

Abstract
Amelogenin is the predominant protein found during enamel development and has been shown to be essential to proper enamel formation. Leucine-rich amelogenin peptide (LRAP) is a naturally occurring splice variant that preserves the charged N- and C-termini of full length amelogenin, regions thought to be crucial in interacting with hydroxaypatite. Particularly, the highly charged C-terminal hexapeptide (KREEVD) is thought to be the region most intimately interacting with hydroxyapatite (HAP). The structure of this charged region was investigated, along with the proximity to the surface and the mobility of two of the residues. The structure was found to be consistent with a random coil or more extended structure, as has been seen for more internalized residues in the C-terminus. The backbone K54(13C′), V58(13C′), and V58(15N) were all found to be close to the surface of HAP, ∼ 6.0 Å from the nearest 31P atom, suggesting a strong interaction and emphasizing the importance of these residues in interacting with HAP. However, both ends of the hexapeptide at residues K54 and V58 experience significant mobility under hydrated conditions, implying that another portion of the protein helps to stabilize the strong LRAP-HAP interaction. Interestingly, the backbone of the C-terminal third of the protein is consistently 6.0 Å from the HAP surface, providing a model in this region of the protein lying flat on the surface with no three-dimensional folding. The combination of these features, that is, a random coil structure, a significant mobility, and a lack of three-dimensional folding in this region of the protein, may have an important functional role, possibly allowing maximum crystal inhibition at low protein concentrations.
Introduction
Biomineralization proteins have been found to be critical in the formation of hard tissues such as bone, teeth, and mollusk shells and result in materials with highly controlled properties such as crystal phase, crystal morphology, and high strength and hardness.1 Enamel is a unique example of biomineralization, where the protein matrix that controls the formation of the highly elongated hydroxyapatite crystals is enzymatically removed prior to final maturation, resulting in crystals which are >99.9% mineral.2 Amelogenin is a highly hydrophobic biomineralization protein that controls the formation of enamel.3 It has been demonstrated to be critical in the proper development of enamel using ribosomal,4 genetic,5 and antisense oligodeoxynucleotide6 knockout mice. It is observed that single amino acid mutations result in amelogenisis imperfectas,7 a group of naturally occurring diseases which result in enamel defects, yielding a wide range of anomalies from discoloration to the wearing away of the enamel. The dramatic loss of function resulting from this small change in primary structure suggests a role for structure in the amelogenin-hydroxyapatite (HAP) interaction and also suggests a very specific interaction between amelogenin and HAP.
A great deal of experimental effort has been invested in understanding the role that amelogenin plays in enamel development, and roles in nucleation,8 growth inhibition,9,10 crystal aggregation,10 and crystal spacing have been proposed based on these studies.11 The specific interaction with HAP, the predominant phase of developing enamel,12 is not well defined; however, the charged residues, which are thought to interact with charged surfaces such as HAP, are localized primarily in the C-terminus (Table 1) of the protein. The resulting charge distribution is thought to contribute to an important functional quaternary structure,3 called nanospheres, which place the charged C-terminus on the nanosphere surface,13 enabling protein–HAP interactions. Removal of the C-terminus reduces the affinity for HAP14 and also reduces HAP inhibition,10 strongly implicating this region as a HAP binding site. Unfortunately, the protein–crystal interface is largely uncharacterized at a molecular level, including little secondary or tertiary structural information for amelogenin bound to hydroxyapatite. The secondary and tertiary structures, as well as the protein orientation are thought to govern the formation of biominerals and their determination is essential to revealing these fundamental formation mechanisms.1
TABLE 1. The Primary Structures of Human, Bovine, Mouse, and Pig Amelogenin23,24 and LRAP Show the Conservation of the Primary Structure of the N and C-Terminusa.
| 1 | 5 | 10 | 15 | 20 | 25 | 30 | 35 | 40 | 45 | 50 | 55 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LRAP-K54V58 | MPLPPHPGSPGYINLpSYEVLTPLKWYQSMIRQP PLSPILPELPLEAWPATDKT REE REE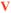 D D |
|||||||||||
| LRAP-V58 | MPLPPHPGSPGYINL SYEVLTPLKWYQSMIRQP PLSPILPELPLEAWPATDKTKREE D D |
|||||||||||
| Bovine | MPLPPHPGHPGYINFpSYEVLTPLKWYQSMIRHP#PLPPMLPDLPLEAWPATDKTKREEVD | |||||||||||
| Human | MPLPPHPGHPGYINFpSYEVLTPLKWYQSMIRPP…PLPPILPDLHLEAWPATDKTKREEVD | |||||||||||
| Mouse | MPLPPHPGSPGYINLpSYEVLTPLKWYQSMIRQP…PLSPILPELPLEAWPATDKTKREEVD | |||||||||||
| Pig | MPLPPHPGHPGYINFpSYEVLTPLKWYQNMIRHP……. SLLPDLPLEAWPATDKTKREEVD | |||||||||||
The central portion of amelogenin is indicated by # (YPSYGYEPMGGWLHHQIIPVLSQQHPPSHTLQPHHHLPVVPAQQPVAPQQPMMPVPGHHSMTPTQHHQPNIPPSAQQPFQQPFQPQAIPPQSHQPMQPQSPLHPMQPLAPQPPLPPLFSMQ). Charged residues are shown in bold type. Blue residues indicate backbone 13C′ labels, and red residues indicate backbone 15N labels incorporated for SSNMR studies.
Leucine-rich amelogenin peptide (LRAP) is a naturally occurring splice variant of amelogenin with only 59 residues.15 LRAP consists of the charged N- and C-termini of the full protein while lacking the hydrophobic polyproline rich region (Table 1). These two regions contain 13 of the 14 charged residues from the full protein, are highly conserved across many species, and are thought to be responsible for protein–protein and protein–surface interactions, respectively.11 These characteristics suggest a specific function for LRAP in the biomineralization process, though experimental data provide conflicting evidence. LRAP was not seen to recover the phenotype for an amelogenin null mouse;16 however, mouse molar explant studies showed a role for LRAP in enamel growth.17 For the present study, LRAP serves as a model for amelogenin. LRAP shares many of the features of the parent protein. LRAP has the ability to form nanospheres,18 is seen to bind similar amounts of calcium per protein as amelogenin,19 and both proteins inhibit HAP crystal growth.10 More importantly for our studies, the smaller size allows easy incorporation of selectively labeled isotopes which are essential for site specific investigation of structure, dynamic, and orientation studies of an immobilized biomineralization protein.20–22
Solid-state NMR (SSNMR) has emerged as a technique uniquely suited to investigating biomineralization proteins bound to surfaces and allows the investigation of these systems under biologically relevant, hydrated conditions.20,21,25,26 Previously, SSNMR was used to aid in identifying the structure and orientation of a large region of the C-terminus of LRAP (L42 through T53) bound to the surface of HAP.21,22 These studies demonstrated that this portion of the C-terminus was interacting closely with the surface and was also unstructured. Investigation of the dynamics of surface-immobilized LRAP revealed that the backbone was very mobile, suggesting a less rigid interaction than might be expected if this region were the only region critical to binding.22 Rather than collocalized charged residues, LRAP has 9 charged residues dispersed throughout the C-terminal 15 residues of the protein, which may play a role in LRAP's observed mobility. However there is a region with higher charge density in the far C-terminus, -KREEVD, which has not previously been investigated. Studies with the biomineralization protein statherin revealed a motionally restricted backbone in its highly charged, acidic binding region (DpSpSEEK), but dynamic mobility outside of that region.26,27 To address the question of the importance of a high concentration of consecutive charged residues to the binding interface, this work extends the previous studies to the most charge concentrated, and possibly the most important crystal interaction region of the C-terminus, the terminal hexapeptide, KREEVD. The structure was investigated from K54 to V58, the dynamics of the backbone at K54(13C′) and V58(13C′) and the distance of the K54(13C′), V58(13C′), and V58(15N) atoms from the nearest 31P atoms in the surface of HAP.
Experimental Methods
Materials
Labeled amino acids were purchased from Cambridge Isotopes and used as received. Solvents were used without further purification. FMOC-protected labeled amino acids were prepared according to standard procedures28,29 and used without further purification.
Protein Preparation, Purification, and Characterization
Proteins were prepared using standard FMOC chemistry by United Biochemical Research (Seattle, WA) and Alpha Diagnostics Intl. Inc. (San Antonio, TX). Proteins were purified using prep. scale reverse phase HPLC, buffer A, 0.1% trifluoroacetic acid in water and buffer B, 0.1% trifluoroacetic acid in acetonitrile. LRAP eluted at 54% B. Proteins were analyzed for molecular weight and purity using electrospray MS.
Sample Preparation of the Free Protein
To prepare a solid state sample of the free protein, 30 mg of LRAP was dissolved into 1 mL of phosphate buffer, consisting of a solution of 0.15 M NaCl and saturated with respect to hydroxyapatite (PB), and diluted to 20 mL with water. This was frozen in liquid nitrogen, lyophilized, and the entirety of the resulting powder packed into the NMR rotor.
Sample Preparation of the Protein Bound to HAP
The protein sample was bound to HAP as described previously.22 Briefly, 0.33 mg/mL LRAP at pH 7.4 was bound to 100 mg of 94 m2/g HAP for 1 h. The amount of protein bound was determined by measuring the change in concentration before and after binding and for each wash using UV absorbance measurements (λ = 275 nm). Typically, 10–14 mg of protein was bound to 100 mg of HAP. The sample was packed into an NMR rotor as a wet paste for the hydrated, surface bound sample.
For the lyophilized, surface-bound sample, the packed hydrated sample was frozen with liquid nitrogen in the rotor and lyophilized. The NMR experiments were always done on the hydrated sample first, followed by the lyophilized sample.
NMR Experiments
NMR experiments were performed on a 3-channel Chemagnetics Infinity console operating at 300 MHz proton frequency. A 3-channel, VT Chemagnetics probe was used, employing a 6 μs 90° pulse for 1H and a 0.5–1 msec contact time for cross polarization experiments. Temperatures in the rotor were calibrated using 207Pb(NO3)2.30 Chemical shifts were referenced to glycine, 177.0 ppm.31
Dynamics
For Herzfeld-Berger (HB) analysis, a spinning speed of 1.5 kHz was used and 28 800 scans were taken for each sample to allow direct intensity comparisons. The hydrated, surface bound samples were also run with additional signal averaging to allow more accurate fitting of the CSA parameters. For 13C T1ρ analysis, 10 lock times were used from 0.05 to 4.55 msec with a radio frequency field of 42 kHz and were fit to the following equation: Mt = M0e−(t/T1ρ). Spectra and 13C T1ρ measurements were taken at both −80 °C (frozen) and at RT (20 °C).
REDOR
For Rotational Echo DOuble Resonance (REDOR) experiments,32,33 XY8 phase cycling was used on both observe and dephasing channels. For all three types of REDOR experiments, 13C{31P}, 15N{31P}, and 13C{15N}, 180° pulses of 13.0–15.0 μs were used for both the observe and dephasing nuclei and samples were spun at 4 kHz. Two Pulse Phase Modulated (TPPM) decoupling34 with a 65 kHz decoupling field was used throughout. Data for the hydrated samples were collected at −80 °C, while lyophilized samples were run at −30 °C. Typically, 4096 scans were taken for shorter dephasing periods and 8192–16 384 scans were taken for longer dephasing times with a 3 s pulse delay. Data was collected at every 8 or every 16 rotor periods, out to 104 rotor periods for 13C{31P} and 13C{15N} REDOR. In all cases, the final dephasing curve represents the average of at least 3–5 repetitions. For the 15N{31P} experiments only, data was collected every 24 rotor periods out to 104 rotor periods with a 1 s pulse delay. A total of 8192–16 384 scans were collected for early dephasing times and 65 538 scans for longer dephasing times. REDOR dephasing curves were fit by simulations generated using SIMPSON.35 The contribution of the natural abundance background (58 backbone carbonyls and 7 side chain carbonyls, or 40%) was removed from the 13C{31P} and 13C{15N} dephasing curves.
Results and Discussion
Two samples were prepared to investigate the structure, dynamics and orientation of the C-terminal hexapeptide of LRAP (Table 1). LRAP-K54V58 was used to determine the structure of LRAP in the charged binding region (from K54 through V58), the dynamics at K54(13C′), and the distances from K54(13C′) and V58(15N) to the surface. LRAP-V58 was used to measure the dynamics at V58(13C′) and to measure the distance from V58(13C′) to the surface.
Secondary Structure
The secondary structure was determined using REDOR by measuring the distance from the backbone 13C′ and backbone 15N, incorporated at the i and i + 4 residues, respectively. This represents the distance between backbone atoms of residues involved in a hydrogen bond for a protein in an α-helical structure. The REDOR dephasing curve for an α-helix is easily distinguished from random coil and β-sheet structures (Figure 1). The structure across the charged hexapeptide region of LRAP (K54 to V58) for both the protein bound to HAP and hydrated, as well as bound to HAP and lyophilized, was found to be in a random coil or extended structure based on the measured distance of 5.5 Å, as shown in Figure 1. The distance for LRAP-K54V58 off the surface of HAP was also found to be 5.5 Å (data not shown). The relatively constant chemical shifts for these residues as a function of preparation (Table 2) are also indicative of little change in structure whether bound to the surface or lyophilized from solution, and their linewidths, 4–6 ppm, are consistent with the interpretation of a disordered structure. These results are consistent with previous structural studies of LRAP from L42 through T53 which also show a largely unstructured protein, with no observable change in structure when LRAP is bound to the surface of HAP. Often, it is proposed that a specific structural motif is necessary for biomineralization proteins to interact with their biologically relevant surface, or for proteins to function in general. However, there is a growing body of evidence that suggests that unstructured proteins are also functional and in fact critical to function.36–40 Unstructured regions in proteins have been proposed to serve many functions such as allowing flexibility in a catalytic site,36 allowing binding site recognition,37–39 providing the ability to recognize a wide range of proteins,37 or allowing access to a site that would be prevented by the steric hindrance of a globular protein.40 In this case, the lack of structure may allow a necessary protein–protein interaction, expose a binding site for protein–crystal interaction, or allow structural flexibility to optimize the interaction of the dispersed carbonyls with the oppositely charged Ca2+ in the surface. It is also possible that a relaxed structure in this region works in concert with a more structured region elsewhere in the protein to serve a functional purpose, as has been observed elsewhere.39 While the functional purpose for disorder in LRAP's backbone remains unclear, it is clear that a specific traditional structure is not necessary for orienting the C-terminus of LRAP next to the HAP surface.
Figure 1.
13C–15N REDOR of LRAP-K54V58 on HAP, hydrated (open blue circles) and lyophilized (closed red diamonds). The dephasing curves for a β-sheet structure (black line), α-helical structure (long dashed green line), and random coil structure (dashed black line) are also shown. The structure of both preparations have measured distances of 5.5 Å (solid red line) and are most consistent with a random coil or more extended structure.
TABLE 2. Chemical Shift, Relaxation Parameters, and Distance from the Surface for the C-Terminus of LRAPa.
| LRAP-K54(C′) | LRAP-V58(C′) | LRAP-V58(N) | |
|---|---|---|---|
| T1ρ (msec) | |||
| hydrated | 6.2 ± 1 | 10.7 ± 1 | |
| frozen | >20 ± 1 | >20 ± 1 | |
| Ω (ppm) | |||
| hydrated | 88.4 ± 20 | 105 ± 20 | |
| frozen | 139.2 ± 20 | 142.0 ± 20 | |
| η | |||
| hydrated | 0.7 ± 0.1 | 0.7 ± 0.1 | |
| frozen | 0.7 ± 0.1 | 0.7 ± 0.1 | |
| σiso (ppm) | |||
| hydrated | 174.9 ± 0.5 | 173.3 ± 0.5 | |
| frozen | 175.1 ± 0.5 | 173.6 ± 0.5 | |
| lyophilized | 174.1 ± 0.5 | 173.2 ± 0.5 | |
| linewidth (ppm) | |||
| hydrated | 5.0 ± 0.3 | 4.0 ± 0.3 | |
| frozen | 5.4 ± 0.3 | 4.6 ± 0.3 | |
| lyophilized | 5.9 ± 0.3 | 6.3 ± 0.3 | |
| distance to HAP (Å) | |||
| hydrated | 6.5 ± 0.5 | 5.8 ± 0.5 | |
| lyophilized | 6.0 ± 0.5 | 5.4 ± 0.5 | 5.8 ± 0.5 |
σiso = (1/3)(σ11 + σ22 + σ33), η = (σ22 − σ11)/(σ33 − σiso), where |σ11 − σiso| < |σ33 − σiso|, and Ω = σ11 − σ33.
Dynamics
The mobility of a protein can reveal important functional roles. For the charged hexapeptide region with a potential role in protein–crystal interaction, less motion is expected for regions or residues which are more strongly interacting with HAP.26,27 By investigating the isotopically labeled, backbone carbonyl carbons, relative comparisons of the strength of the interaction of the protein with the surface can be made. The mobility of the backbone carbonyls K54(13C′) and V58(13C′) was investigated with three techniques: 13C T1ρ relaxation, cross polarization efficiency, and Herzfeld–Berger (HB) analysis. These techniques can provide insight into motion on the time scale of 10−3 to 10−5 sec for carbonyl carbons, specifically by observing a faster relaxation time, a loss in cross polarization efficiency, and/or an averaged spinning sideband pattern. Previous studies have found that these motions in the protein backbone can be on the NMR time scale,22,26 making it an effective diagnostic tool for protein–surface interaction strength.
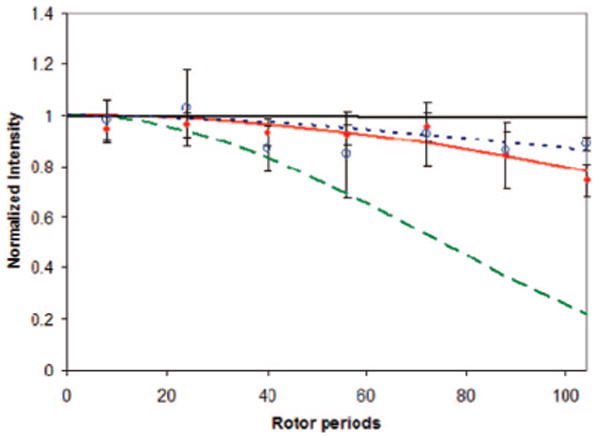
The data are summarized in Table 2 and Figure 2. Both of the samples were measured under hydrated, room temperature conditions, as well as hydrated and frozen conditions, which provides a “rigid” comparison. On the basis of all three types of experiments, both K54(13C′) and V58(13C′) are undergoing significant motional averaging. The significant loss in signal-to-noise ratio, which represents a loss in cross polarization efficiency, is observed when comparing the spectra of the frozen samples (Figure 2A,C) to the spectra of the hydrated samples (Figure 2B,D). This is a strong indication of dynamics on the NMR time scale. The 13C T1ρ relaxation time is faster in the presence of water (Table 2) and provides a more quantitative measure of motion, placing the motion on the kilohertz time scale. The spinning sideband pattern is also motionally averaged for both K54(13C′) and V58(13C′) under hydrated conditions (Figure 2B,D). The frozen spectra (Figure 2A,C) demonstrate the expected spinning sideband pattern of a rigid backbone, while a single isotropic line would represent a completely motionally averaged spectrum (i.e., a liquid like spectrum). A particularly useful parameter obtained from analyzing the spinning sideband pattern is Ω, or span, a parameter that defines the width of the spinning sideband pattern (Table 2). As the spectrum becomes motionally averaged, the value will become smaller, until in the fast motion limit (i.e., a liquid, or isotropically rotating nucleus) the spinning sideband pattern would be completely absent. The partial averaging seen here, where the span is reduced by 30–50% and the isotropic line begins to dominate the spectrum, suggests motion with a significant amplitude on the kilohertz time scale.26,27
Figure 2.
Spectra from top to bottom: (A) LRAP-K54(13C′) hydrated frozen, (B) LRAP-K54(13C′) hydrated room temperature, (C) LRAP-V58(13C′) hydrated frozen, and (D) LRAP-V58(13C′) hydrated room temperature. The arrow indicates the backbone carbonyl isotropic resonance, and the asterisks indicate the spinning side bands. The loss in CP intensity (evidenced by the decrease in signal-to-noise) and the change in the spinning sideband pattern for LRAP-K54(13C′) and LRAP-V58(13C′) in the presence of water all indicate mobility on the NMR time scale (10−3–10−5 sec).
The experimental observations from all three of these techniques provide evidence of mobility in the backbone at these residues of the protein. The 13C T1ρ and cross polarization efficiency for K54(13C′) and V58(13C′) are consistent with previous dynamics results at residues L42(13C′) and A49(13C′). Qualitatively, however, the spinning sideband pattern is not as motionally averaged at K54(13C′) and V58(13C′), based on the larger Ω values (88 and 105 ppm) observed for these residues than for residues further from the charged hexapeptide (∼80 ppm).22 Because the 13C T1ρ is most sensitive to the frequency of motion and the spinning sideband pattern is sensitive to both the frequency and amplitude of motion,26,27 this observation suggests that while the frequency of motion is similar, the amplitude of the motion becomes somewhat more restricted toward the charge dense portion of the protein, as might be expected for a region with multiple acidic residues.
Orientation
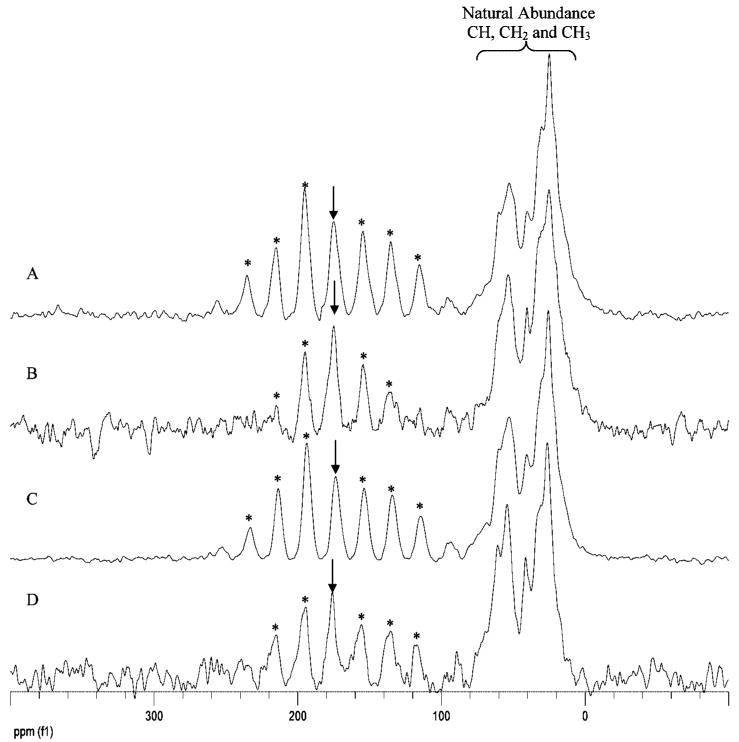
To provide direct insight into the importance of the hexapeptide region in interacting with HAP, the distances of the backbone K54(13C′), V58(13C′), and V58(15N) were measured. Measurement of the distance of a particular residue from the surface provides a quantitative measure of the orientation of the protein on the surface, yielding insight into a possible role in protein–HAP interaction. Clearly, if a particular residue is oriented away from the surface, it is not important in protein-surface interactions. If, however, it is oriented next to the HAP surface, this suggests that it, or nearby residues, are important in the protein–surface recognition mechanism. Using the SSNMR REDOR technique, a distance of up to 10 Å can be measured from a 13C isotopic label to a 31P atom in the surface of HAP. Table 2 and Figure 3 show that the distances from the three backbone positions investigated to the nearest 31P in the surface of HAP were nearly identical, at 6.0 ± 0.5 Å under lyophilized conditions, placing the charged hexapeptide region close enough to HAP to allow this region to influence crystal growth. (The reported distances consider only 1 nearby 31P. Previous studies have shown that at longer distances (>5.5–6 Å), multiple 31P groups do not change the value of the shortest measured distance.22) This is consistent with previous work which also found the backbone at A49(13Cα) to be 6.0 Å from the surface under lyophilized conditions. The slightly shorter distance seen for LRAP-V58(13C′) may indicate a stronger interaction with the surface or may be the result of the backbone conformation naturally aligning this residue closer to the surface. Under hydrated conditions, the distances for both LRAP-K54(13C′) and LRAP-V58(13C′) are slightly longer. The effect of hydration on the surface interaction has been observed before22 and suggests that water plays a role in the interaction mechanism of LRAP with HAP. The lack of change in secondary structure with hydration level suggests that the primary role of water is in stabilizing the protein–surface interaction, and not stabilizing the structure.
Figure 3.
Top: 13C–31P dephasing curves for LRAP-K54(13C′) and K58(13C′) to 31P in the surface of HAP. Hydrated, the backbone 13C′ are 6.5 Å (closed blue circles) and 5.8 Å (open black circles) from the nearest 31P nucleus in the surface of HAP, respectively. Lyophilized, these distances are shortened to 6.0 Å (closed red triangles) and 5.4 Å (open green triangles). A fit with multiple 31P nuclei at 6.0 and 8.0 Å away is shown for comparison (dashed red line) and fits the lyophilized K58(13C′)-31P data well. There is no observed dephasing for either residue lyophilized from PBS (LRAP-K54(13C′) shown, dark blue triangles), as expected. Bottom: 15N–31P dephasing curves for LRAP-V58(15N) to the surface of HAP. Lyophilized (red diamonds), the backbone 15N is 5.8 Å (dashed red line) from the nearest surface 31P. There is no observed dephasing for LRAP-V58(15N) lyophilized from PBS (blue diamonds).
Collectively and considering previous data,22 these data provide a three-dimensional picture of the protein placing the backbone of the protein from residues 49 to 58 equidistant from and very closely associated with the surface of HAP. The side chain of residue A46 was also close to the surface of HAP,21 and the lack of a major structural change from residues 42 to 58 suggests that this close association continues through residue 42. This model would place the entire C-terminal third of the protein flat on the surface, as summarized in Figure 4. The lack of vertical folding in this region of the protein would maximize the interaction of the dispersed charged residues in the C-terminal 1/3 of the protein with the surface.
Figure 4.
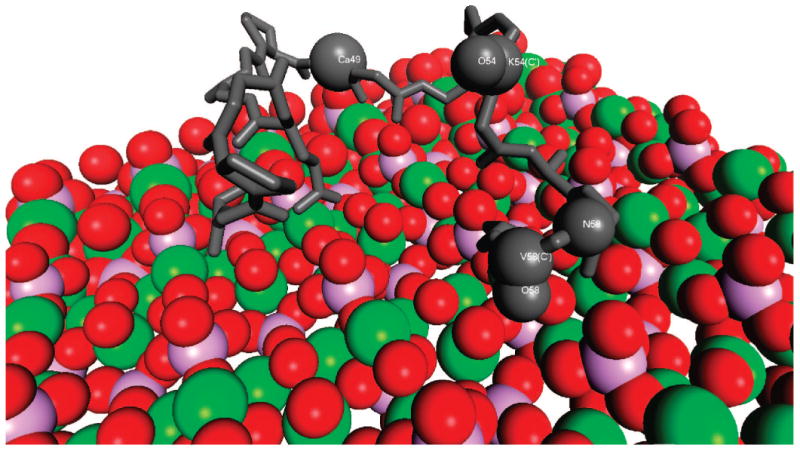
The structural and orientation data combine to show that the C-terminal third of LRAP (shown in gray) is lying flat on the surface of HAP (red = oxygen, green = calcium, purple = phosphate) in a random coil structure. One of the possible structural configurations is shown. The lack of vertical folding in this region, along with the proteins mobility and the disordered structure, suggest a specific functional role, possibly allowing the protein to effectively cover more sites to enhance crystal inhibition, or to promote protein–protein interactions. The role of the remainder of the protein (not shown) is currently under investigation to reveal contributions to protein–crystal interactions.
Interaction Mechanism
Interestingly, there was not a significant change in structure, orientation, or dynamics in the charged hexapeptide region, as compared to the entire C-terminal third of LRAP.21,22 Despite the slightly restricted amplitude of motion of LRAP in the charged hexapeptide, the protein is still experiencing significant mobility, even in this charge dense region. It was postulated that this region with three acidic residues (-KREEVD) might have less motional freedom due to stronger interactions with HAP. This was based on the observation that statherin, a protein with similar binding affinity as amelogenin,22 was found to be very immobile on the surface of HAP in its binding region,27 which contains five consecutive acidic residues (DpSpSEEK-). The smaller number of acidic residues, combined with the presence of two basic residues, may contribute to the increased motional freedom seen for amelogenin.
The C-terminus of amelogenin is generally thought to be one, if not the, controlling factor for the binding of LRAP and amelogenin to HAP,11,14 and the present data confirm that it is lying on the surface of HAP, implying an important role in crystal regulation. This is consistent with previous experimental evidence that has shown that the C-terminus is placed on the surface of the nanosphere for protein–crystal interactions,41,42 and removing it results in decreased protein–crystal interactions,10 providing strong evidence for the importance of this region. However, the mobility in the C-terminus is suprising when compared to previous work showing restricted mobility in the binding site of the biomineralization protein, statherin.26,27 This may point to another region contributing to the binding of LRAP to HAP. In agreement with this interpretation, a study of the C-terminal decapeptide alone showed reduced adsorption and inhibition capacity when compared to the full amelogenin protein,43 suggesting that the charge dense C-terminal region was not the only contributor to binding. Another interpretation is that the six acidic residues dispersed throughout the C-terminus are able to provide similar affinity for HAP as seen for statherin's five consecutive acidic residues,22 while still allowing mobility in the binding region which may serve an important function. Further studies are underway to distinguish between these interpretations.
The present data clearly show that the C-terminus is important in binding the protein to HAP based on the close proximity of the entire C-terminus to the surface of HAP. However, the data are also consistent with the interpretation that the entire C-terminal third of the protein is critical to binding, not just the charged hexapeptide region. This data also implies that motional and structural flexibility serve important functional roles, perhaps to block more crystal growth sites with a lower protein coverage or to allow structural freedom for the protein–protein interactions which are important for amelogenin's function.
Conclusions
The charged hexapeptide binding region in the C-terminus of LRAP (KREEVD) was investigated to understand the role of charge concentration in stabilizing the protein on HAP. Suprisingly, the charge concentrated hexapeptide was not found to have a significantly different secondary structure, protein–crystal interaction or mobility when compared to the charge disperse C-terminal third of LRAP. These results suggest that all of the charges dispersed throughout the C-terminal third of the protein are equally responsible for the interaction of LRAP with the HAP surface. The proximity of the C-terminal third of the backbone to the surface suggests that this portion of LRAP is interacting closely with the surface and is laying flat with no three-dimensional folding. We postulate that this elongated orientation, along with the observed random coil structure and backbone dynamics combine to allow maximal interaction with the surface, important in maximizing crystal inhibition. The mobility throughout the region also suggests that another region of the protein may contribute to the strong LRAP-HAP interaction.
Acknowledgments
This work was supported by NIH-NIDCR Grant DE-015347 and was performed at Pacific Northwest National Laboratory, operated by Battelle for the US-DOE.
References and Notes
- 1.Lowenstam HA, Weiner S. On Biomineralization. Oxford University Press; New York: 1989. [Google Scholar]
- 2.Diekwisch TGH, Berman BJ, Gentner S, Slavkin HC. Cell Tissue Res. 1995;279:149. doi: 10.1007/BF00300701. [DOI] [PubMed] [Google Scholar]
- 3.Margolis HC, Beniash E, Fowler CE. J Dent Res. 2006;85:775. doi: 10.1177/154405910608500902. [DOI] [PubMed] [Google Scholar]
- 4.Lyngstadaas SP, Risnes S, Sproat BS, Thrane PS, Prydz HP. EMBO J. 1995;14:5224. doi: 10.1002/j.1460-2075.1995.tb00207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gibson CW, Yuan ZA, Hall B, Longenecker G, Chen EH, Thyagarajan T, Sreenath T, Wright JT, Decker S, Piddington R, Harrison G, Kulkarni AB. J Biol Chem. 2001;276:31871. doi: 10.1074/jbc.M104624200. [DOI] [PubMed] [Google Scholar]
- 6.Diekwisch T, David S, Jr, Santos V, Slavkin HC. Development. 1993;117:471. doi: 10.1242/dev.117.2.471. [DOI] [PubMed] [Google Scholar]
- 7.Ravassipour DB, Hart PS, Hart TC, Ritter AV, Yamauchi M, Gibson C, Wright JT. J Dent Res. 2000;79:1476. doi: 10.1177/00220345000790070801. [DOI] [PubMed] [Google Scholar]
- 8.Tarasevich BJ, Howard CJ, Larson JL, Snead ML, Simmer JP, Paine M, Shaw WJ. J Cryst Growth. 2007;304:407. doi: 10.1016/j.jcrysgro.2007.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doi Y, Eanes ED, shimokawa H, Termine JD. J Dent Res. 1984;63:98. doi: 10.1177/00220345840630021801. [DOI] [PubMed] [Google Scholar]
- 10.Moradian-Oldak J, Tan J, Fincham AG. Biopolymers. 1998;46:225. doi: 10.1002/(SICI)1097-0282(19981005)46:4<225::AID-BIP4>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 11.Moradian-Oldak J. Matrix Biol. 2001;20:293. doi: 10.1016/s0945-053x(01)00154-8. [DOI] [PubMed] [Google Scholar]
- 12.Robinson C, Briggs HD, Atkinson PJ, Weatherell JA. J Dent Res. 1979;58:871. doi: 10.1177/00220345790580024101. [DOI] [PubMed] [Google Scholar]
- 13.Moradian-Oldak J, Jiminez I, Maltby D, Fincham AG. Biopolymers. 2001;58:606. doi: 10.1002/1097-0282(200106)58:7<606::AID-BIP1034>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 14.Moradian-Oldak J, Bouropoulos N, Wang L, Gharakhanian N. Matrix Biol. 2002;21:197. doi: 10.1016/s0945-053x(01)00190-1. [DOI] [PubMed] [Google Scholar]
- 15.Gibson CW, Golub E, Ding W, Shimokawa H, Young M, Termine J, Rosenbloom J. Biochem Biophys Res Commun. 1991;174:1306. doi: 10.1016/0006-291x(91)91564-s. [DOI] [PubMed] [Google Scholar]
- 16.Chen E, Yuan ZA, Wright JT, Hong SP, Li Y, Collier PM, Hall B, D’Angelo M, Decker S, Piddington R, Abrams WR, Kulkarni AB, Gibson CW. Calcif Tissue Int. 2003;73:487. doi: 10.1007/s00223-002-0036-7. [DOI] [PubMed] [Google Scholar]
- 17.Ravindranath RMH, Devarajan A., Jr Arch Oral Biol. 2007;52:1161. doi: 10.1016/j.archoralbio.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 18.Habelitz S, DenBesten PK, Marshall SJ, Marshall GW, Li W. Eur J Oral Sci. 2006;114(Suppl 1):315. doi: 10.1111/j.1600-0722.2006.00312.x. [DOI] [PubMed] [Google Scholar]
- 19.Le TQ, Gochin M, Featherstone JDB, Li W, DenBesten PK. Eur J Oral Sci. 2006;114(Suppl 1):320. doi: 10.1111/j.1600-0722.2006.00313.x. [DOI] [PubMed] [Google Scholar]
- 20.Long JR, Shaw WJ, Stayton PS, Drobny GP. Biochemistry. 2002;40:15451. doi: 10.1021/bi010864c. [DOI] [PubMed] [Google Scholar]
- 21.Shaw W, Campbell A, Paine M, Snead M. J Biol Chem. 2004;279:40263. doi: 10.1074/jbc.C400322200. [DOI] [PubMed] [Google Scholar]
- 22.Shaw WJ, Ferris K, Tarasevich B, Larson JL. Biophys J. 2008;94:3247. doi: 10.1529/biophysj.107.119636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fincham AG, Belcourt AB, Termine JD, Butler WT, Cothran WC. Biochem J. 1983;211:149. doi: 10.1042/bj2110149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fincham AG, Moradian-Oldak J, Simmer JP. J Struct Biol. 1999;136:270. doi: 10.1006/jsbi.1999.4130. [DOI] [PubMed] [Google Scholar]
- 25.Shaw W, Long J, Dindot J, Campbell A, Stayton P, Drobny G. J Am Chem Soc. 2000;122:1709. [Google Scholar]
- 26.Shaw WJ, Long JR, Campbell AA, Stayton PS, Drobny GP. J Am Chem Soc. 2000;122:7118. [Google Scholar]
- 27.Long JR, Shaw WJ, Stayton PS, Drobny GP. Biochemistry. 2001;40:15451. doi: 10.1021/bi010864c. [DOI] [PubMed] [Google Scholar]
- 28.Carpino LA, Han GY. J Org Chem. 1972;37:3404. [Google Scholar]
- 29.Wiejak S, Masiukiewicz E, Rzeszotarska B. Chem Pharm Bull. 1999;47:1489. doi: 10.1248/cpb.49.1189. [DOI] [PubMed] [Google Scholar]
- 30.Bielecki A, Burum DP. J Magn Reson, Ser A. 1995;116:215. [Google Scholar]
- 31.Duncan TM. Chemical Shift Tensors. 2nd. The Farragut Press; Ithaca, NY: 1997. [Google Scholar]
- 32.Gullion T, Schaefer J. J Magn Reson. 1989;81:196. [Google Scholar]
- 33.Gullion T, Schaefer J. J Magn Reson. 1991;92:439. [Google Scholar]
- 34.Bennett A, Rienstra C, Auger M, Lakshmi KV, Griffen RG. J Chem Phys. 1995;103:6951. [Google Scholar]
- 35.Bak M, Rasmussen JT, Nielsen NC. J Magn Reson. 2000;147:296. doi: 10.1006/jmre.2000.2179. [DOI] [PubMed] [Google Scholar]
- 36.Bemporad F, Gsponer J, Hopearuoho HI, Plakoutsi G, Stati B, Stefani M, Taddei M, Vendruscolo M, Chiti F. EMBO J. 2008;27:1525. doi: 10.1038/emboj.2008.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chatterjee A, Kumar A, Chugh J, Srivastava S, Bhavesh NS, Hosur RV. J Chem Sci. 2005;117:3. [Google Scholar]
- 38.Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Biochemistry. 2002;41:6573. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- 39.Kiss R, Kovacs D, Tompa P, Perczel A. Biochemistry. 2008;47:6936. doi: 10.1021/bi800201a. [DOI] [PubMed] [Google Scholar]
- 40.Plaxco KW, GroB M. Nature. 1997;386:657. doi: 10.1038/386657a0. [DOI] [PubMed] [Google Scholar]
- 41.Du C, Falini G, Fermani S, Abbott C, Moradian-Oldak J. Science. 2005;307:1450. doi: 10.1126/science.1105675. [DOI] [PubMed] [Google Scholar]
- 42.Aichmayer B, Margolis HC, Sigel R, Yamakoshi Y, Simmer JP, Fratzl P. J Struct Biol. 2005;151:239. doi: 10.1016/j.jsb.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 43.Aoba T, Moreno EC, Kresak M, Tanabe T. J Dent Res. 1989;68:1331. doi: 10.1177/00220345890680090901. [DOI] [PubMed] [Google Scholar]