaque number and improved memory deficits when administered early (starting at seven months) [103]. These effects of valproic acid were attributed to inhibition of GSK-3β-mediated γ-secretase cleavage of APP. However, the involvement of HDAC inhibition by valproic acid in these neuroprotective effects was not rigorously examined under the experimental conditions employed. In another study, treatment of 3xTg-AD mice with nicotinamide, a Class III HDAC inhibitor, prevented memory impairments and decreased Tau pathology without affecting Aβ load or production [20]. Interestingly, nicotinamide treatment also induced a chronic, but low-level, increase in endogenous p25, which surprisingly was linked to improved learning and memory. Although nicotinamide could exhibit its beneficial effects through both sirtuin-dependent and independent mechanisms, these results suggest that Class III HDACs are involved in the pathology of AD, and that oral nicotinamide may prove potentially useful as a treatment for this disease.
Conclusions and future directions
Accumulating evidence supports the notion that histone hypoacetylation and transcriptional dysfunction are involved in a large number of neurodegenerative conditions in vivo and in vitro. In most cases, treatment with Class I and II HDAC inhibitors normalizes these deficiencies and protects against neurodegeneration. Multiple genes regulated by HDAC inhibition and involved in neuroprotection and neurotrophicity have been identified (Figure 2). HDAC inhibition-induced neurotrophins were found not only in neurons, but also in astrocytes, suggesting that glia are also an important target for therapeutic intervention. As noted above, Class I and II inhibitors suppress neuroinflammation by inhibiting microglia activation in cultured cells and stroke models. This raises the possibility that HDAC inhibitors may be developed as anti-inflammatory drugs in the treatment of brain disorders. This is an important area for future research in view that presently there is no clinical drug that robustly mitigates neuroinflammation in the brain. In addition to transcriptional regulation, non-transcriptional events, such as improvement of microtubule stability via enhanced acetylation, have been implicated in the neuroprotective effects elicited by HDAC inhibition in a number of disease models. HAT-mediated acetylation of HTT is an important mechanism for removing accumulated toxic protein in the HD model. It remains to be investigated whether this is a general mechanism for selective trafficking of proteins for lysosomal degradation and clearance, and whether this is a regulatory target of HDAC inhibitors.
Figure 2. The actions of HDAC inhibitors in neurodegenerative conditions.

A large number of neurodegenerative conditions in vivo and in vitro involve functional imbalance in HATs and HDACs, resulting in histone hypoacetylation and transcriptional dysfunction. Treatment with Class I, II and, more recently, III HDAC inhibitors restores these deficiencies. These effects appear to be mediated by multiple HDAC-regulated gene products including BDNF, GDNF, HSP70, α-synuclein, Bcl-2, Bcl-XL, p21, and gelsolin, among others. Non-transcriptional effects of HDAC inhibitors, such as hyperacetylation and stabilization of microtubule proteins, have also been shown in many neurodegenerative disease models. Studies suggest that HDAC inhibitors have neuroprotective, neurotrophic, and anti-inflammatory effects, as well as improve neurological performance and learning/memory in various neurodegenerative conditions. Bcl-2: B-cell lymphoma 2; BDNF: brain-derived neurotrophic factor; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; GDNF: glial cell line-derived neurotrophic factor; HAT: histone acetyltransferase; HDAC: histone deacetylase; HSP70: heat shock protein 70.
Among the six animal models of neurodegenerative diseases discussed above, treatments with broad-spectrum, pan-inhibitors of Class I and II HDACs demonstrate various degrees of effectiveness in combating neuronal cell death and improving neurological outcome (Table 2). Emerging evidence also suggests that blocking Class III HDACs is neuroprotective in HD, PD, and AD models. In some cases (e.g., HD, PD, SMA, and possibly stroke models), the main HDAC isoforms involved in the pathology and potential treatment of these diseases have been identified. Paradoxically, in rare cases, the activity rather than the inhibition of certain HDAC isoforms has neuroprotective effects, and these include HDAC1 in the AD model, and SIRT1 in AD and ALS models [104]. Identification of specific genes, whose expressions are regulated by these HDACs and crucial for the pathological conditions, may shed light on the mechanisms underlying these paradoxical effects.
Table 2.
Effects of treatment with HDAC inhibitors in models of neurodegenerative diseases.
| Disease models | Histone hypo- acetylation/ transcriptional dysfunction |
Microtubule dysfunction |
HDAC inhibitors examined | Beneficial effects after treatment | References |
|---|---|---|---|---|---|
| Stroke | Yes | Yes | Valproic acid, vorinostat, sodium butyrate, TSA, 4-phenylbutyrate | Restored histone hypoacetylation and transcriptional dysfunction; enhanced neurogenesis; decreased infarct volume,neuroinflammation and neurological deficits | 41–45, 47–49 |
| Huntington’s disease (HD) | Yes | Yes | Vorinostat, sodium butyrate, 4-phenylbutyrate, TSA, HDACi 4b, nicotinamide | Restored histone hypoacetylation and transcriptional dysfunction; normalized striatal atrophy and degeneration; increased BDNF vesicular transport; improved motor performance and survival | 54–56, 58–62, 66 |
| Amyotrophic lateral sclerosis (ALS) | Yes | ? | 4-phenylbutyrate, valproic acid, 4-phenylbutyrate+antioxidant, valproic acid+lithium, 4-phenylbutyrate+riluzole | Restored histone hypoacetylation and CBP loss; suppressed motor neuronal death; improved motor function and survival | 69–71,73–76 |
| Spinal muscular atrophy (SMA) | Yes | ? | Sodium butyrate, 4-phenylbutyrate, valproic acid, vorinostat, TSA, romidepsin (FK-228) | Increased SMN2 expression; induced Bcl-2, Bcl-XL and BDNF; suppressed spinal motor neuronal degeneration and muscle atrophy; prolonged life span | 77–84, 86–93 |
| Parkinson’s disease (PD) | Yes | Yes | Valproic acid, sodium butyrate, TSA, vorinostat, AGK2 | Increased GDNF and BDNF expression; reduced neuroinflammation and dopaminergic neuronal death; increased acetylation of α-tubulin | 33, 36, 37, 94, 96 |
| Alzheimer’s disease (AD) | Yes | Yes | Valproic acid, sodium butyrate, 4-phenylbutyrate, nicotinamide, vorinostat | Restored histone hypoacetylation; increased synaptic plasticity; decreased Aβ production and Tau hyperphosphorylation; reinstated learning and memory; reversed spatial memory deficits | 20, 97–100, 102, 103 |
A few isoform-specific HDAC inhibitors are now available. Additional HDAC inhibitors, including both isoform-specific and nonspecific drugs, need to be developed. The BBB permeability and cytotoxic profiles of existing HDAC inhibitors require more critical evaluations. The clinical toxicity and side effects of HDAC inhibitors in cancer treatment have been well-documented [105]. These adverse effects of HDAC inhibitors were often detected after relatively short-term therapy in cancer patients, and might be exacerbated after longer-term treatment required in patients with neurodegenerative diseases. It seems worthwhile to explore whether the reduced neurotoxicity and beneficial effects of “pulse treatment” with HDAC inhibitors found in an in vitro experimental setting [25] could be replicated in the in vivo preclinical models or clinical studies. HDAC isoform-specific drugs would be anticipated to have fewer adverse effects and be better tolerated. However, it is presently unknown whether isoform-specific, or pan-HDAC inhibitors would be more efficacious in treating any given neurodegenerative disease. There are indications that combined treatment of an HDAC inhibitor with another neuroprotective drug has additive or even synergistic effects, suggesting that combinatorial approaches should be pursued. Despite the fact that research in this area is still in its infancy, HDAC inhibition is a promising new avenue for therapeutic intervention in CNS neurodegenerative disorders. The development of potent and effective HDAC inhibitors with excellent BBB permeability, less cytotoxicity and fewer undesirable side effects remains a major challenge, and many crucial issues will need to be addressed before these promising drugs can be effectively used in the clinic.
Acknowledgments
Conflict of Interest and Acknowledgements
This work was supported by the Intramural Research Program of the NIMH, NIH. The authors have no conflicts of interest, financial or otherwise, to disclose. We thank Jau-Shyong Hong and his colleagues at the NIEHS, NIH for their collaboration. The critical comments of Li-Kai Tsai and Zhifei Wang (Molecular Neurobiology Section, NIMH, NIH) and the editorial assistance of Peter Leeds (NIMH) and Ioline Henter (NIMH) are appreciated. We also wish to apologize to investigators whose related papers are not cited in this review due to space constraints.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mai A, et al. Histone deacetylation in epigenetics: an attractive target for anticancer therapy. Med Res Rev. 2005;25:261–309. doi: 10.1002/med.20024. [DOI] [PubMed] [Google Scholar]
- 2.Langley B, et al. Remodeling chromatin and stress resistance in the central nervous system: histone deacetylase inhibitors as novel and broadly effective neuroprotective agents. Curr Drug Targets CNS Neurol Disord. 2005;4:41–50. doi: 10.2174/1568007053005091. [DOI] [PubMed] [Google Scholar]
- 3.Abel T, Zukin RS. Epigenetic targets of HDAC inhibition in neurodegenerative and psychiatric disorders. Curr Opin Pharmacol. 2008;8:57–64. doi: 10.1016/j.coph.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov. 2008;7:854–868. doi: 10.1038/nrd2681. [DOI] [PubMed] [Google Scholar]
- 5.Bartsch O, et al. DNA sequencing of CREBBP demonstrates mutations in 56% of patients with Rubinstein-Taybi syndrome (RSTS) and in another patient with incomplete RSTS. Hum Genet. 2005;117:485–493. doi: 10.1007/s00439-005-1331-y. [DOI] [PubMed] [Google Scholar]
- 6.Itoh Y, et al. Isoform-selective histone deacetylase inhibitors. Curr Pharm Des. 2008;14:529–544. doi: 10.2174/138161208783885335. [DOI] [PubMed] [Google Scholar]
- 7.Marks PA, Dokmanovic M. Histone deacetylase inhibitors: discovery and development as anticancer agents. Expert Opin Investig Drugs. 2005;14:1497–1511. doi: 10.1517/13543784.14.12.1497. [DOI] [PubMed] [Google Scholar]
- 8.Carey N, La Thangue NB. Histone deacetylase inhibitors: gathering pace. Curr Opin Pharmacol. 2006;6:369–375. doi: 10.1016/j.coph.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 9.Michishita E, et al. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell. 2005;16:4623–4635. doi: 10.1091/mbc.E05-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Voelter-Mahlknecht S, et al. Chromosomal organization and localization of the novel class IV human histone deacetylase 11 gene. Int J Mol Med. 2005;16:589–598. [PubMed] [Google Scholar]
- 11.Hubbert C, et al. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 12.Phiel CJ, et al. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001;276:36734–36741. doi: 10.1074/jbc.M101287200. [DOI] [PubMed] [Google Scholar]
- 13.Göttlicher M, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001;20:6969–6978. doi: 10.1093/emboj/20.24.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gurvich N, et al. Histone deacetylase is a target of valproic acid-mediated cellular differentiation. Cancer Res. 2004;64:1079–1086. doi: 10.1158/0008-5472.can-03-0799. [DOI] [PubMed] [Google Scholar]
- 15.Butler R, Bates GP. Histone deacetylase inhibitors as therapeutics for polyglutamine disorders. Nat Rev Neurosci. 2006;7:784–796. doi: 10.1038/nrn1989. [DOI] [PubMed] [Google Scholar]
- 16.Khan N, et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J. 2008;409:581–589. doi: 10.1042/BJ20070779. [DOI] [PubMed] [Google Scholar]
- 17.Haggarty SJ, et al. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci U S A. 2003;100:4389–4394. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mai A, et al. Class II (IIa)-selective histone deacetylase inhibitors. 1. Synthesis and biological evaluation of novel (aryloxopropenyl)pyrrolyl hydroxyamides. J Med Chem. 2005;48:3344–3353. doi: 10.1021/jm049002a. [DOI] [PubMed] [Google Scholar]
- 19.Trapp J, et al. Structure-activity studies on suramin analogues as inhibitors of NAD+-dependent histone deacetylases (sirtuins) ChemMedChem. 2007;2:1419–1431. doi: 10.1002/cmdc.200700003. [DOI] [PubMed] [Google Scholar]
- 20.Green KN, et al. Nicotinamide restores cognition in Alzheimer's disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231-phosphotau. J Neurosci. 2008;28:11500–11510. doi: 10.1523/JNEUROSCI.3203-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rouaux C, et al. Critical loss of CBP/p300 histone acetylase activity by caspase-6 during neurodegeneration. EMBO J. 2003;22:6537–6549. doi: 10.1093/emboj/cdg615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ryu H, et al. Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1-dependent pathway. Proc Natl Acad Sci U S A. 2003;100:4281–4286. doi: 10.1073/pnas.0737363100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jeong MR, et al. Valproic acid, a mood stabilizer and anticonvulsant, protects rat cerebral cortical neurons from spontaneous cell death: a role of histone deacetylase inhibition. FEBS Lett. 2003;542:74–78. doi: 10.1016/s0014-5793(03)00350-8. [DOI] [PubMed] [Google Scholar]
- 24.Biswas SC, et al. Bim is a direct target of a neuronal E2F-dependent apoptotic pathway. J Neurosci. 2005;25:8349–8358. doi: 10.1523/JNEUROSCI.1570-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Langley B, et al. Pulse inhibition of histone deacetylases induces complete resistance to oxidative death in cortical neurons without toxicity and reveals a role for cytoplasmic p21(waf1/cip1) in cell cycle-independent neuroprotection. J Neurosci. 2008;28:163–176. doi: 10.1523/JNEUROSCI.3200-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uo T, et al. Histone deacetylase inhibitors prevent p53-dependent and p53-independent Bax-mediated neuronal apoptosis through two distinct mechanisms. J Neurosci. 2009;29:2824–2832. doi: 10.1523/JNEUROSCI.6186-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chuang D-M. Lithium protection from glutamate excitotoxicity: therapeutic implications. Clinical Neuroscience Research. 2004;4:243–252. [Google Scholar]
- 28.Leng Y, Chuang DM. Endogenous alpha-synuclein is induced by valproic acid through histone deacetylase inhibition and participates in neuroprotection against glutamate-induced excitotoxicity. J Neurosci. 2006;26:7502–7512. doi: 10.1523/JNEUROSCI.0096-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanai H, et al. Valproic acid inhibits histone deacetylase activity and suppresses excitotoxicity-induced GAPDH nuclear accumulation and apoptotic death in neurons. Pharmacogenomics J. 2004;4:336–344. doi: 10.1038/sj.tpj.6500269. [DOI] [PubMed] [Google Scholar]
- 30.Chuang DM, et al. Glyceraldehyde-3-phosphate dehydrogenase, apoptosis, and neurodegenerative diseases. Annu Rev Pharmacol Toxicol. 2005;45:269–290. doi: 10.1146/annurev.pharmtox.45.120403.095902. [DOI] [PubMed] [Google Scholar]
- 31.Sen N, et al. Nitric oxide-induced nuclear GAPDH activates p300/CBP and mediates apoptosis. Nat Cell Biol. 2008;10:866–873. doi: 10.1038/ncb1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Monti B, et al. Alpha-synuclein protects cerebellar granule neurons against 6-hydroxydopamine-induced death. J Neurochem. 2007;103:518–530. doi: 10.1111/j.1471-4159.2007.04778.x. [DOI] [PubMed] [Google Scholar]
- 33.Kontopoulos E, et al. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet. 2006;15:3012–3023. doi: 10.1093/hmg/ddl243. [DOI] [PubMed] [Google Scholar]
- 34.Leng Y, et al. Synergistic neuroprotective effects of lithium and valproic acid or other histone deacetylase inhibitors in neurons: roles of glycogen synthase kinase-3 inhibition. J Neurosci. 2008;28:2576–2588. doi: 10.1523/JNEUROSCI.5467-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yasuda S, et al. The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol Psychiatry. 2009;14:51–59. doi: 10.1038/sj.mp.4002099. [DOI] [PubMed] [Google Scholar]
- 36.Chen PS, et al. Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurotrophic factors from astrocytes. Mol Psychiatry. 2006;11:1116–1125. doi: 10.1038/sj.mp.4001893. [DOI] [PubMed] [Google Scholar]
- 37.Wu X, et al. Histone deacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int J Neuropsychopharmacol. 2008;11:1123–1134. doi: 10.1017/S1461145708009024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peng GS, et al. Valproate pretreatment protects dopaminergic neurons from LPS-induced neurotoxicity in rat primary midbrain cultures: role of microglia. Brain Res Mol Brain Res. 2005;134:162–169. doi: 10.1016/j.molbrainres.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 39.Chen PS, et al. Valproic acid and other histone deacetylase inhibitors induce microglial apoptosis and attenuate lipopolysaccharide-induced dopaminergic neurotoxicity. Neuroscience. 2007;149:203–212. doi: 10.1016/j.neuroscience.2007.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huuskonen J, et al. Regulation of microglial inflammatory response by sodium butyrate and short-chain fatty acids. Br J Pharmacol. 2004;141:874–880. doi: 10.1038/sj.bjp.0705682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ren M, et al. Valproic acid reduces brain damage induced by transient focal cerebral ischemia in rats: potential roles of histone deacetylase inhibition and heat shock protein induction. J Neurochem. 2004;89:1358–1367. doi: 10.1111/j.1471-4159.2004.02406.x. [DOI] [PubMed] [Google Scholar]
- 42.Kim HJ, et al. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. J Pharmacol Exp Ther. 2007;321:892–901. doi: 10.1124/jpet.107.120188. [DOI] [PubMed] [Google Scholar]
- 43.Faraco G, et al. Pharmacological inhibition of histone deacetylases by suberoylanilide hydroxamic acid specifically alters gene expression and reduces ischemic injury in the mouse brain. Mol Pharmacol. 2006;70:1876–1884. doi: 10.1124/mol.106.027912. [DOI] [PubMed] [Google Scholar]
- 44.Kim H, et al. The histone deacetylase inhibitor, sodium butyrate, stimulates cell proliferation in the ischemic brain: roles of BDNF-TrkB signaling. J Neurochem. 2009 doi: 10.1111/j.1471-4159.2009.06212.x. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qi X, et al. Sodium 4-phenylbutyrate protects against cerebral ischemic injury. Mol Pharmacol. 2004;66:899–908. doi: 10.1124/mol.104.001339. [DOI] [PubMed] [Google Scholar]
- 46.Zheng Z, et al. Anti-inflammatory effects of the 70 kDa heat shock protein in experimental stroke. J Cereb Blood Flow Metab. 2008;28:53–63. doi: 10.1038/sj.jcbfm.9600502. [DOI] [PubMed] [Google Scholar]
- 47.Meisel A, et al. Inhibition of histone deacetylation protects wild-type but not gelsolin-deficient neurons from oxygen/glucose deprivation. J Neurochem. 2006;98:1019–1031. doi: 10.1111/j.1471-4159.2006.04016.x. [DOI] [PubMed] [Google Scholar]
- 48.Yildirim F, et al. Inhibition of histone deacetylation protects wildtype but not gelsolin-deficient mice from ischemic brain injury. Exp Neurol. 2008;210:531–542. doi: 10.1016/j.expneurol.2007.11.031. [DOI] [PubMed] [Google Scholar]
- 49.Sinn DI, et al. Valproic acid-mediated neuroprotection in intracerebral hemorrhage via histone deacetylase inhibition and transcriptional activation. Neurobiol Dis. 2007;26:464–472. doi: 10.1016/j.nbd.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 50.Granger A, et al. Histone deacetylase inhibition reduces myocardial ischemia-reperfusion injury in mice. FASEB J. 2008;22:3549–3560. doi: 10.1096/fj.08-108548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hodges A, et al. Regional and cellular gene expression changes in human Huntington's disease brain. Hum Mol Genet. 2006;15:965–977. doi: 10.1093/hmg/ddl013. [DOI] [PubMed] [Google Scholar]
- 52.Sugars KL, Rubinsztein DC. Transcriptional abnormalities in Huntington disease. Trends Genet. 2003;19:233–238. doi: 10.1016/S0168-9525(03)00074-X. [DOI] [PubMed] [Google Scholar]
- 53.Hahnen E, et al. Histone deacetylase inhibitors: possible implications for neurodegenerative disorders. Expert Opin Investig Drugs. 2008;17:169–184. doi: 10.1517/13543784.17.2.169. [DOI] [PubMed] [Google Scholar]
- 54.Steffan JS, et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- 55.Bates EA, et al. Differential contributions of Caenorhabditis elegans histone deacetylases to huntingtin polyglutamine toxicity. J Neurosci. 2006;26:2830–2838. doi: 10.1523/JNEUROSCI.3344-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pallos J, et al. Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington's disease. Hum Mol Genet. 2008;17:3767–3775. doi: 10.1093/hmg/ddn273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jeong H, et al. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell. 2009;137:60–72. doi: 10.1016/j.cell.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hockly E, et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc Natl Acad Sci U S A. 2003;100:2041–2046. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ferrante RJ, et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J Neurosci. 2003;23:9418–9427. doi: 10.1523/JNEUROSCI.23-28-09418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sadri-Vakili G, et al. Histones associated with downregulated genes are hypo-acetylated in Huntington's disease models. Hum Mol Genet. 2007;16:1293–1306. doi: 10.1093/hmg/ddm078. [DOI] [PubMed] [Google Scholar]
- 61.Thomas EA, et al. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington's disease transgenic mice. Proc Natl Acad Sci U S A. 2008;105:15564–15569. doi: 10.1073/pnas.0804249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gardian G, et al. Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington's disease. J Biol Chem. 2005;280:556–563. doi: 10.1074/jbc.M410210200. [DOI] [PubMed] [Google Scholar]
- 63.Zuccato C, et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- 64.Hay DG, et al. Progressive decrease in chaperone protein levels in a mouse model of Huntington's disease and induction of stress proteins as a therapeutic approach. Hum Mol Genet. 2004;13:1389–1405. doi: 10.1093/hmg/ddh144. [DOI] [PubMed] [Google Scholar]
- 65.Tagawa K, et al. The induction levels of heat shock protein 70 differentiate the vulnerabilities to mutant huntingtin among neuronal subtypes. J Neurosci. 2007;27:868–880. doi: 10.1523/JNEUROSCI.4522-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dompierre JP, et al. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation. J Neurosci. 2007;27:3571–3583. doi: 10.1523/JNEUROSCI.0037-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Iwata A, et al. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280:40282–40292. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- 68.Pandey UB, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 69.Ryu H, et al. Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J Neurochem. 2005;93:1087–1098. doi: 10.1111/j.1471-4159.2005.03077.x. [DOI] [PubMed] [Google Scholar]
- 70.Del Signore SJ, et al. Combined riluzole and sodium phenylbutyrate therapy in transgenic amyotrophic lateral sclerosis mice. Amyotroph Lateral Scler. 2009;10:85–94. doi: 10.1080/17482960802226148. [DOI] [PubMed] [Google Scholar]
- 71.Petri S, et al. Additive neuroprotective effects of a histone deacetylase inhibitor and a catalytic antioxidant in a transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2006;22:40–49. doi: 10.1016/j.nbd.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 72.Ferraiuolo L, et al. Microarray analysis of the cellular pathways involved in the adaptation to and progression of motor neuron injury in the SOD1 G93A mouse model of familial ALS. J Neurosci. 2007;27:9201–9219. doi: 10.1523/JNEUROSCI.1470-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sugai F, et al. Benefit of valproic acid in suppressing disease progression of ALS model mice. Eur J Neurosci. 2004;20:3179–3183. doi: 10.1111/j.1460-9568.2004.03765.x. [DOI] [PubMed] [Google Scholar]
- 74.Rouaux C, et al. Sodium valproate exerts neuroprotective effects in vivo through CREB-binding protein-dependent mechanisms but does not improve survival in an amyotrophic lateral sclerosis mouse model. J Neurosci. 2007;27:5535–5545. doi: 10.1523/JNEUROSCI.1139-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Feng HL, et al. Combined lithium and valproate treatment delays disease onset, reduces neurological deficits and prolongs survival in an amyotrophic lateral sclerosis mouse model. Neuroscience. 2008;155:567–572. doi: 10.1016/j.neuroscience.2008.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cudkowicz ME, et al. Phase 2 study of sodium phenylbutyrate in ALS. Amyotroph Lateral Scler. 2009;10:99–106. doi: 10.1080/17482960802320487. [DOI] [PubMed] [Google Scholar]
- 77.Chang JG, et al. Treatment of spinal muscular atrophy by sodium butyrate. Proc Natl Acad Sci U S A. 2001;98:9808–9813. doi: 10.1073/pnas.171105098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Andreassi C, et al. Phenylbutyrate increases SMN expression in vitro: relevance for treatment of spinal muscular atrophy. Eur J Hum Genet. 2004;12:59–65. doi: 10.1038/sj.ejhg.5201102. [DOI] [PubMed] [Google Scholar]
- 79.Sumner CJ, et al. Valproic acid increases SMN levels in spinal muscular atrophy patient cells. Ann Neurol. 2003;54:647–654. doi: 10.1002/ana.10743. [DOI] [PubMed] [Google Scholar]
- 80.Brichta L, et al. Valproic acid increases the SMN2 protein level: a well-known drug as a potential therapy for spinal muscular atrophy. Hum Mol Genet. 2003;12:2481–2489. doi: 10.1093/hmg/ddg256. [DOI] [PubMed] [Google Scholar]
- 81.Hahnen E, et al. In vitro and ex vivo evaluation of second-generation histone deacetylase inhibitors for the treatment of spinal muscular atrophy. J Neurochem. 2006;98:193–202. doi: 10.1111/j.1471-4159.2006.03868.x. [DOI] [PubMed] [Google Scholar]
- 82.Riessland M, et al. The benzamide M344, a novel histone deacetylase inhibitor, significantly increases SMN2 RNA/protein levels in spinal muscular atrophy cells. Hum Genet. 2006;120:101–110. doi: 10.1007/s00439-006-0186-1. [DOI] [PubMed] [Google Scholar]
- 83.Hauke J, et al. Survival motor neuron gene 2 silencing by DNA methylation correlates with spinal muscular atrophy disease severity and can be bypassed by histone deacetylase inhibition. Hum Mol Genet. 2009;18:304–317. doi: 10.1093/hmg/ddn357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Avila AM, et al. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J Clin Invest. 2007;117:659–671. doi: 10.1172/JCI29562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kernochan LE, et al. The role of histone acetylation in SMN gene expression. Hum Mol Genet. 2005;14:1171–1182. doi: 10.1093/hmg/ddi130. [DOI] [PubMed] [Google Scholar]
- 86.Tsai LK, et al. Establishing a standardized therapeutic testing protocol for spinal muscular atrophy. Neurobiol Dis. 2006;24:286–295. doi: 10.1016/j.nbd.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 87.Tsai LK, et al. Multiple therapeutic effects of valproic acid in spinal muscular atrophy model mice. J Mol Med. 2008;86:1243–1254. doi: 10.1007/s00109-008-0388-1. [DOI] [PubMed] [Google Scholar]
- 88.Tsai LK, et al. Restoring Bcl-x(L) levels benefits a mouse model of spinal muscular atrophy. Neurobiol Dis. 2008;31:361–367. doi: 10.1016/j.nbd.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 89.Mercuri E, et al. Pilot trial of phenylbutyrate in spinal muscular atrophy. Neuromuscul Disord. 2004;14:130–135. doi: 10.1016/j.nmd.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 90.Brahe C, et al. Phenylbutyrate increases SMN gene expression in spinal muscular atrophy patients. Eur J Hum Genet. 2005;13:256–259. doi: 10.1038/sj.ejhg.5201320. [DOI] [PubMed] [Google Scholar]
- 91.Brichta L, et al. In vivo activation of SMN in spinal muscular atrophy carriers and patients treated with valproate. Ann Neurol. 2006;59:970–975. doi: 10.1002/ana.20836. [DOI] [PubMed] [Google Scholar]
- 92.Weihl CC, et al. Valproate may improve strength and function in patients with type III/IV spinal muscle atrophy. Neurology. 2006;67:500–501. doi: 10.1212/01.wnl.0000231139.26253.d0. [DOI] [PubMed] [Google Scholar]
- 93.Tsai LK, et al. Valproic acid treatment in six patients with spinal muscular atrophy. Eur J Neurol. 2007;14:e8–e9. doi: 10.1111/j.1468-1331.2007.01992.x. [DOI] [PubMed] [Google Scholar]
- 94.Gardian G, et al. Neuroprotective effects of phenylbutyrate against MPTP neurotoxicity. Neuromolecular Med. 2004;5:235–241. doi: 10.1385/NMM:5:3:235. [DOI] [PubMed] [Google Scholar]
- 95.Airaksinen MS, Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci. 2002;3:383–394. doi: 10.1038/nrn812. [DOI] [PubMed] [Google Scholar]
- 96.Outeiro TF, et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson's disease. Science. 2007;317:516–519. doi: 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
- 97.Su Y, et al. Lithium, a common drug for bipolar disorder treatment, regulates amyloid-beta precursor protein processing. Biochemistry. 2004;43:6899–6908. doi: 10.1021/bi035627j. [DOI] [PubMed] [Google Scholar]
- 98.Fischer A, et al. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447:178–182. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- 99.Kim D, et al. Deregulation of HDAC1 by p25/Cdk5 in neurotoxicity. Neuron. 2008;60:803–817. doi: 10.1016/j.neuron.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Guan JS, et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Barrett RM, Wood MA. Beyond transcription factors: the role of chromatin modifying enzymes in regulating transcription required for memory. Learn Mem. 2008;15:460–467. doi: 10.1101/lm.917508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ricobaraza A, et al. Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer's disease mouse model. Neuropsychopharmacology. 2009;34:1721–1732. doi: 10.1038/npp.2008.229. [DOI] [PubMed] [Google Scholar]
- 103.Qing H, et al. Valproic acid inhibits Abeta production, neuritic plaque formation, and behavioral deficits in Alzheimer's disease mouse models. J Exp Med. 2008;205:2781–2789. doi: 10.1084/jem.20081588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kim D, et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer's disease and amyotrophic lateral sclerosis. EMBO J. 2007;26:3169–3179. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bruserud O, et al. Histone deacetylase inhibitors in cancer treatment: a review of the clinical toxicity and the modulation of gene expression in cancer cell. Curr Pharm Biotechnol. 2007;8:388–400. doi: 10.2174/138920107783018417. [DOI] [PubMed] [Google Scholar]