

Abstract
Incorporation of the herpes simplex virus 1 (HSV-1) portal vertex into the capsid requires interaction with a 12-amino-acid hydrophobic domain within capsid scaffold proteins. The goal of this work was to identify domains and residues in the UL6-encoded portal protein pUL6 critical to the interaction with scaffold proteins. We show that whereas the wild-type portal and scaffold proteins readily coimmunoprecipitated with one another in the absence of other viral proteins, truncation beyond the first 18 or last 36 amino acids of the portal protein precluded this coimmunoprecipitation. The coimmunoprecipitation was also precluded by mutation of conserved tryptophan (W) residues to alanine (A) at positions 27, 90, 127, 163, 241, 262, 532, and 596 of UL6. All of these W-to-A mutations precluded the rescue of a viral deletion mutant lacking UL6, except W163A, which supported replication poorly, and W596A, which fully rescued replication. A recombinant virus bearing the W596A mutation replicated and packaged DNA normally, and scaffold proteins readily coimmunoprecipitated with portal protein from lysates of infected cells. Thus, viral functions compensated for the W596A mutation's detrimental effects on the portal-scaffold interaction seen during transient expression of portal and scaffold proteins. In contrast, the W27A mutation precluded portal-scaffold interactions in infected cell lysates, reduced the solubility of pUL6, decreased incorporation of the portal into capsids, and abrogated viral-DNA cleavage and packaging.
Immature herpesvirus capsids or procapsids consist of two shells: an inner shell, or scaffold, and an outer shell that is roughly spherical and largely composed of the major capsid protein VP5 (24, 38).
The capsid scaffold consists of a mixture of the UL26.5 and UL26 gene products, with the UL26.5 gene product (pUL26.5, ICP35, or VP22a) being the most abundant (1, 12, 20, 21, 32, 38). The UL26.5 open reading frame shares its coding frame and C terminus with the UL26 gene but initiates at codon 307 of UL26 (17). The extreme C termini of both VP22a and the UL26-encoded protein (pUL26) interact with the N terminus of VP5 (7, 14, 26, 40, 41). Capsid assembly likely initiates when the portal binds VP5/VP22a and/or VP5/pUL26 complexes (22, 25). The addition of more of these complexes to growing capsid shells eventually produces a closed sphere bearing a single portal. pUL26 within the scaffold contains a protease that cleaves itself between amino acids 247 and 248, separating pUL26 into an N-terminal protease domain called VP24 and a C-terminal domain termed VP21 (4, 5, 8, 9, 28, 42). The protease also cleaves 25 amino acids from pUL26 and VP22a to release VP5 (5, 8, 9). VP21 and VP22a are replaced with DNA when the DNA is packaged (12, 29).
When capsids undergo maturation, the outer protein shell angularizes to become icosahedral (13). One fivefold-symmetrical vertex in the angularized outer capsid shell is biochemically distinct from the other 11 and is called the portal vertex because it serves as the channel through which DNA is inserted as it is packaged (23). In herpes simplex virus (HSV), the portal vertex is composed of 12 copies of the portal protein encoded by UL6 (2, 23, 39). We and others have shown that interactions between scaffold and portal proteins are critical for incorporation of the portal into the capsid (15, 33, 44, 45). Twelve amino acids of scaffold proteins are sufficient to interact with the portal protein, and tyrosine and proline resides within this domain are critical for the interaction with scaffold proteins and incorporation of the portal into capsids (45).
One goal of the current study was to map domains and residues within the UL6-encoded portal protein that mediate interaction with scaffold proteins. We show that the portal-scaffold interaction requires all but the first 18 and last 36 amino acids of pUL6, as well as several tryptophan residues positioned throughout the portal protein.
MATERIALS AND METHODS
Viruses and cells.
CV1 and rabbit skin cells were obtained from the American Type Culture Collection and maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% newborn calf serum, 100 U penicillin per ml, and 100 μg of streptomycin per ml. CV6 cell lines expressing pUL6 were cultured in DMEM supplemented with 10% fetal bovine serum, 100 U/ml of penicillin, 100 μg/ml of streptomycin, and 200 μg/ml of hygromycin B as described previously (46). HSV-1 strain F [HSV-1(F)] and a UL6 null virus derived from HSV-1 strain 17 were described previously (11, 27). Recombinant viruses vJB30 and vJB31 and the restored virus vJB30R are described below.
Plasmids.
Plasmids pJB448, expressing full-length pUL26 and VP22a; pJB437, containing the entire UL6 coding sequence; pJB444, encoding pUL6 with an N-terminal Flag epitope; and pJB445, encoding a C-terminal Flag epitope fused to pUL6, were described previously (43). Truncations of pUL6 were generated by one-step PCR, and point mutations were generated by two-step PCR using pJB437 as a template. Since codons 12 to 18 of UL6 were known to be dispensable for virus replication (19), the primers for generating W27A in a one-step PCR started at codon 16, and the mutation was embedded in the primer. The sequences of the PCR primer used in these studies are listed in Table 1. PCR-generated amplicons were cloned into the BamHI and EcoRI sites located in the multiple cloning site of expression vector pcDNA3. The designations of the resulting plasmids and their corresponding mutations are listed in Fig. 1. All plasmid constructs were confirmed by DNA sequencing performed by the Cornell University DNA sequencing and genotyping core facility (data not shown) and by immunoblotting after transient expression in mammalian cells using pUL6-specific or Flag-specific antibodies.
TABLE 1.
Primers used for generation of mutant plasmid or recombinant virusa
| Plasmid or BAC | Mutation | Primer sequence |
|---|---|---|
| Plasmids | ||
| pJB451 | Flag1-321 | 5′TGACGGATCCGCCACCATGGATTACAAGGATGACGACGATAAGACCGCACCACGCTCGCGGGCCCCCACT3′, 5′CGATGAATTCTCAGACCCGCGCGGCACCGGCCAC3′ |
| pJB450 | 314-676flag | 5′TGACGGATCCGCCATGGTGGCCGGTGCCGCGCGGGTCGTT3′, 5′CGATGAATTCCTACTTATCGTCGTCATCCTTGTAATCTCGTCGGCCGTCGCGGCGGCCATCCCG3′ |
| pJB601 | 51-676flag | 5′TGACGGATCCACCATGGAGGGCCAGGGGGTGTACAACGTC3′, 5′CGATGAATTCCTACTTATCGTCGTCATCCTTGTAATCTCGTCGGCCGTCGCGGCGGCCATCCCG3′ |
| pJB604 | 19-676flag | 5′TGACGGATCCACCATGGCGCTGTGCTCCCCCGAGGACGGC3′, 5′CGATGAATTCCTACTTATCGTCGTCATCCTTGTAATCTCGTCGGCCGTCGCGGCGGCCATCCCG3′ |
| pJB605 | 30-676flag | 5′TGACGGATCCACCATGGTTCACCCCAGCCCCGGTACGATG3′, 5′CGATGAATTCCTACTTATCGTCGTCATCCTTGTAATCTCGTCGGCCGTCGCGGCGGCCATCCCG3′ |
| pJB611 | Flag1-410 | 5′TGACGGATCCGCCACCATGGATTACAAGGATGACGACGATAAGACCGCACCACGCTCGCGGGCCCCCACT3′, 5′TCGAGAATTCTCAGAAGGCCTGGCGAAGCTGCGGCGC3′ |
| pJB612 | Flag1-530 | 5′TGACGGATCCGCCACCATGGATTACAAGGATGACGACGATAAGACCGCACCACGCTCGCGGGCCCCCACT3′, 5′TCGAGAATTCTCAGCGCGACGAGCGCTCCAGGTCCTG3′ |
| pJB613 | Flag1-640 | 5′TGACGGATCCGCCACCATGGATTACAAGGATGACGACGATAAGACCGCACCACGCTCGCGGGCCCCCACT3′, 5′TCGAGAATTCTCACGCGCCGGGCCGGAAATCGGCGCC3′ |
| pJB623 | W27A | 5′TGACGGATCCACCATGGACACGGAAGCGCTGTGCTCCCCCGAGGACGGCgccGTAAAGGTTCACCCCAGCCCCGGTACG3′, 5′CGATGAATTCTCATCGTCGGCCGTCGCGGCGGCCATC3′ |
| pJB624 | W90A | 5′TACCGGGACCTCGAGGCGGACgccCTCGGCCACGTGGCGGCCCGC3′, 5′GCGGGCCGCCACGTGGCCGAGggcGTCCGCCTCGAGGTCCCGGTA3′ |
| pJB625 | W241A | 5′TACAACCACCGCCGGGGCGACgccCTCGTGCGAGACCCCATCAGC3′, 5′GCTGATGGGGTCTCGCACGAGggcGTCGCCCCGGCGGTGGTTGTA3′ |
| pJB626 | W596A | 5′ATCCTGGGGGAGGAGGAGTTAgccGATGCGGTGTTTAAGAAAACC3′, 5′GGTTTTCTTAAACACCGCATCggcTAACTCCTCCTCCCCCAGGAT3′ |
| pJB631 | W127A | 5′GCCGAGCGGGTGTTCGACACGgccCGGAACACGCTTAGGACGACG3′, 5′CGTCGTCCTAAGCGTGTTCCGggcCGTGTCGAACACCCGCTCGGC3′ |
| pJB632 | W163A | 5′AGCTTCCCCAAATATATCGACgccCTGACGTGCCTGGGGCTGGTC3′, 5′GACCAGCCCCAGGCACGTCAGggcGTCGATATATTTGGGGAAGCT3′ |
| pJB634 | W262A | 5′CTGGTGCTGTGGCCCCCCTTGgcaACCGGGGACCGTCTGGTCTTC3′, 5′GAAGACCAGACGGTCCCCGGTtgcCAAGGGGGGCCACAGCACCAG3′ |
| pJB641 | W532A | 5′CTGGAGCGCCTGTCGCGCCTCgcaGAGCACGAGCTGGTGCGCTGT3′, 5′ACAGCGCACCAGCTCGTGCTCtgcGAGGCGCGACAGGCGCTCCAG3′ |
| BACs | ||
| bJB30 | W27A | 5′GCGCGGGGGGACACGGAAGCGCTGTGCTCCCCCGAGGACGGCgcaGTAAAGGTTCACCCCAGCCCCTAGGGATAACAGGGTAATCGATTT3′, 5′CTCGCGGAACAGCATCGTACCGGGGCTGCTGGGGTGAACCTTTACtgcGCCGTCCTCGGGGGAGCACAGGCCAGTGTTACAACCAATTAACC3′ |
| bJB31 | W596A | 5′CCCATCACGGGCTCCGATGTCATCCTGGGGGAGGAGGAGTTAgcaGATGCGGTGTTTAAGAAAACCTAGGGATAACAGGGTAATCGATTT3′, 5′TGTCAGGTACGTTTGCAGGCGGGTTTTCTTAAACACCGCATCtgcTAACTCCTCCTCCCCCAGGATGCCAGTGTTACAACCAATTAACC |
Lowercase letters represent introduced mutations.
FIG. 1.

Diagrams of pUL6 expression plasmid (A) and recombinant virus (B) with mutations in the UL6 gene. The designation of the plasmid (A) or recombinant virus (B) is indicated to the right of each line. The number above each line is the codon number. Letters plus numbers above each line indicate single-amino-acid substitutions. The stars designate the positions of the Flag epitope tags.
Plasmid pCAGGS-nlsCre, expressing Cre recombinase, was a gift from Michael Kotlikoff, Cornell University. Plasmids pBAD-I-SceI, containing the gene encoding the Saccharomyces cerevisiae I-SceI endonuclease, and pEPkan-S, containing aphAI (encoding kanamycin resistance) adjacent to an I-SceI restriction site, were gifts from Nikolaus Osterrieder, Cornell University.
Construction of recombinant viruses.
Recombinant viruses were constructed by en passant mutagenesis, a two-step Red-mediated recombinant system described by B. K. Tischer et al. (37). The bacterial artificial chromosome (BAC) containing the entire HSV-1(F) genome was described previously (34). The primers for production of a PCR amplicon for eventual mutation of W27A or W596A of UL6 in the HSV-1(F)-containing BAC are listed in Table 1. The expected mutations in HSV-1 BAC DNA were confirmed by DNA sequencing, and the resulting recombinant BACs were designated bJB30 and bJB31. Purified BAC DNA was cotransfected with a Cre expression plasmid into CV6 cells expressing pUL6. The presence of viable recombinant virus was indicated by the formation of cytopathic effects, and the resulting viruses were subjected to two further rounds of plaque purification. The genotypes of the recombinant viruses, designated vJB30 and vJB31, were confirmed by PCR and DNA sequencing, whereas the viral phenotype was characterized as described in Results. To repair the mutated UL6 gene of vJB30, rabbit skin cells were cotransfected with vJB30 viral DNA and linearized pJB132, which contains the gene fragment from positions 11820 to 21655 of the HSV-1F genome (18). The virus arising from homologous recombination was able to form plaques in rabbit skin cells and was designated vJB30R.
Immunoprecipitation and immunoblotting.
Appropriately 2 × 106 CV1 cells were either transfected with 4 μg each of expression plasmids containing UL26 and wild-type UL6 or its derivatives, as shown in Fig. 1, or infected with wild-type and recombinant viruses. At 24 h after transfection or 18 h after infection, the cells were washed with cold phosphate-buffered saline (PBS) and lysed in cold RIPA buffer containing 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholate, 1 mM EDTA, and 1× protease inhibitor cocktail (Roche). Mouse anti-VP22a monoclonal antibody (MCA406; 1:200 dilution; AbD SeroTEC) was used for immunoprecipitation. Immune complexes, RIPA buffer-solubilized clarified lysates, total lysates solubilized in 1% sodium dodecyl sulfate (SDS) and beta mercaptoethanol, and, in some experiments, SDS-denatured purified B capsids were separated on SDS-12% polyacrylamide gels, and transferred to nitrocellulose membranes for immunoblotting. The immunoblots were probed with anti-VP22a antibodies diluted 1:2,000 and/or anti-pUL6 polyclonal antiserum diluted 1:1,000. The bound immunoglobulins were revealed by reaction with horseradish peroxidase-conjugated anti-rabbit or -mouse immunoglobulin G and visualized by enhanced chemiluminescence (Amersham Pharmacia Biotech).
Capsid purification.
Capsid purification procedures were performed as described previously (43). Briefly, CV1 cell monolayers from two 850-cm2 roller bottles were infected with either HSV-1(F) or mutant viruses at a multiplicity of infection of 5 PFU/cell. The cells were harvested 18 h later and washed with cold PBS. The cell pellets were suspended in 25 ml of lysis buffer (1 mM dithiothreitol, 1 mM EDTA, 20 mM Tris, pH 7.6, 500 mM NaCl, 1% Triton X-100, and protease inhibitor), sonicated briefly, and precleared by spinning them at 10,000 × g for 15 min. Capsids in the supernatant were pelleted through a 5-ml 35% sucrose cushion in TNE buffer (20 mM Tris-HCl [pH 7.6], 500 mM NaCl, 1 mM EDTA) in an SW28 ultracentrifuge tube at 24,000 rpm for 1 h. The pellets were resuspended in TNE buffer and applied to 20% to 50% sucrose gradients in SW41 ultracentrifuge tubes, followed by centrifugation at 24,500 rpm for 1 h. After centrifugation, the light-refracting B-capsid band was collected with a Pasteur pipette. The purity of capsid preparations was evaluated by transmission electron microscopy and negative staining (data not shown).
Southern blotting.
Approximately 2 × 106 CV1 cells were mock infected or infected with HSV-1(F), JB30, vJB30R, or vJB31. At 18 h postinfection, viral DNA was extracted, digested with BamHI, separated on 0.8% agarose gels, and transferred to nylon membranes as described previously (46). The bound DNA was UV cross-linked to the membrane and hybridized with [32P]dCTP-labeled BamHI P fragment of HSV-1(F) DNA at 42°C for 24 h according to standard procedures (30). The membrane was washed twice with 1× SSC (3.0 M NaCl, 0.3 M sodium citrate, pH, 7.0) at 42°C for 15 min each time and once with 0.1× SSC, 0.1% SDS for 1 h at 64°C and then autoradiographed by exposure to X-ray film.
Transient-complementation assay.
Approximately 1.2 × 106 CV1 cells in six-well plates were transfected with 4 μg of the plasmids listed in Table 2 by use of Lipofectamine 2000 according to the manufacture's protocol (Invitrogen). Twenty-four hours after transfection, the cells were either harvested and subjected to immunoblotting (data not shown) or superinfected with 1 PFU per cell of UL6 null virus. After adsorption for 2 hours at 37°C with shaking, the inoculates were removed, and the cells were washed with CBS buffer (40 mM citric acid, 10 mM KCl, 135 mM NaCl, pH 3.0) to remove residual infectivity. The cells were then washed once with PBS and overlaid with 5 ml DMEM supplemented with 5% newborn calf serum. Progeny virus was harvested 24 hours postinfection by three cycles of freezing and thawing, and the amount of infectivity was determined by plaque assay in CV6 cells, which contain the UL6 gene and complement the replication of UL6 null viruses. This experiment was repeated three times.
TABLE 2.
Rescue of UL6 null mutant propagation by transiently expressed UL6 mutantsa
| Plasmid | Genotype of pUL6 in tested plasmid | Yield of UL6 null virusb (PFU/ml) |
|---|---|---|
| pcDNA3 | <102 | |
| pJB448 | Wild type | (3.2 ± 0.23) × 105 |
| pJB623 | W27A | <102 |
| pJB624 | W90A | <102 |
| pJB631 | W127A | <102 |
| pJB632 | W163A | (3 ± 2.5) × 102 |
| pJB625 | W241A | <102 |
| pJB634 | W262A | <102 |
| pJB641 | W532A | <102 |
| pJB626 | W596A | (4.4 ± 4.8) × 105 |
The transient-complementation assay was performed as described in Materials and Methods.
Infectious titers were determined on CV6 cells expressing pUL6, and the data represent means ± standard deviations from three independent experiments.
Virus replication assay.
Approximately 2 × 106 CV1 cells in 25-cm2 flasks were infected with the viruses indicated in Table 3 at a multiplicity of infection of 0.1 PFU/cell. After adsorption for 2 hours at 37°C with shaking, the inoculates were removed, and the cells were washed with CBS buffer to remove residual infectivity. The cells were then washed with PBS once and overlaid with 5 ml DMEM supplemented with 5% newborn calf serum. Twenty-four hours after infection, virus was harvested by three cycles of freezing and thawing, and infectious yields were determined by plaque assay on the cell monolayers indicated in Table 3. This experiment was repeated three times.
TABLE 3.
Replication of wild-type and mutant virusesa
| Virus | Genotype of UL6 | Cell line for propagation/cell line for determining titer | Infectious titerb (PFU/ml) |
|---|---|---|---|
| 6 null | CV1/CV6 | <102 | |
| F | Wild type | CV1/CV1 | (1.9 ± 0.65) × 107 |
| vJB30 | W27A | CV1/CV6 | <102 |
| vJB30 | W27A | CV6/CV6 | (8 ± 0.2) × 106 |
| vJB30R | Wild type | CV1/CV6 | (1.7 ± 1.1) × 107 |
| vJB31 | W596A | CV1/CV6 | (2.0 ± 0.45) × 107 |
The virus replication assay was performed as described in Materials and Methods.
The data represent means ± standard deviations from three independent experiments.
RESULTS
To map the domains of portal protein pUL6 that are critical for interaction with scaffold proteins, UL6 was cloned in frame with a Flag epitope. Full-length and truncated versions of this plasmid were transfected into CV1 cells with a pUL26 expression plasmid. Lysates of the transfected cells were then subjected to immunoprecipitation with scaffold-specific antibody, and the presence or absence of scaffold and portal proteins in electrophoretically separated immunoprecipitated material was determined by immunoblotting.
As shown in Fig. 2, the anti-scaffold antibody immunoprecipitated VP22a, the major scaffold protein, in all cases and pUL26 in most cases. Moreover, Flag-pUL6 was expressed in contransfected cells to readily detectable levels, as revealed on immunoblots of the cellular lysates probed with M2 antibody. Most importantly for the purposes of this experiment, full-length pUL6 was coimmunoprecipitated with anti-scaffold antibody whether the Flag epitope was located at the N or the C terminus. Examination of truncated pUL6 revealed that amino acids 1 to 18 and 641 to 676 were dispensable for coimmunoprecipitation with scaffold proteins, whereas more extreme truncations than these precluded the interaction with scaffold proteins. We concluded that amino acids 19 to 640 of pUL6 were sufficient to interact with scaffold proteins as determined by immunoprecipitation.
FIG. 2.

Mapping regions of pUL6 required for scaffold protein interactions. CV1 cells were cotransfected with expression plasmids encoding full-length pUL26 and Flag-tagged pUL6 or its derivatives. (A) Twenty-four hours after transfection, cells were harvested and lysed in RIPA buffer. The lysates were electrophoretically separated, transferred to nitrocellulose, and probed with anti-Flag antibody to monitor pUL6-Flag expression. (B) The lysates were reacted with anti-scaffold protein monoclonal antibody. The presence of pUL6 in immunoprecipitated material was detected by immunoblotting. (C) Immunoprecipitation reactions were performed with anti-scaffold antibody followed by immunoblotting with the same antibody. In all cases, bound immunoglobulins were revealed by enhanced chemiluminescence. The positions of pUL6, scaffold proteins, and the mouse immunoglobulin G heavy chain are indicated on the right. The positions of protein mass standards are indicated on the left. IP, immunoprecipitation; IB, immunoblotting.
We next focused on residues within the identified scaffold interaction region of pUL6 that might be critical to its interaction with scaffold proteins. Given our previous results showing that a hydrophobic domain in scaffold proteins mediated interaction with the portal protein, we reasoned that hydrophobic residues within the portal might be important. We noted several highly conserved tryptophan residues within the portal protein (see Fig. 7). To test whether these amino acids were important for scaffold protein interaction, we mutated each tryptophan codon to alanine and tested each mutant protein for its ability to interact with the scaffold protein by coimmunoprecipitation.
FIG. 7.

Sequence alignments showing that tryptophan residues are conserved in the portal proteins of alphaherpesvirus. BHV, bovine herpesvirus; CHV, cercopithecine herpesvirus; EHV, equine herpesvirus; GaHV, gallid herpesvirus; HSV, herpes simplex virus; MeHV-1, meleagrid herpesvirus 1 (herpesvirus of turkeys); PRV, pseudorabies virus; VZV, varicella zoster virus. The sequences are from references 3, 6, 10, 16, 18, 31, 35, and 36.
As shown in Fig. 3, mutation of tryptophan residues at position 27, 90, 127, 163, 241, 262, 532, or 596 completely precluded coimmunoprecipitation by scaffold-specific antibody despite ample levels of the mutated portal proteins in the lysates. We concluded that these residues are critical to interaction with the scaffold protein in the absence of other viral proteins.
FIG. 3.

Replacement of tryptophan by alanine within pUL6 abolishes its interaction with scaffold protein in transfected cells. CV1 cells were cotransfected with expression plasmids bearing full-length UL26 and wild-type UL6 or UL6 genes containing substitutions of alanine for tryptophan codons. Twenty-four hours after transfection, cells were harvested and lysed in RIPA buffer, and coimmunoprecipitation with anti-ICP35 monoclonal antibodies was performed. (A) Immunoblots of transfected cell lysates probed with pUL6-specific antibody. (B) Immunoblot of proteins immunoprecipitated with anti-scaffold antibody and probed with pUL6-specific antibody. (C) Immunoblot of proteins immunoprecipitated with anti-scaffold antibody and probed with the same antibody. The positions of pUL6 and scaffold proteins are indicated on the right. Bound immunoglobulins were revealed by enhanced chemiluminescence. IP, immunoprecipitation; IB, immunoblotting.
To test for the effects of the W/A mutations on viral replication, CV1 cells were transfected with plasmids bearing vector DNA, wild-type pUL6, or pUL6 bearing the W/A mutations at position 27, 90, 127, 163, 241, 262, 532, or 596. Expression of mutant and wild-type pUL6 was verified by immunoblotting 24 h later (not shown), and the pUL6-expressing cells were infected with a UL6 null virus. Twenty-four hours after virus infection, the cells were lysed, and the yield of infectious virus was determined on a UL6-expressing cell line, CV6. As shown in Table 2, expression of wild-type pUL6 rescued replication of the UL6 null virus, producing a final infectious titer of approximately 3 × 105 PFU/ml. In contrast, W/A mutations at positions 27, 90, 127, 241, 262, and 532 completely precluded rescue of viral infectivity, indicating that these residues were critical to portal function. Compared to wild-type pUL6, the W163A-bearing plasmid was reduced by approximately 1,000-fold in its ability to rescue replication of the UL6 null mutant, producing an infectious titer of approximately 300 PFU/ml. In contrast, the plasmid bearing W596A rescued viral replication as well as the wild-type plasmid, despite the observation that the mutation precluded coimmunoprecipitation of pUL6 and scaffold proteins when these proteins were coexpressed in the absence of virus infection (Fig. 3). We conclude that other functions in virus-infected cells can compensate for the diminished interaction between the portal and the scaffold in cells expressing pUL6 W596A.
To investigate this idea further, recombinant viruses bearing (i) the W596A mutation and (ii) W27A (as an example of the group of mutations precluding replication in our complementation assay) were constructed using BAC technology and were designated vJB31(W596A) and vJB30(W27A), respectively. A virus derived from vJB30 but bearing a restored UL6 gene was also constructed and designated vJB30R.
As shown in Table 3, vJB31(W596A) replicated to levels similar to those of the wild-type virus in both CV1 cells and CV6 cells, which are derived from CV1 cells but express wild-type pUL6 (46). In contrast to these results, neither vJB30(W27A) nor the UL6 null mutant replicated to an appreciable extent on CV1 cells. vJB30(W27A) was able to propagate on UL6-complementing cells to levels similar to that of wild-type virus, indicating that UL6 was solely responsible for the replication defect in CV1 cells. This conclusion was further confirmed by the observation that vJB30R was able to replicate normally on CV1 cells.
To determine the fate of the portal-scaffold interaction in cells infected with vJB30(W27A) and vJB31(W596A), cells were infected with these viruses or HSV-1(F), and the amount of pUL6 in clarified or total lysates was determined by immunoblotting. As shown in Fig. 4, steady-state levels of pUL6 accumulated to similar levels in cells infected with JB30, JB31, and HSV-1(F) (Fig. 4B). On the other hand, slightly reduced levels of pUL6 were present in clarified lysates of vJB30(W27A)-infected cells compared to the amounts in lysates of cells infected with vJB31(W596A) or HSV-1(F). This observation was potentially significant, because it was noted previously that mutations precluding scaffold-portal interactions inhibit solubilization of pUL6 (36).
FIG. 4.
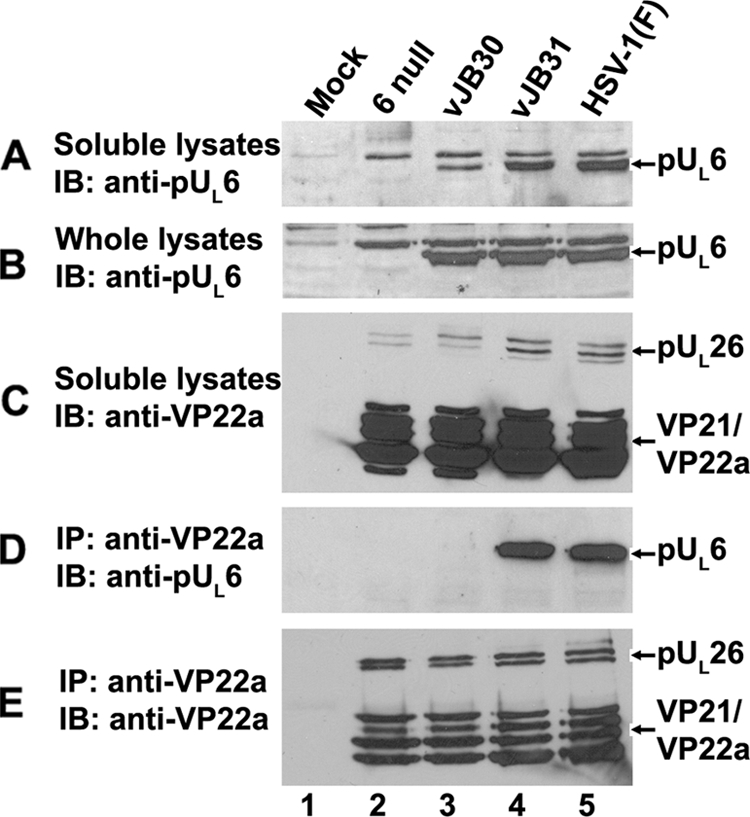
pUL6 and scaffold protein interaction in virus-infected cells. CV1 cells were mock infected or infected with HSV-1(F), vJB30(W27A), vJB31(W596A), or a UL6 null mutant. Eighteen hours after infection, coimmunoprecipitation was performed using anti-scaffold antibodies. (A) Immunoblot of electrophoretically separated lysates probed with anti-pUL6 antibody. (B) Whole-cell lysates probed with anti-pUL6 antibody. (C) Soluble lysates probed with anti-scaffold antibody. (D) Immunoblot of material immunoprecipitated with anti-scaffold antibody and probed with antibody to pUL6. (E) Immunoblot of proteins immunoprecipitated with scaffold-specific antibody and probed with the same antibody. Bound immunoglobulins were revealed by enhanced chemiluminescence. The positions of scaffold proteins and pUL6 are indicated on the right. IP, immunoprecipitation; IB, immunoblotting.
To determine whether the mutations affected portal-scaffold interaction as suspected, the clarified lysates were subjected to immunoprecipitation with scaffold-specific antibody. As shown in Fig. 4E, the scaffold-specific antibody readily immunoprecipitated scaffold proteins from all the infected cell lysates. Most importantly, pUL6 coimmunoprecipitated with VP22a from lysates of cells infected with vJB31(W596A), but not from vJB30(W27A)-infected cell lysates (Fig. 4D). Thus, the inability of vJB30 to replicate in CV1 cells correlated with a failure of its portal and scaffold proteins to interact with infected cells as assessed by immunoprecipitation.
To determine whether the failure of vJB30 scaffold and portal protein interaction affected incorporation of the portal protein into capsids, cells were infected with the UL6 null virus, wild-type HSV-1(F), vJB30(W27A), or vJB31(W596A), and capsids were purified. Capsid proteins were denatured and electrophoretically separated, and an immunoblot of the separated proteins was probed with antibodies to pUL6, scaffold, or VP5 as a loading control. As shown in Fig. 5, approximately equal amounts of capsids were loaded in each lane, as revealed by the roughly equal intensities of the VP5-specific bands. Similar levels of scaffold proteins were also contained within each lane, suggesting that the portal mutations did not affect scaffold incorporation into capsids. Most significantly, less pUL6-specifc signal was present in the vJB30(W27A) capsids than was detected in wild-type capsids or vJB31(W596A) capsids. To ensure that the association of mutant portal with capsids was not a consequence of the protein migrating throughout the sucrose gradient, all gradient fractions were probed with the pUL6 antibody and VP5 antibody was used to mark capsid-containing fractions. pUL6 immunoreactivity was detected only in fractions containing VP5, indicating that the mutant portal protein did not migrate throughout the sucrose gradient (data not shown). Thus, the failure of vJB30(W27A) portal and scaffold to interact, as revealed by immunoprecipitation, correlated with a reduced, but not complete, inhibition of portal incorporation into vJB30 capsids.
FIG. 5.
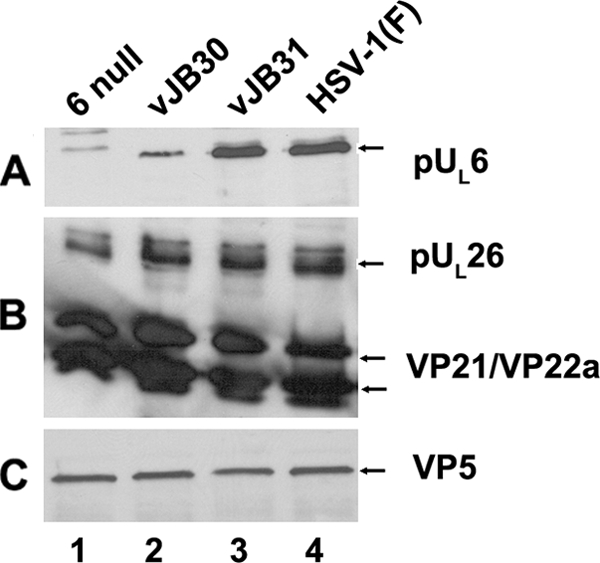
B-capsid proteins probed with pUL6-, VP22a-, or VP5-specific antibodies. CV1 cells (4 × 108) were infected with 5 PFU/cell of HSV-1(F), UL6 null, vJB30(W27A), or vJB31(W596A). Twenty hours after infection, B capsids were purified, denatured, separated by SDS-12% polyacrylamide gel electrophoresis, and transferred onto a nitrocellulose membrane, followed by immunoblotting with anti-pUL6, anti-VP22a, or anti-VP5 antibodies. Bound immunoglobulin was revealed by enhanced chemiluminescence.
Given the absence of a scaffold/portal interaction in cells infected with vJB30, we predicted that, as in the case of UL6 null viruses, genomic DNA would not be cleaved from concatameric DNA. To test this possibility, viral DNA was purified from cells infected with wild-type HSV-1(F), UL6 null virus, vJB30, vJB30R, or vJB31. The purified DNA was then digested with BamHI, transferred to a nylon membrane, and probed with radiolabeled DNA from HSV-1 genomic ends. The results are shown in Fig. 6. In cells infected with wild-type virus, the viral cleavage and packaging machinery cleaves sequences corresponding to the BamHI S-P fragment, yielding, upon digestion with BamHI, the BamHI P fragment. This was observed in lanes containing DNA from HSV-1(F)-, vJB30R-, and, to a lesser extent, vJB31(W596A)-infected cells. In contrast to these results, the BamHI P fragment was not observed in BamHI-digested DNA from cells infected with the UL6 null virus or vJB30(W27A). We concluded that the pUL26 W27A mutation precludes DNA cleavage, whereas the W596A mutation does not. Thus, the failure to cleave DNA correlates with the inability of vJB30(W27A) to incorporate portal efficiently into capsids and to propagate in CV1 cells.
FIG. 6.
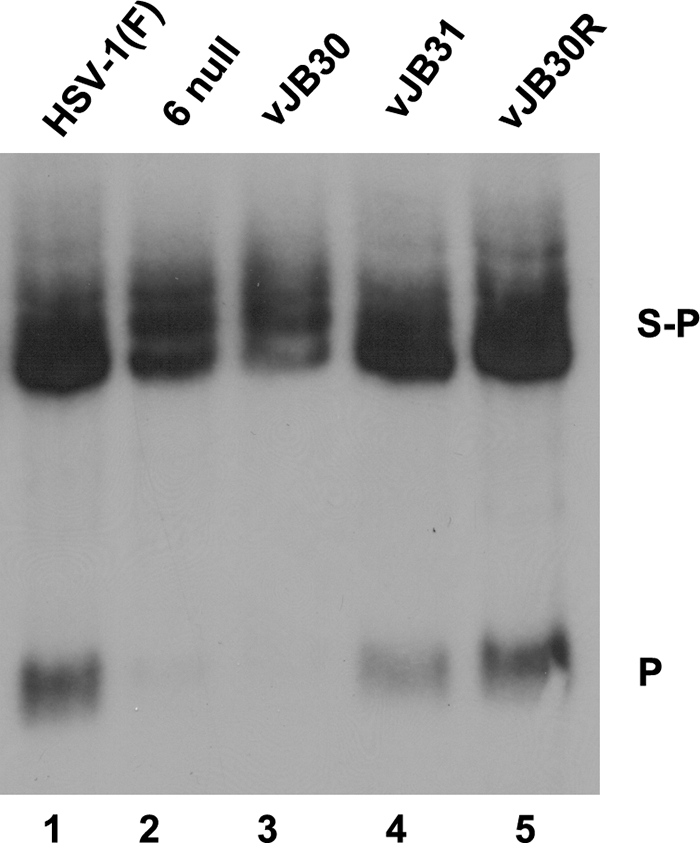
Southern blotting of viral genomes digested with BamHI. CV1 cells (2 × 106) were infected with 5 PFU/cell of HSV-1(F), UL6 null, JB30(W27A), vJB31(W596A), or vJB30R. Fifteen hours after infection, total nuclear DNA was extracted, digested with BamHI, transferred to a nylon membrane (0.45 μM), and hybridized with radiolabeled BamHI P fragment of HSV-1(F) DNA. Bound probe DNA was revealed by autoradiography. The positions of the BamHI S-P and P fragments are indicated on the right.
DISCUSSION
These data indicate that the first 18 and last 36 amino acids of pUL6 are dispensable for interaction with scaffold proteins, as assessed by immunoprecipitation, whereas the rest of the protein is required for this interaction. This observation suggests that UL6 must remain largely intact to interact with scaffold proteins and, by extension, to support viral replication. We also cannot exclude the possibility that one or more of these mutations preclude pUL6-pUL6 interactions and thereby preclude the formation of functional portals.
The relative positions of most of the tryptophan residues in pUL6 are conserved in portal proteins of other alphaherpesviruses (Fig. 7). In the current study, all of these tryptophan resides were found to be essential for interaction with scaffold protein in the absence of other viral proteins, suggesting that they are important to maintain the optimal conformation of the portal. In infected cells, however, W596A was tolerated, so that viral replication, incorporation of the portal into capsids, and viral-DNA cleavage and packaging occurred normally. One interpretation of these results is that our in vitro coimmunoprecipitation assay is a more stringent test of the scaffold-portal interaction, whereas in infected cells other functions compensate for the mutation's effects on the interaction. Such compensating functions might include the proper ratio of VP22a to VP21 in infected cells versus those expressing pUL26 and VP22a transiently, high protein concentrations in assembling capsids that might compensate for low affinity between mutant portal and scaffold proteins, interactions with other capsid proteins that fortify the scaffold-portal interaction, or virus-encoded or virus-induced chaperone functions that augment folding of mutant pUL6 to augment proper portal conformation. It is noteworthy that of the tryptophan residues within pUL6, W596 is the least highly conserved (Fig. 7, W7), and it is completely absent from the portal proteins of pseudorabies virus, bovine herpesviruses I and V, equine herpesvirus 1, gallid herpesviruses 1 to 3, and circopethecine herpesvirus 2.
Acknowledgments
These studies were supported by R01 grant GM50740 from the National Institutes of Health.
We thank Elizabeth Wills for reading the manuscript.
Footnotes
Published ahead of print on 9 September 2009.
REFERENCES
- 1.Baker, T. S., W. W. Newcomb, F. P. Booy, J. C. Brown, and A. C. Steven. 1990. Three-dimensional structures of maturable and abortive capsids of equine herpesvirus 1 from cryoelectron microscopy. J. Virol. 64:563-573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cardone, G., D. C. Winkler, B. L. Trus, N. Cheng, J. E. Heuser, W. W. Newcomb, J. C. Brown, and A. C. Steven. 2007. Visualization of the herpes simplex virus portal in situ by cryo-electron tomography. Virology 361:426-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davison, A. J., and J. E. Scott. 1986. The complete DNA sequence of varicella-zoster virus. J. Gen. Virol. 67:1759-1816. [DOI] [PubMed] [Google Scholar]
- 4.Davison, M. D., F. J. Rixon, and A. J. Davison. 1992. Identification of genes encoding two capsid proteins (VP24 and VP26) of herpes simplex type 1. J. Gen. Virol. 73:2709-2713. [DOI] [PubMed] [Google Scholar]
- 5.Deckman, I. C., M. Hagen, and P. J. McCann III. 1992. Herpes simplex virus type 1 protease expressed in Eschericia coli exhibits autoprocessing and specific cleavage of the ICP35 assembly protein. J. Virol. 66:7362-7367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Delhon, G., M. P. Moraes, Z. Lu, C. L. Afonso, E. F. Flores, R. Weiblen, G. F. Kutish, and D. L. Rock. 2003. Genome of bovine herpesvirus 5. J. Virol. 77:10339-10347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Desai, P., and S. Person. 1999. Second site mutations in the N-terminus of the major capsid protein (VP5) overcome a block at the maturation cleavage site of the capsid scaffold proteins of herpes simplex virus type 1. Virology 261:357-366. [DOI] [PubMed] [Google Scholar]
- 8.DiIanni, C. L., D. A. Drier, I. C. Deckman, P. J. McCann, F. Liu, B. Roizman, R. J. Colonno, and M. G. Cordingley. 1993. Identification of the herpes simplex virus-1 protease cleavage sites by direct sequence analysis of autoproteolytic cleavage products. J. Biol. Chem. 268:2048-2051. [PubMed] [Google Scholar]
- 9.DiIanni, C. L., C. Mapelli, D. A. Drier, J. Tsao, S. Natarajan, D. Riexinger, S. M. Festin, M. Bolgar, G. Yamanaka, and S. P. Weinheimer. 1993. In vitro activity of the herpes simplex virus type 1 protease with peptide substrates. J. Biol. Chem. 268:25449-25454. [PubMed] [Google Scholar]
- 10.Dolan, A., F. E. Jamieson, C. Cunningham, B. C. Barnett, and D. J. McGeoch. 1998. The genome of herpes simplex virus type 2. J. Virol. 72:2010-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ejercito, P. M., E. D. Kieff, and B. Roizman. 1968. Characterization of herpes simplex virus strains differing in their effects on social behavior of infected cells. J. Gen. Virol. 2:357-364. [DOI] [PubMed] [Google Scholar]
- 12.Gibson, W., and B. Roizman. 1972. Proteins specified by herpes simplex virus VIII. Characterization and composition of multiple capsid forms of subtypes 1 and 2. J. Virol. 10:1044-1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heymann, J. B., N. Cheng, W. W. Newcomb, B. L. Trus, J. C. Brown, and A. C. Steven. 2003. Dynamics of herpes simplex virus capsid maturation visualized by time-lapse cryo-electron microscopy. Nat. Struct. Biol. 10:334-341. [DOI] [PubMed] [Google Scholar]
- 14.Hong, Z., M. Beaudet-Miller, J. Durkin, R. Zhang, and A. D. Kwong. 1996. Identification of a minimal hydrophobic domain in the herpes simplex virus type 1 scaffolding protein which is required for interaction with the major capsid protein. J. Virol. 70:533-540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huffman, J. B., W. W. Newcomb, J. C. Brown, and F. L. Homa. 2008. Amino acids 143 to 150 of the herpes simplex virus type 1 scaffold protein are required for the formation of portal-containing capsids. J. Virol. 82:6778-6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klupp, B. G., C. J. Hengartner, T. C. Mettenleiter, and L. W. Enquist. 2004. Complete, annotated sequence of the pseudorabies virus genome. J. Virol. 78:424-440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu, F., and B. Roizman. 1991. The promoter, transcriptional unit, and coding sequences of herpes simplex virus 1 family 35 proteins are contained within and in frame with the UL26 open reading frame. J. Virol. 65:206-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McGeoch, D. J., M. A. Dalrymple, A. J. Davison, A. Dolan, M. C. Frame, D. McNab, L. J. Perry, J. E. Scott, and P. Taylor. 1988. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 69:1531-1574. [DOI] [PubMed] [Google Scholar]
- 19.Nellissery, J. K., R. Szczepaniak, C. Lamberti, and S. K. Weller. 2007. A putative leucine zipper within the herpes simplex virus type 1 UL6 protein is required for portal ring formation. J. Virol. 81:8868-8877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Newcomb, W. W., and J. C. Brown. 1989. Use of Ar+ plasma etching to localize structural proteins in the capsid of herpes simplex virus. J. Virol. 63:4697-4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Newcomb, W. W., and J. C. Brown. 1991. Structure of the herpes simplex virus capsid: effects of extraction with guanidine hydrochloride and partial reconstitution of extracted capsids. J. Virol. 65:613-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newcomb, W. W., F. L. Homa, D. R. Thomsen, B. L. Trus, N. Cheng, A. Steven, F. Booy, and J. C. Brown. 1999. Assembly of the herpes simplex virus procapsid from purified components and identification of small complexes containing the major capsid and scaffolding proteins. J. Virol. 73:4239-4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Newcomb, W. W., R. M. Juhas, D. R. Thomsen, F. L. Homa, A. D. Burch, S. K. Weller, and J. C. Brown. 2001. The UL6 gene product forms the portal for entry of DNA into the herpes simplex virus capsid. J. Virol. 75:10923-10932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Newcomb, W. W., B. L. Trus, N. Cheng, A. C. Steven, A. K. Sheaffer, D. J. Tenney, S. K. Weller, and J. C. Brown. 2000. Isolation of herpes simplex virus procapsids from cells infected with a protease-deficient mutant virus. J. Virol. 74:1663-1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Newcomb, W. W., F. L. Homa, and J. C. Brown. 2005. Involvement of the portal at an early step in herpes simplex virus capsid assembly. J. Virol. 79:10540-10546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nicholson, P., C. Addison, A. M. Cross, J. Kennard, V. G. Preston, and F. J. Rixon. 1994. Localization of the herpes simplex virus type 1 major capsid protein VP5 to the cell nucleus requires the abundant scaffolding protein VP22a. J. Gen. Virol. 75:1091-1099. [DOI] [PubMed] [Google Scholar]
- 27.Patel, A. H., F. J. Rixon, C. Cunningham, and A. J. Davison. 1996. Isolation and characterization of herpes simplex virus type 1 mutants defective in the UL6 gene. Virology 217:111-123. [DOI] [PubMed] [Google Scholar]
- 28.Person, S., S. Laquerre, P. Desai, and J. Hempel. 1993. Herpes simplex virus type 1 capsid protein, VP21, originates within the UL26 open reading frame. J. Gen. Virol. 74:2269-2273. [DOI] [PubMed] [Google Scholar]
- 29.Rixon, F. J., A. M. Cross, C. Addison, and V. G. Preston. 1988. The products of herpes simplex virus type 1 gene UL26 which are involved in DNA packaging are strongly associated with empty but not with full capsids. J. Gen. Virol. 69:2879-2891. [DOI] [PubMed] [Google Scholar]
- 30.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 31.Schwyzer, M., and M. Ackermann. 1996. Molecular virology of ruminant herpesviruses. Vet. Microbiol. 53:17-29. [DOI] [PubMed] [Google Scholar]
- 32.Sheaffer, A. K., W. W. Newcomb, J. C. Brown, M. Gao, S. K. Weller, and D. J. Tenney. 2000. Evidence for controlled incorporation of herpes simplex virus type 1 UL26 protease into capsids. J. Virol. 74:6838-6848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singer, G. P., W. W. Newcomb, D. R. Thomsen, F. L. Homa, and J. C. Brown. 2005. Identification of a region in the herpes simplex virus scaffolding protein required for interaction with the portal. J. Virol. 79:132-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanaka, M., H. Kagawa, Y. Yamanashi, T. Sata, and Y. Kawaguchi. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J. Virol. 77:1382-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Telford, E. A., M. S. Watson, K. McBride, and A. J. Davison. 1992. The DNA sequence of equine herpesvirus-1. Virology 189:304-316. [DOI] [PubMed] [Google Scholar]
- 36.Telford, E. A., M. S. Watson, J. Perry, A. A. Cullinane, and A. J. Davison. 1998. The DNA sequence of equine herpesvirus-4. J. Gen. Virol. 79:1197-1203. [DOI] [PubMed] [Google Scholar]
- 37.Tischer, B. K., J. von Einem, B. Kaufer, and N. Osterrieder. 2006. Two-step RED-mediated recombination for versatile high-efficiency markerless DNA manipulation in Eschericia coli. BioTechniques 40:191-197. [DOI] [PubMed] [Google Scholar]
- 38.Trus, B. L., F. P. Booy, W. W. Newcomb, J. C. Brown, F. L. Homa, D. R. Thomsen, and A. C. Steven. 1996. The herpes simplex virus procapsid: structure, conformational changes upon maturation, and roles of the triplex proteins VP19C and VP23 in assembly. J. Mol. Biol. 263:447-462. [DOI] [PubMed] [Google Scholar]
- 39.Trus, B. L., N. Cheng, W. W. Newcomb, F. L. Homa, J. C. Brown, and A. C. Steven. 2004. Structure and polymorphism of the UL6 portal protein of herpes simplex virus type 1. J. Virol. 78:12668-12671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Warner, S. C., G. Chytrova, P. Desai, and S. Person. 2001. Mutations in the N-terminus of VP5 alter its interaction with the scaffold proteins of herpes simplex virus type 1. Virology 284:308-316. [DOI] [PubMed] [Google Scholar]
- 41.Warner, S. C., P. Desai, and S. Person. 2000. Second-site mutations encoding residues 34 and 78 of the major capsid protein (VP5) of herpes simplex virus type 1 are important for overcoming a blocked maturation cleavage site of the capsid scaffold proteins. Virology 278:217-226. [DOI] [PubMed] [Google Scholar]
- 42.Weinheimer, S. P., P. J. McCann III, D. R. O'Boyle II, J. T. Stevens, B. A. Boyd, D. A. Drier, G. A. Yamanaka, C. L. Dilanni, I. C. Deckman, and M. G. Cordingly. 1993. Autoproteolysis of herpes simplex virus type 1 protease releases an active catalytic domain found in intermediate capsid particles. J. Virol. 67:5813-5822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang, K., F. Homa, and J. D. Baines. 2007. Putative terminase subunits of herpes simplex virus 1 form a complex in the cytoplasm and interact with portal protein in the nucleus. J. Virol. 81:6419-6433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang, K., and J. D. Baines. 2008. Domain within herpes simplex virus 1 scaffold proteins required for interaction with portal protein in infected cells and incorporation of the portal vertex into capsids. J. Virol. 82:5021-5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang, K., and J. D. Baines. 2009. Proline and tyrosine residues in scaffold proteins of herpes simplex virus 1 critical to the interaction with portal protein and its incorporation into capsids. J. Virol. 83:8076-8081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang, K., E. Wills, and J. D. Baines. 2009. The putative leucine zipper of the UL6-encoded portal protein of herpes simplex virus 1 is necessary for interaction with pUL15 and pUL28 and their association with capsids. J. Virol. 83:4557-4564. [DOI] [PMC free article] [PubMed] [Google Scholar]