

Abstract
Block polymers offer an attractive route to densely-packed, monodisperse nanoscale pores. However, their fragility as thin films complicates their use as membranes. By integrating a block polymer film with a thin (100 μm) silicon substrate, we have developed a composite membrane providing both nanoscale size exclusion and fast transport of small molecules. Here we describe the fabrication of this membrane, evaluate its mechanical integrity, and demonstrate its transport properties for model solutes of large and small molecular weight. The ability to block large molecules without hindering smaller ones, coupled with the potential for surface modification of the polymer and the MEMS style of the support make this composite membrane an attractive candidate for interfacing implantable sensing and drug delivery devices with biological hosts.
Keywords: MEMS, nanoporous, membrane, block polymer, size selectivity
Introduction
Nanoporous membranes have long played a role in the industrialized world. Pore sizes and morphologies in such membranes often exhibit significant dispersion, and increasingly stringent demands on pore size, monodispersity, transport resistance, durability, chemical and bio-compatibility, processability and cost, have spurred the development of thin membranes with straight, monodisperse nanopores. Contemporary examples include anodized alumina membranes containing straight, densely packed pores as narrow as 5 nm in diameter, and track-etched polymer membranes with pores also less <10 nm across but limited pore densities. Pores of either material can be narrowed or functionalized by coating the walls.1–4
New materials and processes are being developed to provide alternatives to these conventional films. Aligned carbon nanotubes have been used to form pores with interior diameters as small as 2 nm and smooth walls.5,6 Streimer et al.7 relied on the volume change during amorphous silicon annealing to create thin, rugged polysilicon membranes with maximum pore sizes less than 17 nm. Desai et al.8–10 fabricated silicon membranes with arrayed monodisperse pores as small as 7 nm across and demonstrated immunoisolation and cell support. None of these membranes have porosities much above 1%, however. Higher porosities have been achieved by variations around the anodized alumina theme, either by substituting tantalum for aluminum,11 or by transferring. the nanoporous pattern from alumina onto a silicon nitride substrate.12 The Si3N4 membranes were integrated into a microfluidic device supporting neuronal cells. These membranes share the high porosity of alumina, but also many of its drawbacks.
Block polymers offer another strategy for nanoporous membrane development.13–15 With proper choice of monomers and block lengths, these materials self-assemble into uniform cylinders of the minority component embedded in the majority component. Under proper conditions the cylinders align perpendicular to the plane of a thin film.16–19 Selective removal of the minority block yields films with straight, monodisperse densely-packed pores with diameters down to about 10 nm. The size selectivity of such films has been demonstrated electrochemically20 and they have been used to template nanoscale features in other materials.21–24 These films, however, are typically too thin and fragile to serve as stand-alone membranes. Mechanical properties are improved with thicker nanoporous membranes at the expense of higher transport resistance.25–28 For example, Yang et al.29,30 transferred a PS-PMMA film onto 150 μm thick porous polysulfone, etched the PMMA component and used this assembly to filter viruses, but this support structure also greatly decreased the membrane’s permeability.
To support a nanoporous polymer film with minimal increase in transport resistance, we prepared a composite nanoporous membrane system, integrating a poly(styrene)-poly(isoprene)-poly(lactide) (PS-PI-PLA) triblock polymer31,32 derived membrane with a microfabricated silicon support containing wide, short, straight, densely-packed pores. Here we describe this integrated fabrication, evaluate the viability of the supported nanoporous membrane under various mechanical challenges, and demonstrate its size-based transport selectivity.
Membrane Fabrication
Figure 1 shows the fabrication scheme for the composite membrane. A 100 μm thick double-side-polished silicon wafer is coated on both sides with 40 nm of low stress silicon nitride (Si3N4) by low pressure chemical vapor deposition (A). The top side of the wafer is coated with photoresist and patterned with a darkfield mask containing numerous 50×50 arrays of 20 μm squares spaced 20 μm apart. Following development, the wafer is glued onto a 500 μm thick handle wafer using photoresist. The wafer stack is hard-baked and subjected to reactive ion etching (RIE) to remove the exposed top-side Si3N4 squares (B). Bosch process-directed reactive ion etching (DRIE) anisotropically etches these squares through the silicon wafer with etch rate ≈2 μm/min. The etched wafer is removed from its handle and is cleaned by oxygen RIE. The resulting microporous support is 25% porous, with straight, nearly square pores 100 μm long, each capped on the bottom by the remaining 40 nm layer of Si3N4 (C). Figure 2 shows optical top, cross-section, and bottom views of the microporous support.
Figure 1.

Fabrication scheme for composite nanoporous membrane. Silicon wafer (dark gray, 100 μm thick) is coated on both sides with low stress silicon nitride (light gray, 40 nm) (A). Si3N4 is patterned via photolithography and reactive ion etching (RIE) (B). Pores are anisotropically dry-etched through Si (C). Triblock copolymer (black, 80 nm) is spin-coated onto top-side Si3N4 and annealed (D). PLA polymer domains are selectively removed by NaOH etching, followed by a brief O2 RIE (E) Si3N4 covering Si pores is removed by hydrofluoric acid etching (F). Note that figures are not drawn to scale.
Figure 2.
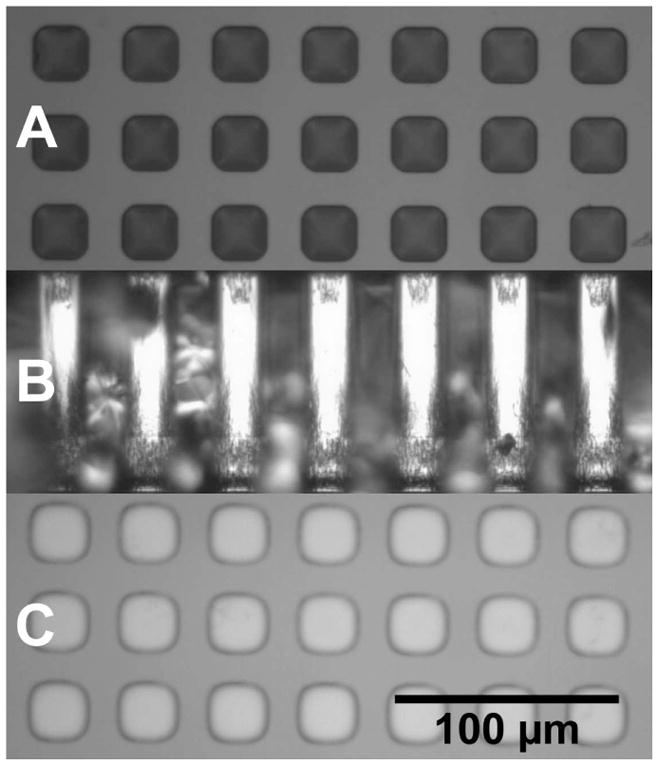
Microporous silicon support as viewed from the top (A), cross-section (B) and bottom (C).
To prepare the nanoporous skin, the Si3N4 surface of the support is immersed in a 5% solution of dichloromethyloctylsilane in toluene, coating the surface with covalently bonded alkyl chains33 to promote polymer adhesion to the Si3N4 surface. A chlorobenzene solution of PS-PI-PLA (Mn 69.5 kg/mol; 56% PS, 10% PI, 34% PLA by weight; polydispersity index 1.14, see refs. 28, 29) is then spin-coated onto the support. PI-sheathed PLA domains spontaneously self-assemble into discrete cylinders aligned perpendicular to the film surface within a continuous planar PS phase31,34–36 (Fig. 1D). The composite device is submerged in 0.05M NaOH in 60%/40% v/v water/methanol solution for 45 min, selectively removing most of the PLA and forming nanoscale PI-lined pores in the PS continuum (Fig. 1E). Pores at this stage are imaged in Figure 3 by tapping mode AFM, and are found to have mean pore size of 43 nm ± 11%RSD. Film thickness was determined by AFM to be 82 nm. Ten seconds of low-power oxygen RIE removes any polymer remaining at the bottom of the pores in the PS-PI film.23
Figure 3.

Tapping mode AFM height image of nanoporous block polymer film on Si3N4. Pore diameters are 43 nm ± 11% RSD. Image is 1 μm square.
To remove the Si3N4 between the micropores and the nanoporous block polymer film, a 30 μL drop of 49% hydrofluoric acid is introduced on the silicon side of the device (Fig. 1F). While HF may affect the PI sheaths, the PS film is largely unaffected by exposure to HF. 37,38
Alignment testing and pore continuity
To confirm the perpendicular domain alignment suggested by Figure 3, devices at the stage depicted in Figure 1E were subjected to an RIE protocol that etches Si3N4 as well as polymer, transferring the polymer’s nanopores into the underlying Si3N4. Remaining polymer was removed by both oxygen RIE and/or piranha bath (4:1 conc. H2SO4:30% H2O2). Figure 4 (SEM) shows that the pore pattern from the polymer is transferred into the nitride by etching, consistent with perpendicularly-aligned pores in the film. The irregular shapes of the etched domains are likely the result of fluoropolymer deposition on the PS film during RIE.
Figure 4.

Demonstration of pore alignment in nanoporous block polymer film. The film was used as a mask to etch the pits shown in this SEM image. The irregular pit shapes are likely due to product deposition during the etch.
To confirm that the nanopores span the entire block polymer film, finished devices as depicted in Figure 1F were used as masks for molecular beam deposition of permalloy (Ni80Fe20) onto a silicon wafer. Prior to deposition, different portions of the membrane were subjected to various O2 RIE etching times, ranging from 0 to 25 s. During deposition, twenty nanometers of permalloy was grown on the substrate at a rate of 0.2 Å/s, and the substrate was subsequently inspected by SEM. Figure 5 shows the permalloy deposition pattern in the absence of O2 RIE. Under each micropore, bright permalloy-coated regions surround a dark cross-shaped pattern. This pattern, as will be seen below, correlates with the topography of the polymer and probably reflects wrinkling and de-alignment of nanopores with the direction of the permalloy beam. Confocal microscopy conducted before and after permalloy deposition confirmed that the polymer film covered the pores throughout the process. (One micropore in Fig. 5 appears to have lost its block polymer film cover, explaining the complete permalloy coverage of the silicon substrate below it.)
Figure 5.

SEM image of permalloy (light) on silicon substrate (dark) following epitaxy using the nanoporous membrane as a mask. Feature size and spacing match those of the membrane’s microporous support. The cross-shaped pattern in each feature corresponds to the topography of the nanoporous polymer film. The bright feature in the lower-right quadrant shows growth under a micropore lacking a nanoporous polymer cover.
Since permalloy grew under the block polymer film with no O2 RIE etching, this etching step may not be required to provide pores that span the film. Exposure to O2 RIE over increasing times tended to erode the block polymer layer. Therefore, RIE etch may be regarded as a “polishing” step which should not be overutilized.
Imaging and Mechanical Evaluation of Nanoporous Membrane
The result of the fabrication steps is a two-layer composite (asymmetric) membrane with both microscale and nanoscale pores. As a free standing membrane, the nanoporous polymer is too thin and fragile for most applications. The underlying microporous silicon membrane provides mechanical support to the film. Morphology and viability of the film, attached to the microporous support, were investigated by a variety of means.
Figure 6 shows a reflectance confocal image of an intact film spanning a 20 μm pore. The film exhibits shallow wavelike wrinkles (~200 nm peak to trough) intersecting at the pore center. Wrinkles correlate with the pattern seen in the permalloy growth of Figure 5, suggesting, as already noted, that line-of-sight transmittance of the molecular beam was altered by the local angle of the film. Prior to spin-coating, the thin layer of Si3N4 (40 nm) over the pores was also wavy, probably a consequence of the tensile stress present in the Si3N4 film. The overlying polymer layer maintained this shape after the nitride was removed. Thicker (90 nm) Si3N4 films provide much flatter pore coverings.
Figure 6.

Confocal microscope topograph of polymer film spanning a 20 μm pore in the silicon support. The z-axis is magnified by a factor of 5 to better view the curvature in these pore coverings.
Stress is expected to be most concentrated near micropore perimeters. To determine whether stress leads to mechanical failure at the perimenter, a “lift and shift” procedure was used, enabling visualization of the patches of membrane covering the micropores. The wrinkle patterns provided visible markers designating these patches. In this case, the composite membrane was formed without surface treating the Si3N4, resulting in reduced adhesion between the nitride and the polymer film. Upon careful immersion in water, a section of the polymer film delaminated from the support. Slight lateral translation of the support caused the delaminated polymer to tear away from the adhered polymer. Lifting the support and carefully wicking away the water, the delaminated polymer laid back onto the support, approximately 30 μm to the left of its original position. This delaminated polymer and adhered polymer are seen, respectively, at the left and right sides of Figure 7, with a bright stripe of newly-exposed nitride in between.
Figure 7.

Optical micrograph of nanoporous block copolymer on microporous silicon support. Polymer on left was lifted and moved ~30 um to the left, allowing the polymer to be scrutinized independently from the pore it had previously covered. The exposed region of the microporous support is bright-colored and runs from upper-left to lower-right. Black uncovered pores are visible in this region. Covered pores are brightly-colored; the ones on the right have their original ‘wavy’ pore coverings. On the left, the original pore coverings are visible as light-colored ‘X’s partially overlaying the next column of pores to their left. Small tears are also visible as bright lines running laterally from the left-hand edge.
In Figure 7, shifted patches of the film that previously covered the micropores are identified by cross-shaped patterns, now overlapping micropores one column to the left. The crosses correlate with both the confocal topography in Fig. 6 and the permalloy deposition patterns in Fig. 5. There is no evidence of fracture inside the patches, and the three horizontal tears from the left edge of the figure do not propagate around or into the patch perimeters as one might expect if the perimeters were particularly weak.
In another test of the system’s robustness, we altered the fabrication protocol to precipitate a solid on the film. Rather than using aqueous HF to etch away the Si3N4 covering the support pores (per Figure 1E) we used gaseous HF. Aqueous HF provides a medium through which the reaction products (specifically NH4F) diffuse away. Since gas phase etching offers no such exit route for NH4F, salt crystals precipitate on the nanoporous polymer film, as shown in Figure 8. Some of these crystals are apparently supported by the polymer film, which conforms to the crystals, as shown in the confocal topograph Figure 8A. The topography persists after dissolving the crystals in excess deionized water. Figure 8B, an AFM relief map, shows a similar crystal-induced prominence along with radial wrinkles. Zooming in (Fig. 8c), the nanoscale pore structure of the polymer is seen to be unaffected by local topography, even along high-relief contours.
Figure 8.

(A) Confocal microscope topograph of polymer pore covering subjected to precipitation of Si3N4 etch products. (B) Tapping mode AFM image (height on left, phase on right) of pore from same sample as top. (C) AFM image of center 1 μm from middle image, showing edge of precipitation zone. The polymer retains its nanoporous morphology over these contours and appears undamaged.
Considering these observations, we conclude that the polymer film covering the micropores withstands localized stress and deformation without either tearing or losing its nanoporous morphology. The immersions and shear-rinses suffered by the samples during this investigation far exceed the physical challenges they would likely receive in most diffusional transport systems.
Transport Studies
Transport resistance and size selectivity of the bilayered composite were evaluated in a side-by-side diffusion cell (Crown Glass, 3 ml on both sides, with 9 mm orifice). A (2 mm)2 composite, embedded in the original wafer, was mounted and clamped between the donor and receptor cells with the block polymer film facing the receptor cell. Methyl orange (MO; Sigma, MW=327 g/mol) and dextran blue (DEX; Sigma, MW=2×106/mol) were introduced into the donor cell and their time dependent concentration in the receptor cell was monitored by UV/Vis spectroscopy. As shown in Figure 9a, cumulative flux of MO increased nearly linearly with time, with almost 15% of the molecules crossing the composite membrane during the first 48 hours. DEX was effectively blocked, however, with less than 1% crossing the composite over 6 weeks, showing a 1500-fold lower permeability.
Figure 9.

Membrane breakthrough curves. A) MO and DEX across nanoporous composite membrane indicating 1500-fold permeability difference between MO and DEX. B) MO and DEX across Anodisc control membrane, indicating 30-fold permeability difference between MO and DEX. C) MO across composite membrane without prior PLA block removal. NaOH was added after 18 hours, prompting dramatic increase in permeability.
As a control, the composite membrane was replaced by a commercial anodized alumina membrane (Anodisc, Whatman), 60 μm thick and with a nominal pore size of 200 nm. As the lateral dimension of these pores well exceeds the hydrodynamic radii of either solute, differences in transport are mostly attributable to differences in aqueous diffusivities of MO and DEX inside the pores. As shown in Fig. 9b, DEX transport across this non-selective membrane is only 30-fold lower than MO transport, as expected from its much larger hydrodynamic radius. The composite membrane, with nanopore diameters ~43 nm, therefore demonstrates a ~50-fold increase in selectivity compared to Anodisc, due to size exclusion of DEX and enhanced hydrodynamic interactions between DEX and the nanopore walls.
It should be noted that the Anodisc membrane was uniformly porous, and its area supporting transport was much larger (~16X) than that of the bilayer composite. Hence fluxes were much higher, necessitating more frequent sampling. In ultimate practice, pore coverage in the bilayer composite could be increased to the same order as Anodisc, and comparable MO fluxes but enhanced selectivity against DEX should be observed. Also, while qualitative changes in size selectivity between the composite membrane and Anodisc are obvious, the extremely low fluxes of DEX preclude precise quantitation. Both membranes are very thin, and unstirred layers may play a significant role in determining flux, particularly for MO. Rough calculations (see Supplementary Information), based on flux of MO through Anodisc and the aqueous diffusion coefficient of MO, suggest boundary layers of order 200 μm, thicker than either membrane. As these effects should be more significant for MO than for DEX, the difference in selectivity between the two membranes may be greater than what was inferred from the slopes of curves in Fig. 9a,b.
In another experiment, a composite membrane was prepared without the PLA removal step, and mounted in the diffusion cell. No MO diffused across the membrane for 18 hours, indicating that all three components of the block polymer membrane are impermeable. The downstream cell was then spiked with 0.5mL 0.37M of NaOH to degrade the PLA block. Methyl orange transport was soon observed downstream, as shown in Figure 9c. No DEX was detected in the receptor cell, however, even after five days of extreme upstream DEX concentration. As shown in Figure 9c, normalized methyl orange flux for the initially plugged membrane was about half that seen in Fig. 9a, possibly due to subtle differences in cell hydrodynamics or to incomplete removal of material from the pore regions (there was no O2 etch step.) These results suggest that PLA domains span the entire block polymer film, and demonstrate that size-selective transport through this membrane can be activated with a simple chemical signal.
Discussion and Conclusions
We have demonstrated fabrication and viability of a composite membrane combining the self-assembled nanoporous morphology of block polymers with the strength and regularity of micromachined silicon. This composite nanoporous membrane should be sufficiently robust for a variety of membrane applications, and the microfabricated membrane support should be amenable to scale-up and integration in MEMS devices.
Transport studies demonstrated minimal resistance of the membrane to small-molecule transport and high resistance to transport of large macromolecules. This combination of large permeability and large size-selectivity may be valuable in a variety of applications such as biomedical sensors and controlled delivery systems.
We have also demonstrated the ability to chemically trigger transport through the membrane, effectively turning the membrane into a controlled release device. This trigger may be tunable by a variety of strategies such as incorporating comonomers (e.g., glycolide) in the PLA block, assuming they do not significantly impact polymer morphology. Alternatively, following etching of PLA one may consider using the PI nanopore lining as a locus for functionalization with polymers that passivate the structure or mediate transport selectivity, perhaps in a stimuli-sensitive manner.32 Similarly, the PI lining can be used to anchor bioactive agents such as enzymes and antibodies.
Finally, by modifying the chain lengths in the block polymer, the size of the nanoscale pores can be tuned. The silicon support can also be optimized. We have already built supports with pore spacings as thin as 5 μm, and thinner wafers (already commercially available) should make that process even easier.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health (F32-HD051366, R21-EB003125). This work was also supported partially by the MRSEC Program of the National Science Foundation under Award Numbers DMR-0212302 and DMR-0819885. The authors also thank Dr. David Olson and Marc Rodwogin for synthesis and assistance with the block polymer.
Footnotes
Supporting Information Available: Fabrication Details, Pattern transfer into silicon nitride, UV/Vis calibration/detection limits, Diaphragm cell experiments, Calculation of boundary layers. Instrumentation. This information is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Martin CR. Science. 1994;266:1961. doi: 10.1126/science.266.5193.1961. [DOI] [PubMed] [Google Scholar]
- 2.Jirage KB, Hulteen JC, Martin CR. Science. 1997;278:655. [Google Scholar]
- 3.Lee SB, Martin CR. Chem Mater. 2001;13:3236. [Google Scholar]
- 4.Yamaguchi A, Uejo F, Yoda T, Uchida T, Tanamura Y, Yamashita T, Teramae N. Nat Mater. 2004;3:337. doi: 10.1038/nmat1107. [DOI] [PubMed] [Google Scholar]
- 5.Hinds BJ, Chopra N, Rantell T, Andrews R, Gavalas V, Bachas LG. Science. 2004;303:62. doi: 10.1126/science.1092048. [DOI] [PubMed] [Google Scholar]
- 6.Holt JK, Park HG, Wang Y, Stadermann M, Artyukhin AB, Grigoropoulos CP, Noy A, Bakajin O. Science. 2006;312:1034. doi: 10.1126/science.1126298. [DOI] [PubMed] [Google Scholar]
- 7.Striemer CC, Gaborski TR, McGrath JL, Fauchet PM. Nature. 2007;445:749. doi: 10.1038/nature05532. [DOI] [PubMed] [Google Scholar]
- 8.Leoni L, Boiarski A, Desai TA. Biomed Microdevices. 2002;4:131. [Google Scholar]
- 9.Desai TA, West T, Cohen M, Boiarski A, Rampersaud A. Adv Drug Delivery Rev. 2004;56:1661. doi: 10.1016/j.addr.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 10.Lopez CA, Fleischman AJ, Roy S, Desai TA. Biomaterials. 2006;27:3075. doi: 10.1016/j.biomaterials.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 11.Singh S, Greiner MT, Kruse P. Nano Lett. 2007;9:2676. doi: 10.1021/nl071061z. [DOI] [PubMed] [Google Scholar]
- 12.Wolfrum B, Mourzina Y, Sommerhage F, Offenhäusser A. Nano Lett. 2006;6:453. doi: 10.1021/nl052370x. [DOI] [PubMed] [Google Scholar]
- 13.Hawker CJ, Russell TP. MRS Bulletin. 2005;30:952. [Google Scholar]
- 14.Shin K, Leach KA, Goldbach JT, Kim DH, Jho JY, Tuominen M, Hawker CJ, Russell TP. Nano Lett. 2002;2:933. [Google Scholar]
- 15.Bang J, Kim SH, Drockenmuller E, Misner MJ, Russell TP, Hawker CJ. J Am Chem Soc. 2006;128:7622. doi: 10.1021/ja0608141. [DOI] [PubMed] [Google Scholar]
- 16.Kim SH, Misner MJ, Xu T, Kimura M, Russell TP. Adv Mater. 2004;16:226. [Google Scholar]
- 17.Cavicchi KA, Russell TP. Macromolecules. 2007;40:1181. [Google Scholar]
- 18.Xiang H, Lin Y, Russell TP. Macromolecules. 2004;37:5358. [Google Scholar]
- 19.Xu T, Zvelindovsky AV, Sevink GJA, Lyakhova KS, Jinnai H, Russell TP. Macromolecules. 2005;38:10788. [Google Scholar]
- 20.Li Y, Ito T. Anal Chem. 2009;81:851. doi: 10.1021/ac802201w. [DOI] [PubMed] [Google Scholar]
- 21.Hillmyer MA. Adv Polym Sci. 2005;190:137. [Google Scholar]
- 22.Black CT, Ruiz R, Breyta G, Cheng JY, Colburn ME, Guarini KW, Kim HC, Zhang Y. IBM J Res & Dev. 2007;51:605. [Google Scholar]
- 23.Kubo T, Parker JS, Hillmyer MA, Leighton C. Appl Phys Lett. 2007;90:233113. [Google Scholar]
- 24.Black CT, Guarini KW, Breyta G, Colburn MC, Ruiz R, Sandstrom RL, Sikorski EM, Zhang Y. J Vac Sci Technol B. 2006;24:3188. [Google Scholar]
- 25.Liu G, Ding J, Stewart S. Angew Chem Int Ed. 1999;38:835. doi: 10.1002/(SICI)1521-3773(19990315)38:6<835::AID-ANIE835>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 26.Liu G, Ding J, Hashimoto T, Kimishima K, Winnik FM, Nigam S. Chem Mater. 1999;11:2233. [Google Scholar]
- 27.Cooney DT, Hillmyer MA, Cussler EL, Moggridge GD. Crystallogr Rev. 2006;12:13. [Google Scholar]
- 28.Phillip WA, Rzayev J, Hillmyer MA, Cussler EL. J Membr Sci. 2006;286:144. [Google Scholar]
- 29.Yang SY, Ryu I, Kim HY, Kim JK, Jang SK, Russell TP. Adv Mater. 2006;18:709. [Google Scholar]
- 30.Yang SY, Park J, Yoon J, Ree M, Jang SK, Kim JK. Adv Func Mater. 2008;18:1371. [Google Scholar]
- 31.Guo S, Rzayev J, Bailey TS, Zalusky AS, Olayo-Valles R, Hillmyer MA. Chem Mater. 2006;18:1719. [Google Scholar]
- 32.Bailey TS, Rzayev J, Hillmyer MA. Macromolecules. 2006;39:8772. [Google Scholar]
- 33.Fadeev AY, McCarthy TJ. Langmuir. 2000;16:7268. [Google Scholar]
- 34.Kubo T, Wang RF, Olson DA, Rodwogin M, Hillmyer MA, Leighton C. Appl Phys Lett. 2008;93:133112. [Google Scholar]
- 35.Olayo-Valles R, Guo S, Lund MS, Leighton C, Hillmyer MA. Macromolecules. 2005;38:10101. [Google Scholar]
- 36.Park C, Cheng JY, Fasolka MJ, Mayes AM, Ross CA, Thomas EL. Appl Phys Lett. 2001;79:848. [Google Scholar]
- 37.Mao H, Hillmyer MA. Macromolecules. 2005;38:4038. [Google Scholar]
- 38.Mao H. Ph.D. Thesis. University of Minnesota; Minneapolis, MN: 2006. [Google Scholar]
- 39.Sigma did not provide a hydrodynamic radius; GE Healthcare lists a stokes radius of 27 nm for its 2 MDa dextran blue
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.